 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic calixarene capsules: the evolution of controlled assembly
Piotr P.
Cholewa
and
Scott J.
Dalgarno
*
Institute of Chemical Sciences, Heriot – Watt University, Riccarton, Edinburgh, EH14 4AS, Scotland, UK. E-mail: S.J.Dalgarno@hw.ac.uk; Fax: +44 (0)131 451 3180; Tel: +44 (0)131 451 8025
First published on 27th February 2014
Abstract
Calixarenes are versatile cyclic host molecules that have been utilised widely across supramolecular chemistry for a variety of reasons. As the title suggests, the main thrust of this Highlight is the metal-directed assembly of calixarene (as well as other related host) capsules. As an introduction for the non-specialist, this article first describes some design strategies that have been successfully employed in the target synthesis of porous materials. The conformational features of calixarenes are then introduced prior to some developments in their (and other cyclic host) functionalisation. Finally we describe the application of the aforementioned design strategies in metal-directed assembly to afford target capsules, highlighting recent developments in the field with p-carboxylatocalix[4 and 5]arenes.
 Piotr Przemysław Cholewa | Piotr Przemysław Cholewa was born in Wroclaw, Poland in 1983. He obtained his MChem at Heriot-Watt University in 2009 under the supervision of Professor Martin J. Paterson. He is currently undertaking a PhD in the Dalgarno research group at Heriot-Watt University, working on controlled self-assembly of supramolecular architectures using functionalised calix[4]arenes as building units. |
 Scott John Dalgarno | Scott John Dalgarno was born in Aberdeen, Scotland in 1978. He obtained his MChem and PhD at The University of Leeds, working with Colin Raston and Michaele Hardie on molecular capsules and coordination networks. Following this he undertook a postdoctoral position with Jerry Atwood, working on the self- and metal-directed assembly of pyrogallol[4]arenes. In 2007 he was appointed a Lecturer at Heriot-Watt University, and in 2010 was promoted to Reader. His research focuses broadly on supramolecular coordination chemistry, as well as self-assembly and host–guest templation. |
Introduction: common assembly strategies for the targeted formation of porous materials
The past few decades have witnessed rapid technology development that has moved hand-in-hand with new challenges which are driven by the demand for novel solutions to everyday problems; in many cases this is becoming the major driving force behind targeted scientific research and it is certainly no different when considering the design of porous materials (PMs). Ever since zeolites found application in industry (due to their porosity and thermal stability) there has been a constant pursuit of the design and further improvement of PMs.1 The volume of publications focusing on the design and modification of PMs grows larger each year, with a number reporting on the application of these materials in chemistry (e.g. catalysis), biology (e.g. drug delivery), separation and gas storage.2 Various design strategies have emerged with a view to synthesising target PMs. One of the more recent examples involves condensation reactions using light materials containing hydrogen, carbon, oxygen and boron (e.g. reaction between phenyl diboronic acid and hexahydroxy-triphenylene). These reactions are under thermodynamic control and afford extended structures called covalent organic frameworks (COFs).3 These rigid crystalline structures contain high surface areas, are held together by strong covalent bonds between B, C and O atoms, and as a result of this connectivity exhibit relatively good thermal stability. There are also means of synthesising COFs under kinetic control, with the resulting materials also displaying good thermal stabilities and large surface areas.4 The pore sizes in some reported COFs are comparable to those found in zeolites, rendering them interesting in terms of industrial application. There are numerous other examples of COFs in the literature where similar approaches have been utilised in synthesis.5More often PM design takes a different approach, whereby additional components such as metal ions are used to direct assembly. The role of the metal ions is to act as ‘joints’ that connect lighter materials (linkers) to form extended networks. By choosing specific metal ions and terminal ligands one can have control over the number of linkers coordinated to the metal as well as their relative orientation, ultimately governing the connectivity and dimensionality of the network. Control can be further enhanced via synthetic means by pre-organising an appropriate metal complex, in which certain sites around the coordination sphere are restricted, leaving only desired positions available to undergo bond formation with functional groups of the linker. This strategy is known as the ‘directional bonding’ approach.
In order to maximise the rate of bond formation between the metal ion and the linker there is a set of functional groups used which are known to form stable coordinate-covalent bonds; examples of these are pyridines and carboxylates. There are numerous examples in the literature where authors construct networks of varying dimensionality by selection of specific metal ions, with the resulting materials often referred to as coordination polymers (CPs). Based on the connectivity of these networks there are a range of 1D6 and 2D7 CPs (Fig. 1A and B respectively). A similar approach is utilised when constructing 3D networks; by choosing an appropriate metal ion and a ditopic linker with functional groups located on both ends, one can synthesise a 3D grid as shown in Fig. 1C.8 Although this is the case, the use of monodentate ditopic linkers (e.g. 4,4′-bipyridine) makes achieving absolute control over the assembly process somewhat difficult. There is a degree of flexibility in the binding of monodentate ligands within the coordination sphere (making the assembly less predictable), and as a result this can lead to formation of undesired topologies exhibiting non-porous character.9
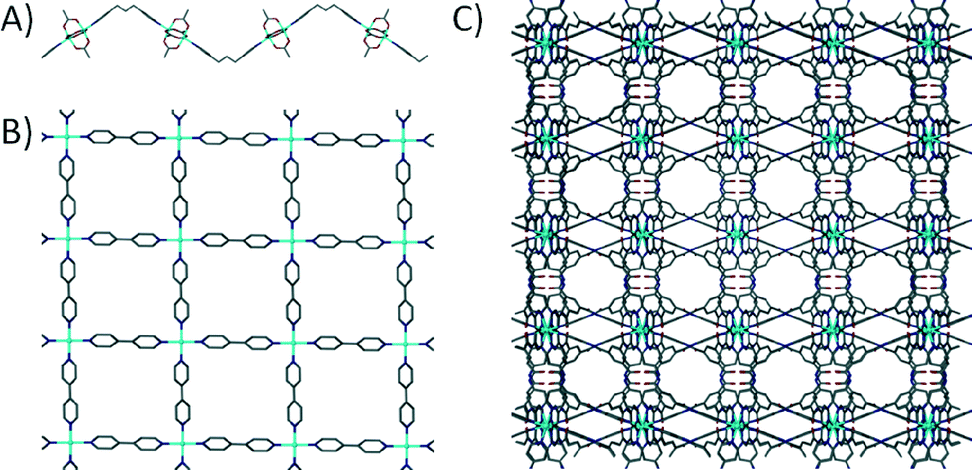 | ||
| Fig. 1 Example 1D6a (A), 2D7d (B) and 3D8f (C) coordination polymers with hydrogen atoms and solvent molecules omitted for clarity. | ||
In order to decrease flexibility and increase control in the design of PMs, linkers with functional groups binding to the metal ion in a multidentate fashion (e.g. carboxylates and phenanthrolines) have been employed. The reason for their superiority is the higher affinity for binding metal ions (chelation), a result being that they are locked within rigid clusters. These rigid inorganic clusters, due to their repetitiveness in the synthesised materials, were termed secondary building units (SBUs).10 Each SBU contains extension points through which they are connected to symmetry equivalent clusters. By targeting a specific SBU one can obtain a cluster with extension points orientated in a chosen direction, making the design and assembly of PMs more predictable. The most commonly occurring SBUs in PMs are: 1) a square ‘paddlewheel’ 2) a tetrahedral cluster 3) an octahedral cluster and 4) a triangular prism (Fig. 2). It has been shown that different framework topologies can be obtained depending on the SBU employed. These PMs are called metal–organic frameworks (MOFs) to denote the inorganic part (often SBU), the organic linker and the high dimensionality of the material (which exhibits porosity). There are many examples in which metal ions and a series of carboxylates form square ‘paddlewheel’ SBUs11 that dictate the topology of the resulting MOFs.12 The second commonly observed SBU in MOFs has extension points based on octahedral geometry. This enables construction of highly ordered systems and allows for precise alteration of porosity by synthetic modification of the organic linkers.13 There are also many other examples of SBUs adopting shapes different to those mentioned here, a number of which are emerging as useful directing centres in the design of new MOFs.14
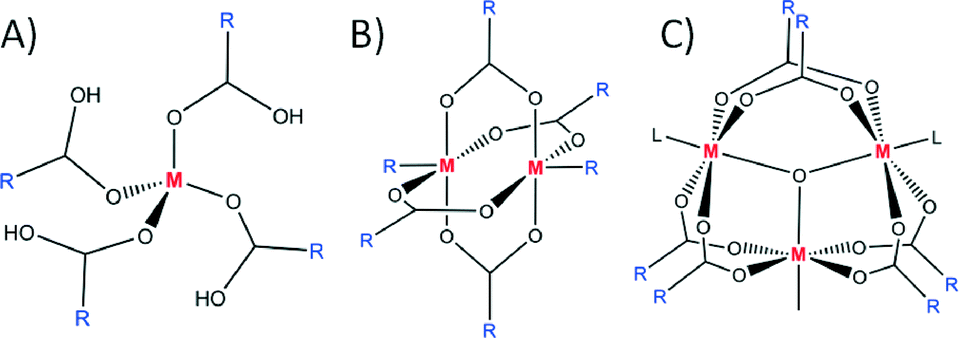 | ||
| Fig. 2 Most commonly occurring SBUs: tetrahedral (A), octahedral (B) and triangular prismatic (C). | ||
Along with the growing interest in the design and construction of PMs, as well as the fact that their topologies rely on the interconnectivity of the network, supramolecular/coordination chemists have also become interested in the design of discrete molecular containers known as metal–organic polyhedra (MOP). The ability to control polyhedron geometry and size has been realised, allowing for the construction of MOPs in a predictable fashion. These systems are potentially very useful in applications such as separation and gas storage, and the ability to design discrete architectures allows for the introduction of specific features; an example would be tailored pore size that either permits or precludes guest uptake within a molecular container based purely on size considerations. As in the case of PMs, extensive research in this field has afforded various strategies for the rational design and construction of MOPs. Since the research areas covering PMs (especially MOFs) and MOPs are closely related, one can find that similar strategies are often applied in the synthesis of these materials. With growing complexity of the components used, along with the topologies of new molecular structures, a classification system has been devised to identify structural similarities, which are not always apparent. The classification system is based on principles of solid geometry, whereby any given polyhedron can be assigned according to its symmetry to either a Platonic or Archimedean Solid. Platonic solids consist of five convex polyhedra, in which each of its faces is made out of a one type of polygon; these consist of the tetrahedron (Td symmetry), the cube and octahedron (Oh symmetry), and the dodecahedron and icosahedron (Ih symmetry). The Archimedean Solids are also convex polyhedra, but their faces consist of two or more regular polygons.15
The work that pioneered MOP design started with construction of 2D discrete molecular structures relying on the ‘directional bonding’ approach. Early examples included the preparation of precursors based on square-planar Pt and Pd complexes with ligands blocking two cis-coordination sites, which when mixed with linear ligands such as 4,4′-bipy, form molecular squares with four metal centres connected by the specifically chosen linker.16 Similar concepts were applied in order to construct differently shaped 2D discrete structures by clipping a pre-organised organic molecule to two trans-Pt complexes. Based on the shape of the molecular clip used the angle between the Pt binding sites available for subsequent bond formation with a linker can be controlled, allowing for synthesis of 2D molecular squares,17 triangles,18 and hexagons (Fig. 3).19 Now the array of linkers and molecular clips employed has been significantly expanded and applied to a variety of metals, yielding a wide range of 2D architectures.20
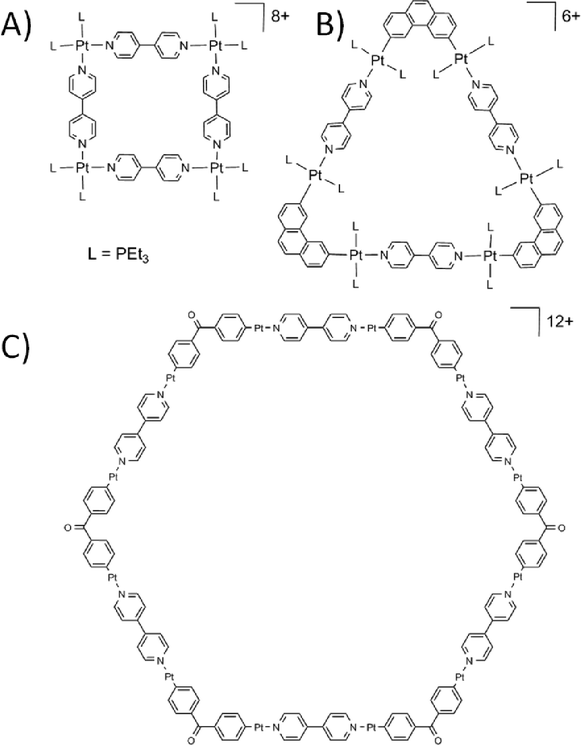 | ||
| Fig. 3 Examples of molecular squares17 (A), triangles18 (B) and hexagons19 (C) constructed using “directional bonding” approach. trans-Pt ligands omitted for clarity in C. | ||
In order to increase the dimensionality and complexity of the structures one can vary the number of available binding sites on the metal and/or introduce more functional groups to the linker. Various MOPs become accessible by assembling pre-organised precursors with synthetically modified linkers.20 The design of these discrete 3D structures is based on angular complementarity of all building blocks. As a result the components form thermodynamically favoured architectures. It has been demonstrated that the same metal precursor can be used to construct differently sized MOPs by changing the angle between the functional groups in a linker, i.e. the bite angle of the ligand coordinating to metal ions (Fig. 4). It has also been shown that the assembly of Pd-based precursors with dipyridyl linkers (bite angle of 90°) results in a cube consisting of six Pd metal ions and twelve linkers.21 The use of a linker with a larger bite angle (127°) results in formation of a molecular sphere possessing cuboctahedral symmetry; this contains thirty six components consisting of twelve Pd containing precursors and twenty four linkers (M12L24).22
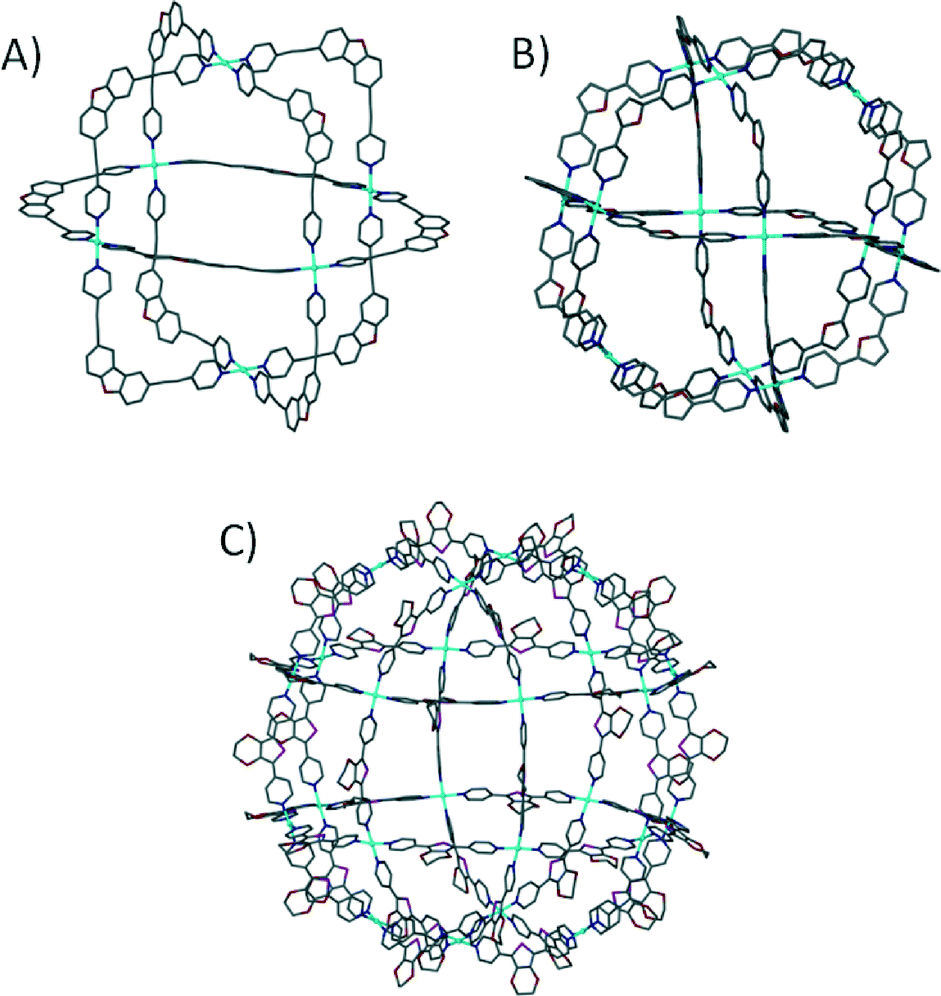 | ||
| Fig. 4 Examples of MOPs designed and constructed using the ‘directional bonding’ approach. The MOPs shown adopt cubic21 (A), cuboctahedral22 (B) and rhombicuboctahedral23a (C) topologies. | ||
A further increase in the linker bite angle (135°) yields the thermodynamically favoured architecture which is an even larger near-spherical MOP based on a rhombicuboctahedral symmetry; this contains seventy two components (M24L48).23 In principle larger MOP formation should be possible by continually increasing the bite angle (up to 180°, when there is no curvature and 2D7a–c CPs form), but this is still to be realised. There are a number of other examples in which similar approaches have been utilised in constructing various molecular containers,24 but as is the case for MOFs, it is outside the scope of this highlight to comprehensively cover the area.
In relation to the above, it has also been demonstrated that one can construct molecular capsules using only two metal centres through rational design of the linker (shape, curvature and length).25 The use of terminal ligands (often chelates) to cap specific coordination sites around metal ions has been proven to be a successful method in controlling a) the directionality of metal centres and b) the topology of constructed architectures when using octahedral metal ions.26 MOPs can be also synthesised by designing linkers with rationally positioned chelating groups. One can assemble molecular cages with metal centres connected by three or more linkers by having the spacing and orientation of these functional groups appropriately pre-organised.27
The concept of using SBUs in the design and construction of PMs has not only found application in assembling MOFs, but has also been successfully used in the construction of various MOPs. However, as opposed to the strategy when designing MOFs, in order to obtain discrete molecular structures, SBUs often have to be truncated with terminal ligands to limit the connectivity. In the literature one can find an array of MOPs possessing symmetry based on Platonic and Archimedean Solids.28
Molecular capsules comprising cyclic hosts: self-assembly and covalent-organic synthesis
The design strategies for molecular capsule formation are not strictly limited to those discussed above. In the pursuit of versatile and directional linkers, a class of molecule emerged as perfect candidates for the construction of discrete molecular containers. This group of molecules consists of a variety of cyclic hosts, which due to their inherent bowl-shape, are ideal for use in the design of molecular capsules. They are synthesised via condensation reactions between readily available aromatic compounds and a series of different precursors that act as a source of bridging atoms. Based on the chosen arene and reaction conditions employed one can obtain a series of differently sized cyclic oligomers with varying functionality positioned across the molecular framework.29 A major advantage of these macrocycles is the relative ease with which each molecule can be modified, allowing for additional functionalisation and directional promotion of specific interactions and/or bond formation. In addition, synthetic modification of the framework generally allows one to change (and in some cases control) the degree of flexibility within these cyclic hosts.30 Variation in both the substituents present and the flexibility of the macrocycle can provide control over the host cavity size, and thus the internal volume of any resulting molecular capsule. The cyclic hosts highlighted here in the design of molecular capsules are shown in Fig. 5.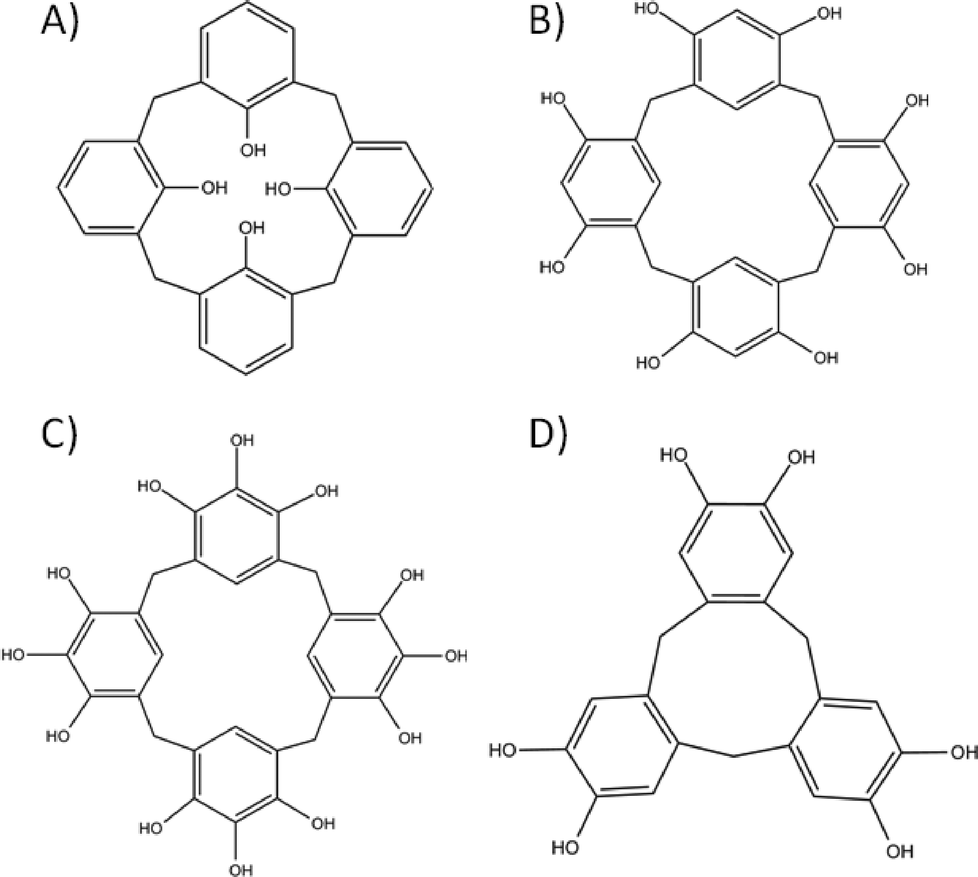 | ||
| Fig. 5 Commonly used macrocycles in supramolecular chemistry; calix[4]arene (A), resorcin[4]arene (B), pyrogallol[4]arene (C) and cyclotricatechylene (D). | ||
Calix[4]arene (Fig. 5A), synthesised from p-tert-butylphenol and formaldehyde with subsequent de-tert-butylation, adopts a cone conformation that has been exploited extensively in supramolecular chemistry. Removal of H-bonding interactions at the lower-rim by sequential or selective alkylation leads to a change in conformation; this results in a shift from the cone (cavity open) through partial cone (cavity open) to pinched cone (cavity closed) conformation. There are viable synthetic strategies to functionalise the para positions of the calix[4]arene aromatic rings, thereby allowing for introduction of different groups (e.g. sulfonato, nitro or carboxylato) or extension of the cavity (e.g. by addition of arenes). This therefore provides the supramolecular chemist access to an almost unlimited library of molecular building blocks.
Resorcin[4]arenes (Fig. 5B) also possess a cavity and these have been used extensively in molecular capsule formation. They are synthesised by condensation of resorcinol with a chosen aldehyde and contain hydroxyl groups at the meta positions rather than at the lower-rim; the para positions therefore remain available for synthetic modification. The resorcin[4]arene framework can be rigidified by the introduction of methylene bridges between the meta hydroxyl groups to afford macrocycles called cavitands. Pyrogallol[4]arenes (Fig. 5C) are structural analogues to resorcin[4]arenes but with one important difference, that being the presence of an additional hydroxyl group at the para position. This can be beneficial in chelating the macrocycle to a metal or in directing self-assembly (vide infra). The final group of cyclic hosts to be covered here are catechol-based cyclotricatechylenes (CTCs, Fig. 5D) and the related veratrole-based cycloctriveratrylenes (CTVs). They are made up of three arenes and possess a shallower and smaller cavity than the three types of macrocycle mentioned above. Their upper-rims are also available for synthetic modification in order to introduce functional groups that are desirable for directed assembly purposes.
The concept of using macrocycles as molecular building blocks in the construction of capsules dates back to the 1980s. This example reported the first synthesis of a covalent organic system (known as a cryptophane) from CTV sub-units (Fig. 6A).31 Various synthetic strategies have since been established for cryptophanes that allow for selective functionalisation at the upper-rim, with subsequent control of the linker length between the constituent CTV building blocks.32 The possibility of altering these topological features has made cryptophanes very useful in molecular encapsulation, recognition, and biosensing.33 A short time after the first reported cryptophanes, C-methyl-resorcin[4]arene-based cavitands were also used to form dimeric covalent organic capsules (Fig. 6B).34 Soon after this discovery it was realised that cavitands were useful for the construction of near-spherical capsules. The synthetic modification of the resorcin[4]arene framework and subsequent use yielded a range of dimeric covalent organic capsules.35
 | ||
| Fig. 6 Dimeric covalent-organic capsules constructed from CTV31 (A) and resorcin[4]arene-based cavitand34b (B) building blocks. | ||
Further pursuit of this research resulted in the synthesis of cyclic tetrameric and hexameric architectures which were found to undergo rearrangements to form bis- and tris-capsules respectively.36 Due to the shape of the resorcin[4]arene-based cavitands it was realised that larger spherical assemblies could be isolated. This theoretical concept came into being with the report of the first ‘superbowl’ container molecule comprising five cavitands,37 followed shortly by a report of a octahedral covalent organic capsule comprising six.38 Further advances have been made in simplifying and improving the synthesis, thereby facilitating the construction of a series of nanocapsules with (amongst others) tetrahedral, octahedral, square antiprismatic and rhombicuboctahedral topologies (Fig. 7).39
 | ||
| Fig. 7 Covalent-organic capsules adopting rhombicuboctahedral (A) and square antiprismatic (B) topologies, both of which are constructed from resorcin[4]arene-based cavitands.39a Adapted with permission from ref. 39a. Copyright (2006) American Chemical Society. | ||
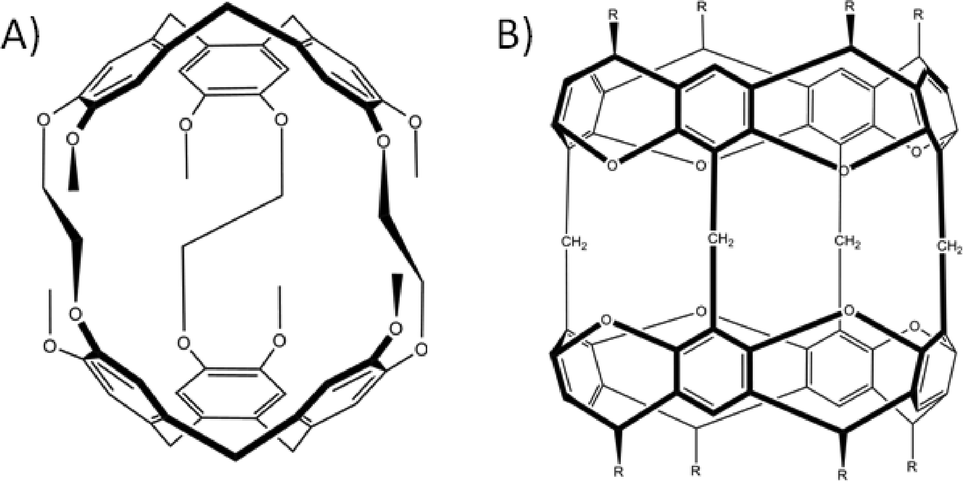
Despite the huge scope for upper-rim alteration there are, to date, few examples of near spherical covalent organic capsules synthesised using calix[4]arenes. The first reported dimeric capsule analogues date back to the end of the 1980s when it was demonstrated that synthetic modification of the calix[4]arene upper-rim could be used to link two macrocycles via one, two or four aliphatic bridges (Fig. 8A).40 There are also examples in which calixarenes have been linked by four ether or thioether bridges (Fig. 8B).41 It has also been demonstrated that flexible/hinged bis-calixarenes can arrange themselves to form capsules through encapsulation of an appropriately sized guest molecule.42 Notably, there is only one example of calix[6]arene used as a building block to construct a dimeric organic capsule, whereby two macrocycles are linked by six thioether bridges.43 This is due to increased conformational versatility upon moving to the calix[6]arene framework, in addition to the fact that the molecule forms a double-cone conformation, all of which impacts on potential capsule formation. Overall little has been reported in this area due to the poor solubility of the capsule assemblies relative to the aforementioned cavitand systems; this can be an insurmountable problem. However, in the quest to construct spherical covalent architectures scientists have also applied the strategy of mixing different pre-organised macrocycles. By introducing complementary functional groups to the respective frameworks they have been able to assemble them together to form hybrid capsules. For example it is possible to find literature examples of hybrid capsules comprising calix[4]arene and resorcin[4]arene sub-units.44
 | ||
| Fig. 8 Dimeric covalent organic capsules formed by the introduction of aliphatic (A)40 or thioether (B)41 groups to calix[4]arene para-positions. | ||
The discovery of non-covalent near-spherical architectures, spectacular capsules held together by many concerted interactions, came a little later than their covalent counterparts.45 In the early 90s a handful of groups reported the existence of large aggregates in solution that were held together by H-bonding between functional groups introduced to the calix[4]arene upper-rim.46 These initial reports were followed by others detailing new solution phase assemblies.47 However, it was not until the first single crystal X-ray structure of a dimeric tetraurea calix[4]arene capsule was reported that the formation of such assemblies became unambiguous (Fig. 9A).48 The field soon flourished, with more groups contributing to this burgeoning area of research.49 It became apparent that in order to successfully assemble these macrocycles one had to carry out complementary functionalisation of the upper-rim in order to promote concerted non-covalent interactions. These were crucial in stabilisation of the capsules, allowing them to remain intact in the solid state; single crystal X-ray diffraction studies became extremely important for structural verification and further understanding of the related host–guest chemistry. Resorcin[4]arenes and pyrogallol[4]arenes (Fig. 9B) were also shown to be very useful in the construction of dimeric capsule assemblies.50
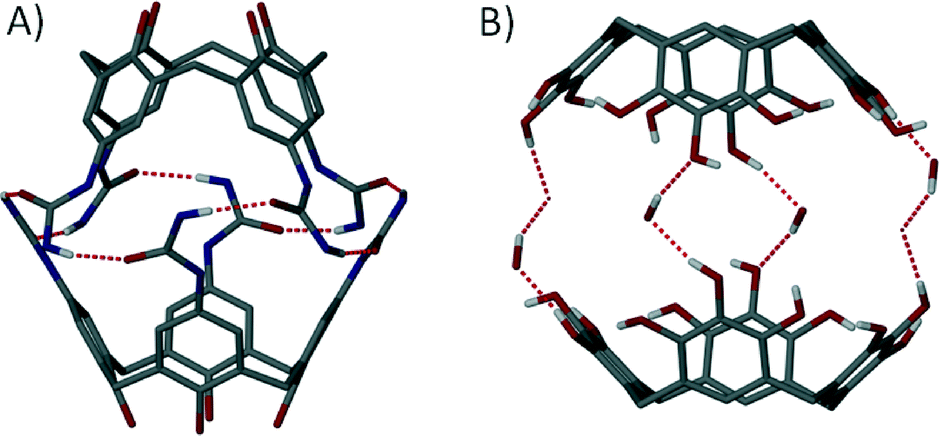 | ||
| Fig. 9 Dimeric capsules held together by non-covalent interactions (dashed lines) constructed from calix[4]arenes48 (A) and pyrogallol[4]arenes50f (B) with additional solvent shown in the latter. | ||
The hydroxyl groups located at the upper-rim were found to be ideal for promoting self-assembly of these molecules and a key development was the structural characterisation of the first nanometre scale hexameric resorcin[4]arene molecular capsule (Fig. 10A).51 This fascinating structure consisted of six resorcin[4]arenes and eight structural waters of crystallisation, all of which were held together by sixty concerted hydrogen bonds. This strategy was enhanced when a similar hexameric capsule was attained using pyrogallol[4]arene, which due to extra hydroxyl groups located at the upper-rim, was found to be held together by seventy two hydrogen bonds (Fig. 10B).52 Based on these studies, numerous reports have shown that cavitands that are synthetically pre-organised with H-bond donor and acceptor atoms strategically positioned at the upper-rim can maximise non-covalent interactions between the macrocycles and often the solvent/guest; these have also been shown to be viable sub-units in the formation of non-covalent capsules.53 Despite ready access to larger macrocycles (calix[n]arenes where n = 5,6,7,8…) there are few examples of dimeric non-covalent capsules using calix[5 and 6]arenes in the literature.54 As for the covalently linked calix[6]arene system described above, this scarcity is attributable to increased flexibility of the framework that is associated with the growing size of the macrocycle, rendering control over the assembly process significantly more challenging. There are also many elegant examples in the literature where the calix[n]arene lower-rim has been modified/selectively functionalised to facilitate the construction of discrete structures that can be covalent or non-covalent in nature;55 the breadth of literature associated with lower-rim alteration is vast and is thus not within the scope of this highlight.
 | ||
| Fig. 10 Non-covalent organic capsules constructed from resorcin[4]arene51 (A) and pyrogallol[4]arene52b (B). | ||
Molecular capsules comprising cyclic hosts: metal-directed assembly
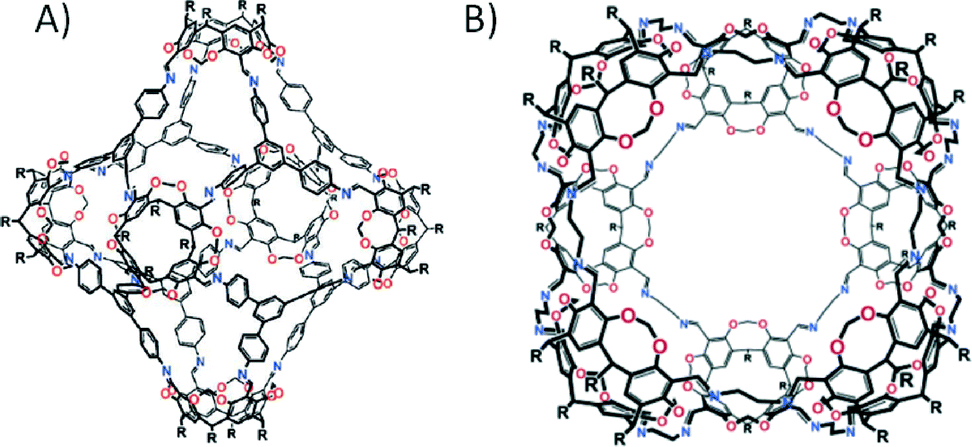
In the examples outlined above we have highlighted how strategies have evolved in comparatively recent times to facilitate the targeted design of architectures with desirable topologies. We have also shown how the approach used to construct MOFs and MOPs is based on coordination chemistry and the geometrical complementarity of the constituent sub-units. Finally we have shown how macrocyclic compounds can be successfully used in the targeted assembly of nanometre scale capsules held together by non-covalent interactions. The final topic of this highlight article brings all of these sub-topics together, focusing on the area of metal–organic calixarene capsules. The principles behind the design of porous materials have been widely utilised in this research as coordination chemistry can be directly applied to suitably functionalised building blocks.When considering the breadth of literature based on cryptophanes, the number of reports of metal–organic capsules constructed from CTCs and CTVs is surprisingly limited.56 One of the few examples involved introduction of a pyridyl moiety to the upper-rim of a CTV. Subsequent linking of two building blocks with cis-protected Pd(II) centres afforded the metal–organic capsule shown in Fig. 11A.57 The functional composition of CTC can be directly utilised in the construction of metal–organic capsules via deprotonation of the upper-rim hydroxyl groups, followed by the introduction of a metal salt or vanadyl ions; the result of this reaction is a tetrahedral cage comprising six metal centres and four CTCs (Fig. 11B).58 Two catecholates (from symmetry equivalent CTCs) coordinate to each metal centre in square planar fashion, and although this is an elegant example of metal-directed assembly, it was not the first time the inherent functionalisation of the upper-rim had been exploited in metal ion binding.
 | ||
| Fig. 11 Metal–organic capsules constructed from CTV/Pd(II) ion57 (A) and CTC/Cu(II) ion58a (B) combinations. | ||
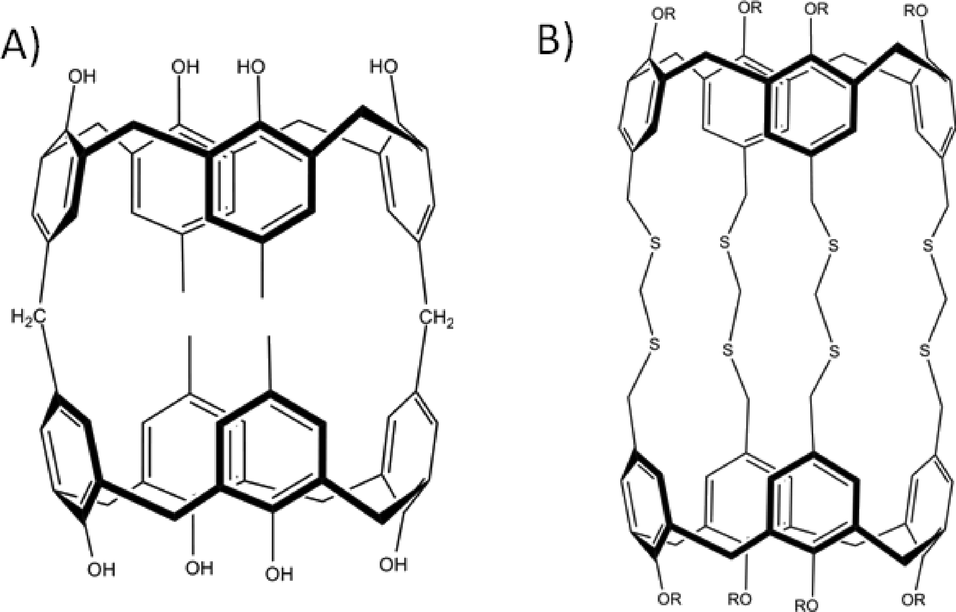
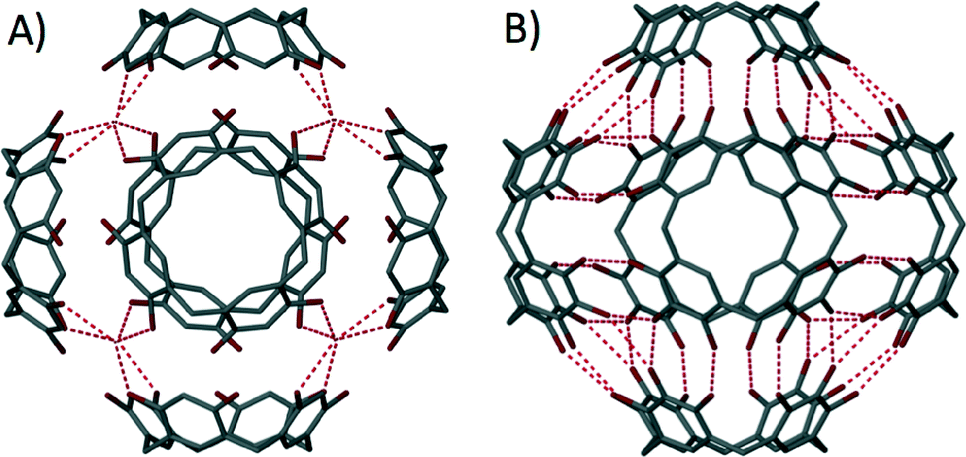
The first report that used upper-rim hydroxyl groups in this way involved the mixing of C-propylpyrogallol[4]arene with cupric nitrate in acetone and water.59 Structural analysis revealed the formation of a nanometre scale capsule based on an octahedral arrangement of pyrogallol[4]arene sub-units (Fig. 12). The assembly process involved removal of forty eight upper-rim hydrogens from the hydroxyl groups, with subsequent formation of ninety six new Cu–O bonds; the resulting assembly comprises six macrocycles and twenty four Cu(II) ions. Interestingly, the supramolecular architecture adopted is very closely related to the non-covalent hexameric capsule shown in Fig. 10,52b proving yet again that functionalised pyrogallol[4]arenes are viable components for the construction of molecular capsules. Metal-directed assembly with pyrogallol[4]arenes has also been demonstrated with Ga(III) ions, affording a metal–organic capsule that is of similar size to the Cu(II)-templated analogue, but that adopts a slightly different shape as shown in Fig. 12B.60 In this case the capsule conforms to a disordered “rugby-ball” consisting of six pyrogallol[4]arenes and twelve Ga(III) ions. Assembly requires removal of thirty six hydrogens from the hydroxyl groups, with concomitant formation of forty eight new Ga–O bonds. There are also examples where the pyrogallol[4]arene upper-rim hydroxyl groups undergo binding to metal ions to form dimeric capsules connected by a seam of eight metal (Fig. 13).61 More recently it has been shown that alteration of the reaction conditions employed allows for the synthesis of dimeric capsules from either Zn(II), Cu(II), Co(II) or Ni(II) ions.62
 | ||
| Fig. 12 Metal–organic capsules constructed from six pyrogallol[4]arenes and either twenty four Cu(II)59 (A) or twelve Ga(III)60 ions (B). | ||
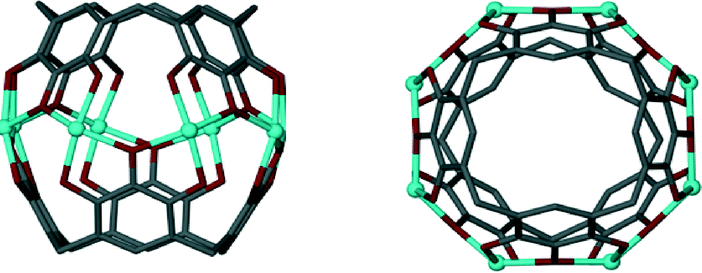 | ||
| Fig. 13 A dimeric metal–organic capsule constructed from two pyrogallol[4]arenes and eight transition metal ions (Zn(II), Cu(II), Co(II) or Ni(II)).61,62 | ||
With respect to the general modification to the cyclic host molecule framework, an advantage resorcin[4]arenes and the related cavitands have over pyrogallol[4]arenes is the presence of vacant positions at the upper-rim. This availability allows for the introduction of functional groups such as pyridyls, nitriles, chelates (e.g. dithiocarbamates) and carboxylic acids, a number of which have already been discussed with respect to their importance in the design of MOFs and MOPs. Early examples involved the functionalisation of these positions with nitrile groups and their subsequent use as monodentate ligands for coordination to cis-protected Pt(II) and Pd(II) ions.63 This afforded dimeric capsules consisting of two macrocycles and four metal centres as shown in Fig. 14A. Similar dimeric metal–organic capsules were reported when using pyridyl rather than nitrile functionalised cavitands.64 Synthetic variation in the length of the upper-rim appended pyridyl moiety was shown to provide concomitant control over the volume of the resulting capsule interior.65 Groups that can act as bidentate ligands were explored in order to further promote the formation of near-spherical architectures. A cavitand functionalised with a 2,2-bipyridine derivative, upon mixing with Ag(I) ions, assembled as a dimeric capsule with four metal centres, each with two chelating ligands coordinated in a tetrahedral fashion (Fig. 14B).66 Dithiocarbamate was also shown to act as a bidentate upper-rim metal binding site. Reaction of this ligand with Zn(II) or Cd(II) ions produced a trimeric capsule motif comprising six metal ions and three cavitands (Fig. 15A).67 Each of the metal centres in this assembly adopt five coordinate square pyramidal geometry, with two dithiocarbamates coordinated in a square plane and a pyridine ligand occupying each axial site. By changing the metal ion template to either Cu(II) or Au(III) the resulting assembly conforms to tetrahedral topology (Fig. 15B).67 These alternative metal–organic cages consist of eight square planar metal centres and four cavitands. There are also a handful of examples in the literature in which iminodiacetate moieties occupy the upper-rim cavitand positions. These functionalised cavitands were found to coordinate to metal ions (Co(II) and Fe(II)) via iminodiacetate groups in a tridentate fashion, resulting in a dimeric metal–organic capsule comprising two cavitands and four metal centres as shown in Fig. 15C.68
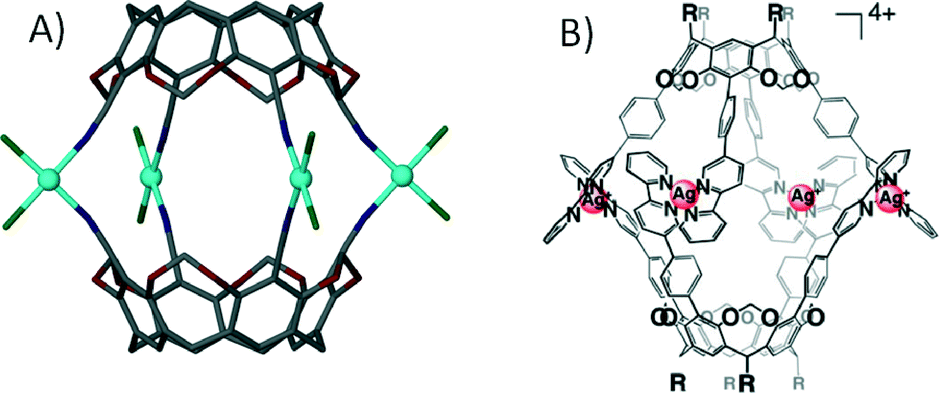 | ||
| Fig. 14 Dimeric metal–organic capsules constructed from functionalised63b (A)/extended (B) cavitands.66 | ||
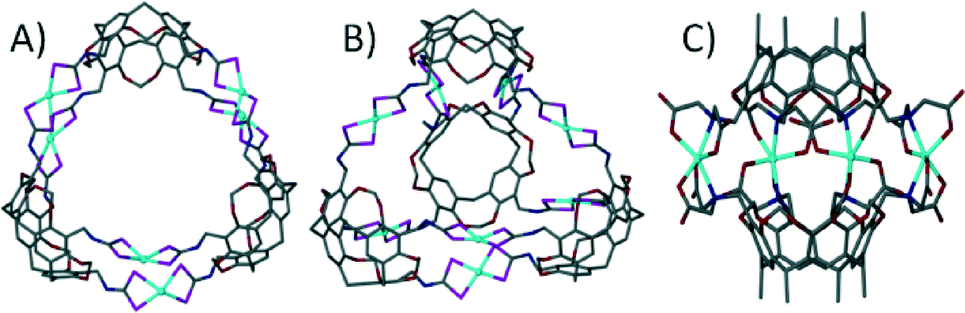 | ||
| Fig. 15 Utilisation of bidentate67a (A and B) and tridentate68b (C) functional groups in the design of cavitand-based metal–organic capsules. | ||
When one considers the importance of the benzoate moiety in the formation of SBUs, and thus in the design and construction of MOFs and MOPs, it is surprising that little has been reported on metal–organic capsule formation with calixarenes possessing upper-rim carboxylic acid functionality. This is especially true given that synthetic pathways for the introduction of upper-rim CO2H groups to the general macrocyclic framework are well established. One of the few examples of a metal–organic capsule constructed from a calix[4]arene containing carboxylates at the para-position beautifully illustrates the potential of these molecules as building blocks in the rational design of novel supramolecular architectures.69 Reaction of tetra-p-carboxylato-calix[4]arene with a pre-organised Rh(II) complex yielded a dimeric capsule with four dirhodium centres fastening two calixarenes together as shown in Fig. 16A. Each dirhodium complex comprises two formamidinates and two carboxylates, all of which bridge the two rhodium centres to form a paddlewheel structure, a SBU which (as we have shown above) is very important in the synthesis of MOFs and MOPs. Another example where the formation of an SBU dictates the outcome of an assembly is the reaction of upper-rim carboxylate functionalised cavitand with Zn(II) ions, which results in formation of a nanometre scale capsule comprising sixteen metal centres and six cavitands (Fig. 16B).70 Each of the dinuclear metal clusters has three-fold symmetry, and comprises two Zn(II) ions and three carboxylates that coordinate in a bridging fashion. All of the metal centres possess apical aquo ligands, of which two act as linear linkers between neighbouring capsules, producing a 1D coordination polymer of metal–organic calixarene capsules.
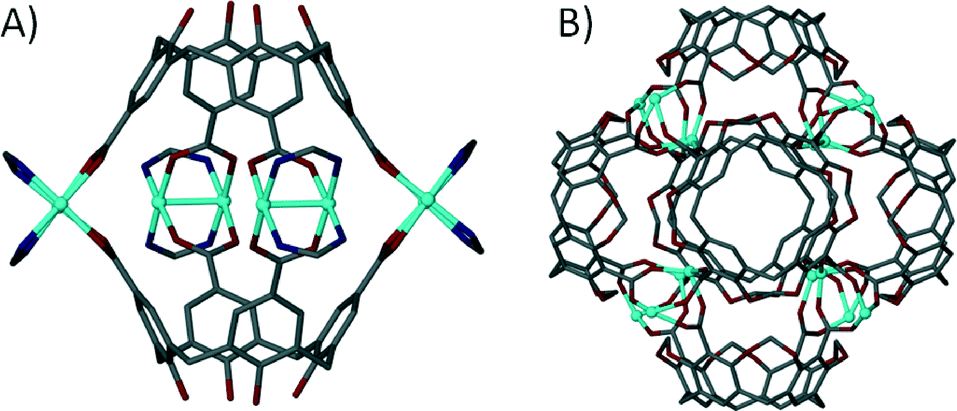 | ||
| Fig. 16 Dimeric (A) and hexameric (B) metal–organic capsules constructed from p-carboxylato functioanlised calix[4]arene69 and cavitand70 respectively. | ||
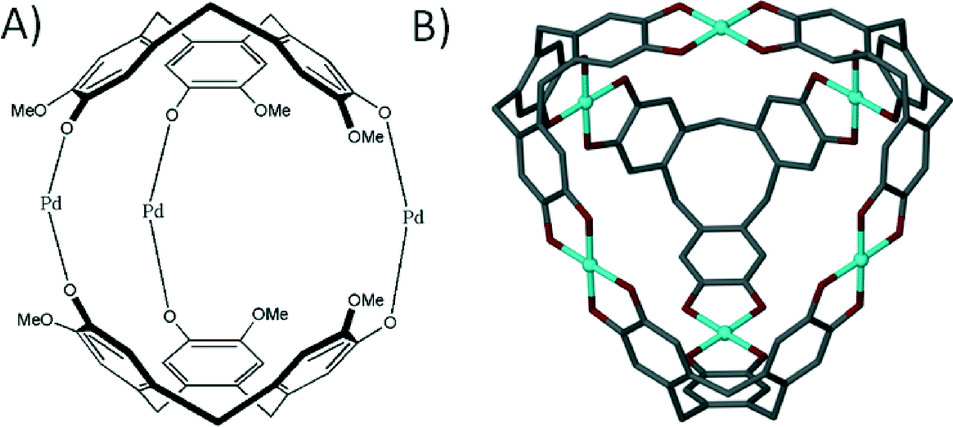
The ‘directional bonding’ approach can be also used to design calixarene-based capsules. One can construct different Platonic solids by choosing sub-units with complementary symmetry. This approach was utilised and elegantly demonstrated by reacting a C3-symmetric directing centre (a uranyl ion, UO22+) with a C4-symmetric ligand (p-carboxylatocalix[4]arene) to produce a metal–organic calixarene capsule conforming to octahedral topology (Fig. 17A).71
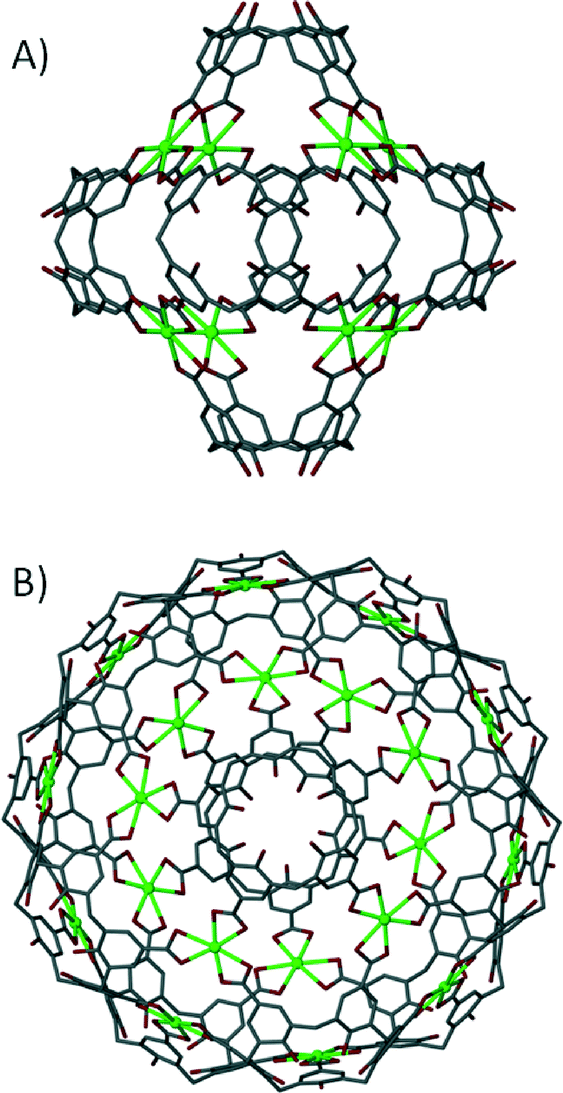 | ||
| Fig. 17 Metal–organic calixarene capsules adopting octahedral (A) and icosahedral (B) topologies.71 | ||
The capsule comprises six calixarenes and eight uranyl sub-units, bearing an overall negative charge of 8− which is counterbalanced by the presence of two fully protonated 1,4,7,10-tetraazacyclododecanes. The same strategy was employed to greatly enhance the size/volume of the capsule by using a C3-symmetric directing centre, but with a larger C5-symmetric ligand analogue (p-carboxylatocalix[5]arene). As a result of using a ligand of higher symmetry, twelve calixarenes and twenty uranyl ions form a capsule conforming to icosahedral topology with an overall negative charge of 20− that is counterbalanced by associated pyridinium ions (Fig. 17B).71
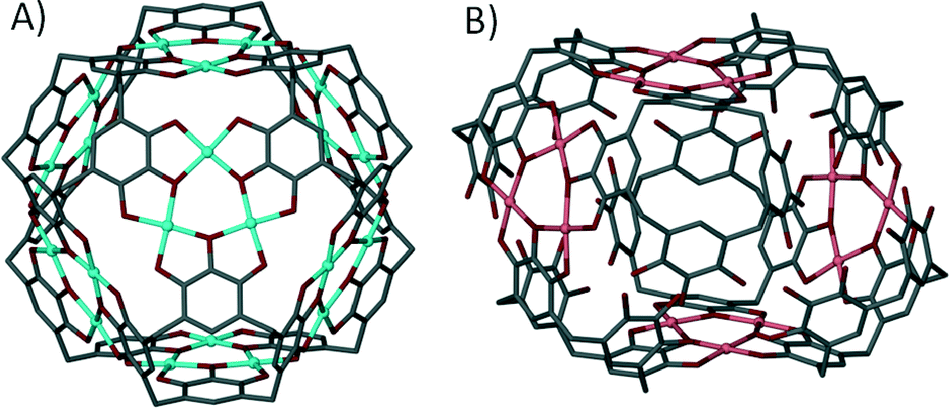
We have also been applying a ‘directional bonding’ approach in the design of metal–organic calixarene capsules. The strategy we employed is based on selective functionalisation of the calix[4]arene framework to pre-organise it as a sub-unit possessing desired shape and symmetry characteristics. The upper-rim is di-functionalised with carboxylic acid functionality for metal binding, whilst the lower-rim is di-O-alkylated to preserve the (albeit partial) cone conformation through the two remaining hydrogen bonding interactions; in this arrangement the molecular cleft remains open for guest occupation. To direct the bond formation we chose to restrict the coordination sites around the metal centres by introducing chelating ligands such as 1,10-phenanthroline (1,10-Phen) during the assembly process. Through the use of stoichiometric control we established reaction conditions that afforded dimeric metal–organic calixarene capsules. Mixing Cd(II) ions with di-p-carboxylatocalix[4]arene and 1,10-Phen results in the formation of a tilted capsule comprising two metal centres, two calixarenes and two chelates as shown in Fig. 18.72 The 1,10-Phen chelates restrict the coordination chemistry as anticipated and these point away from the capsule periphery. The interior of the assembly is occupied by two dimethylformamide molecules (one of which is coordinated to a Cd(II) centre) and an aquo ligand from one Cd(II) centre. The tilt angle found between calixarene lower-rim centroids and one generated between the two Cd(II) centres was found to be ~128°, showing that this is far from linear (Fig. 18B).
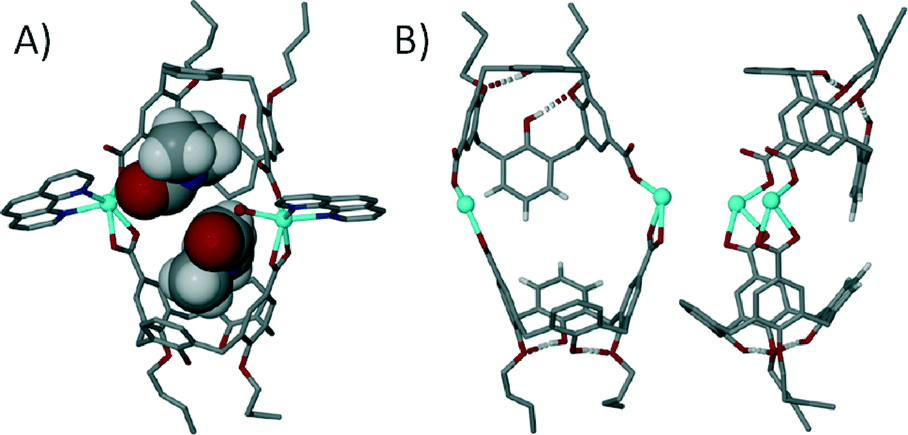 | ||
| Fig. 18 Views of the tilted dimeric metal–organic calixarene capsule containing two dmf molecules and an aquo ligand. A) Space filling representation showing guest/ligand occupation in the cavity. B) Guest/ligand free view emphasising tilt within the capsule framework.72 | ||
We investigated the influence of substituted 1,10-phenanthrolines on this assembly motif and found that steric factors between capsules play an important role in determining the resulting shape of the discrete structure.73 When using either 2-methyl-1,10-phenanthroline or 3,4,7,8-tetramethyl-1,10-phenanthroline as a co-ligand the resulting dimeric capsules are orientated in a head-to-head rather than tilted fashion, with the angle between lower-rim centroids and one generated between the two Cd(II) centres found to be ~180° (Fig. 19). From our experiments we concluded that steric factors of the phenanthroline methyl groups caused rearrangements in the coordination sphere. A result of this is that the composition of the capsule interior is markedly different; there are no ligated solvents on the capsule interior, which is occupied by two guest dmf molecules of crystallisation. In all of these cases the angles between the upper-rim carboxylates are crucial to drive assembly of a discrete structure; placement at the alternative upper-rim positions (at a more obtuse angle) results in 1-D coordination polymer rather than discrete 6assembly formation.
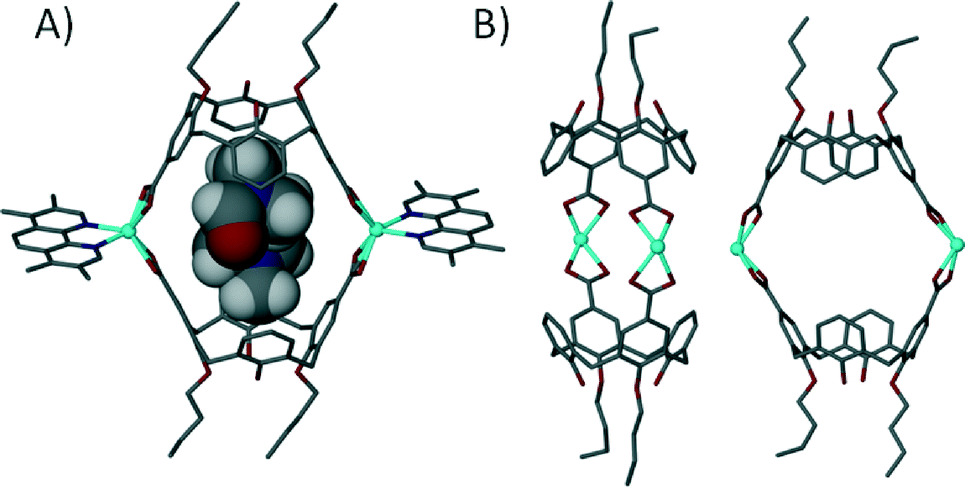 | ||
| Fig. 19 Views of the linear dimeric metal–organic calixarene capsule containing two dmf molecules. A) Space filling representation showing guest occupation in the cavity. B) Guest/ligand free view emphasising the linear nature of the capsule framework.73 | ||
Conclusions
Various strategies have emerged for the rational construction of extended structures through well-developed coordination chemistry. Cyclic host molecules hold huge potential for the application of these principles to afford discrete polyhedral species. These may have interesting properties such as inherent porosity, or the ability to selectively bind particular guests from mixtures. The p-carboxylato hosts described towards the end of this highlight are gaining importance as they offer an advantage in that they should afford libraries of stable materials for exploitation. When one considers directed assembly and the interesting host–guest chemistry associated with these building blocks, as well as their associated nanoscale metal–organic architectures, it is clear that there remains much to explore and understand.Notes and references
- R. Szostak, Molecular Sieves, Van Nostrand Reinhold, New York, 1989 Search PubMed.
- L. MacGillivray, Metal-Organic Frameworks, John Wiley & Sons, 2010, and references therein Search PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS PubMed.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS PubMed.
- M. Mastalerz, Angew. Chem., Int. Ed., 2008, 47, 445 CrossRef CAS PubMed , and references therein.
- (a) W. J. Belcher, C. A. Longstaff, M. R. Neckenig and J. W. Steed, Chem. Commun., 2002, 1602 RSC; (b) S. A. Baudron and M. W. Hosseini, Inorg. Chem., 2006, 45, 5260 CrossRef CAS PubMed; (c) P. P. Cholewa, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2013, 13, 2703 CrossRef CAS; (d) W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688 CrossRef CAS PubMed.
- (a) M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151 CrossRef CAS; (b) R. W. Gable, B. F. Hoskins and R. Robson, J. Chem. Soc., Chem. Commun., 1990, 1677 RSC; (c) K. Biradha, Y. Hongo and M. Fujita, Angew. Chem., Int. Ed., 2000, 39, 3843 CrossRef CAS; (d) S. Noro, R. Kitaura, M. Kondo, S. Kitagawa, T. Ishii, H. Matsuzaka and M. Yamashita, J. Am. Chem. Soc., 2002, 124, 2568 CrossRef CAS PubMed; (e) X. Q. Liang, X. H. Zhou, C. Chen, H. P. Xiao, Y. Z. Li, J. L. Zuo and X. Z. You, Cryst. Growth Des., 2009, 9, 1041 CrossRef CAS; (f) K. Biradha, Y. Hongo and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3395 CrossRef CAS; (g) R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428 CrossRef CAS PubMed.
- (a) S. Noro, S. Kitagawa, M. Kondo and K. Seki, Angew. Chem., Int. Ed., 2000, 39, 2081 CrossRef; (b) O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096 CrossRef CAS; (c) J. F. Ma, J. Yang, G. L. Zheng, L. Li and J. F. Liu, Inorg. Chem., 2003, 42, 7531 CrossRef CAS PubMed; (d) O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1996, 118, 295 CrossRef CAS; (e) J. Zhang, R. Liu, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2007, 46, 8388 CrossRef CAS PubMed; (f) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607 CrossRef CAS PubMed; (g) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490 CrossRef CAS PubMed; (h) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
- O. M. Yaghi, H. Li and T. L. Groy, Inorg. Chem., 1997, 36, 4292 CrossRef CAS.
- J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239 CrossRef CAS PubMed.
- J. N. Van Niekerk and F. R. L. Schoening, Acta Crystallogr., 1953, 6, 227 CrossRef CAS.
- (a) B. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021 CrossRef CAS PubMed; (b) S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS; (c) H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650 CrossRef CAS PubMed; (d) Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296 CrossRef CAS PubMed; (e) H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS PubMed; (f) X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schroder, J. Am. Chem. Soc., 2009, 131, 2159 CrossRef CAS PubMed; (g) B. Rather and M. J. Zaworotko, Chem. Commun., 2003, 830 RSC; (h) W. Mori and S. Takamizawa, J. Solid State Chem., 2000, 152, 120 CrossRef CAS; (i) S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC; (j) M. Köberl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Dalton Trans., 2011, 40, 6834 RSC.
- (a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed; (b) T. M. Reineke, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 1999, 38, 2590 CrossRef CAS; (c) J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS PubMed.
- (a) Z. Zhang, L. Zhang, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 928 CrossRef CAS PubMed; (b) P. S. Nugent, V. L. Rhodus, T. Pham, K. Forrest, L. Wojtas, B. Space and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 10950 CrossRef CAS PubMed; (c) W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443 CrossRef CAS PubMed; (d) A. Schoedel, A. J. Cairns, Y. Belmabkhout, L. Wojtas, M. Mohamed, Z. Zhang, D. M. Proserpio, M. Eddaoudi and M. J. Zaworotko, Angew. Chem., Int. Ed., 2013, 52, 2902 CrossRef CAS PubMed; (e) D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC.
- L. R. MacGillivray and J. L. Atwood, Angew. Chem., Int. Ed., 1999, 38, 1018 CrossRef CAS.
- (a) M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645 CrossRef CAS; (b) P. J. Stang and D. H. Cao, J. Am. Chem. Soc., 1994, 116, 4981 CrossRef CAS.
- (a) P. J. Stang, D. H. Cao, S. Saito and A. M. Arif, J. Am. Chem. Soc., 1995, 117, 6213 Search PubMed; (b) C. J. Kuehl, C. L. Mayne, A. M. Arif and P. J. Stang, Org. Lett., 2000, 2, 3727 CrossRef CAS PubMed.
- Y. K. Kryschenko, S. R. Seidel, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2003, 125, 5193 CrossRef CAS PubMed.
- S. Leininger, M. Schmitz and P. J. Stang, Org. Lett., 1999, 1, 1921 CrossRef CAS.
- (a) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- K. Suzuki, M. Tominaga, M. Kawano and M. Fujita, Chem. Commun., 2009, 1638 RSC.
- M. Tominaga, K. Suzuki, M. Kawano, T. Kusukawa, T. Ozeki, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5621 CrossRef CAS PubMed.
- (a) Q. F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144 CrossRef CAS PubMed; (b) J. Bunzen, J. Iwasa, P. Bonakdarzadeh, E. Numata, K. Rissanen, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 3161 CrossRef CAS PubMed.
- (a) B. Olenyuk, J. A. Whiteford, A. Fechtenkotter and P. J. Stang, Nature, 1999, 398, 796 CrossRef CAS PubMed; (b) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS; (c) M. Fujita, M. Tominga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 371 CrossRef PubMed; (d) M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509 RSC; (e) M. Schweiger, S. R. Seidel, M. Schmitz and P. J. Stang, Org. Lett., 2000, 2, 1255 CrossRef CAS; (f) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS PubMed.
- (a) G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010 CrossRef CAS PubMed; (b) S. Freye, J. Hey, A. Torras-Galan, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed.
- (a) B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773 CrossRef CAS PubMed; (b) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754 CrossRef CAS PubMed; (c) V. Vajpayee, Y. J. Yang, S. C. Kang, H. Kim, I. S. Kim, M. Wang, P. J. Stang and K. W. Chi, Chem. Commun., 2011, 47, 5184 RSC.
- (a) Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 2002, 41, 2515 CrossRef CAS; (b) N. K. Al-Rasbi, I. S. Tidmarsh, S. P. Argent, H. Adams, L. P. Harding and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 11641 CrossRef CAS PubMed; (c) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2011, 133, 858 CrossRef CAS PubMed; (d) T. D. Hamilton, G. S. Papaefstathiou and L. R. MacGillivray, J. Am. Chem. Soc., 2002, 124, 11606 CrossRef CAS PubMed; (e) T. D. Hamilton, G. S. Papaefstathiou, T. Friscic, D. K. Bucar and L. R. MacGillivray, J. Am. Chem. Soc., 2008, 130, 14366 CrossRef CAS PubMed; (f) M. H. Alkordi, J. L. Belof, E. Rivera, L. Wojtas and M. Eddaoudi, Chem. Sci., 2011, 2, 1695 RSC; (g) Y. Liu, V. C. Kravtsov, D. A. Beauchamp, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2005, 127, 7266 CrossRef CAS PubMed; (h) M. D. Ward, Chem. Commun., 2009, 4487 RSC.
- (a) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136 CrossRef CAS PubMed; (b) F. A. Cotton, L. M. Daniels, C. Lin and C. A. Murillo, Chem. Commun., 1999, 841 RSC; (c) G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS PubMed; (d) A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Cote, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110 CrossRef CAS PubMed; (e) M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368 CrossRef CAS; (f) M. J. Byrnes, N. J. Patmore and M. H. Chisholm, Inorg. Chem., 2005, 44, 9347 CrossRef CAS PubMed; (g) J. R. Li, D. J. Timmons and H. C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368 CrossRef CAS PubMed; (h) Y. Yan, I. Telepeni, S. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schroder, J. Am. Chem. Soc., 2010, 132, 4092 CrossRef CAS PubMed.
- C. D. Gutsche, Calixarenes - An Introduction, RSC, 2nd edn, 2008 Search PubMed.
- Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Calixarenes 2001, Kluwer Academic Publishers, 2001 Search PubMed.
- (a) J. Gabard and A. Colle, J. Chem. Soc., Chem. Commun., 1981, 1137 RSC; (b) A. Colle, Tetrahedron, 1987, 43, 5725 CrossRef.
- T. Brotin and J. P. Dutasta, Chem. Rev., 2009, 109, 88 CrossRef CAS PubMed.
- (a) K. Bartik, M. Luhmer, J. P. Dutasta, A. Collet and J. Reisse, J. Am. Chem. Soc., 1998, 120, 784 CrossRef CAS; (b) M. Luhmer, B. M. Goodson, Y. Q. Song, D. D. Laws, L. Kaiser, M. C. Cyrier and A. Pines, J. Am. Chem. Soc., 1999, 121, 3502 CrossRef CAS; (c) Q. Wei, G. K. Seward, P. A. Hill, B. Patton, I. E. Dimitrov, N. N. Kuzma and I. J. Dmochowski, J. Am. Chem. Soc., 2006, 128, 13274 CrossRef CAS PubMed; (d) C. Hilty, T. J. Lowery, D. E. Wemmer and A. Pines, Angew. Chem., Int. Ed., 2006, 45, 70 CrossRef CAS PubMed; (e) R. M. Fairchild, A. I. Joseph, K. T. Holman, H. A. Fogarty, T. Brotin, J. P. Dutasta, C. Boutin, G. Huber and P. Berthault, J. Am. Chem. Soc., 2010, 132, 15505 CrossRef CAS PubMed; (f) R. M. Fairchild and K. T. Holman, J. Am. Chem. Soc., 2005, 127, 16364 CrossRef CAS PubMed; (g) S. T. Mough, J. C. Goeltz and K. T. Holman, Angew. Chem., Int. Ed., 2004, 43, 5631 CrossRef CAS PubMed.
- (a) D. J. Cram, S. Karbach, Y. H. Kim, L. Baczynskyj and G. W. Kalleymeyn, J. Am. Chem. Soc., 1985, 107, 2575 CrossRef CAS; (b) D. J. Cram, S. Karbach, Y. H. Kim, L. Baczynskyj, K. Marti, R. M. Sampson and G. W. Kalleymeyn, J. Am. Chem. Soc., 1988, 110, 2554 CrossRef CAS.
- (a) J. C. Sherman and D. J. Cram, J. Am. Chem. Soc., 1989, 4527 CrossRef CAS; (b) J. C. Sherman, C. B. Knobler and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 2194 CrossRef CAS; (c) R. G. Chapman, G. Olovsson, J. Trotter and J. C. Sherman, J. Am. Chem. Soc., 1998, 120, 6252 CrossRef CAS; (d) S. Mendoza, P. D. Davidov and A. E. Kaifer, Chem.–Eur. J., 1998, 4, 864 CrossRef CAS; (e) N. Nishimura and K. Kobayashi, Angew. Chem., Int. Ed., 2008, 47, 6255 CrossRef CAS PubMed.
- (a) N. Chopra, C. Naumann and J. C. Sherman, Angew. Chem., Int. Ed., 2000, 398, 194 CrossRef; (b) R. Mungaroo and J. C. Sherman, Chem. Commun., 2002, 1672 RSC.
- E. S. Barrett, J. L. Irwin, A. J. Edwards and M. S. Sherburn, J. Am. Chem. Soc., 2004, 126, 16747 CrossRef CAS PubMed.
- D. A. Makeiff and J. C. Sherman, J. Am. Chem. Soc., 2005, 127, 12363 CrossRef CAS PubMed.
- (a) X. Liu and R. Warmuth, J. Am. Chem. Soc., 2006, 128, 14120 CrossRef CAS PubMed; (b) X. Liu, Y. Liu, G. Li and R. Warmuth, Angew. Chem., Int. Ed., 2006, 45, 901 CrossRef CAS PubMed; (c) J. Sun and R. Warmuth, Chem. Commun., 2011, 47, 9351 RSC; (d) Y. Liu, X. Liu and R. Warmuth, Chem.–Eur. J., 2007, 13, 8953 CrossRef CAS PubMed; (e) J. Sun, J. L. Bennett, T. J. Emge and R. Warmuth, J. Am. Chem. Soc., 2011, 133, 3268 CrossRef CAS PubMed; (f) X. Liu, Y. Liu and R. Warmuth, Supramol. Chem., 2008, 20, 41 CrossRef; (g) J. Sun, B. O. Patrick and J. C. Sherman, Tetrahedron, 2009, 65, 7296 CrossRef CAS PubMed.
- V. Böhmer, H. Goldmann, W. Vogt, J. Vicens and Z. Asfari, Tetrahedron Lett., 1989, 30, 1391 CrossRef.
- (a) K. Araki, K. Sisido, K. Hisaichi and S. Shinkai, Tetrahedron Lett., 1993, 34, 8297 CrossRef CAS; (b) M. T. Blanda and K. E. Griswold, J. Org. Chem., 1994, 59, 4313 CrossRef CAS.
- (a) P. Neri, A. Bottino, F. Cunsolo, M. Piattelli and E. Gavuzzo, Angew. Chem., Int. Ed., 1998, 37, 166 CrossRef CAS; (b) M. S. Brody, C. A. Schalley, D. M. Rudkevich and J. Rebek, Jr., Angew. Chem., Int. Ed., 1999, 38, 1640 CrossRef CAS; (c) A. Arduini, A. Pochini and A. Secchi, Eur. J. Org. Chem., 2000, 2325 CrossRef CAS.
- T. Arimura, S. Matsumoto, O. Teshima, T. Nagasaki and S. Shinkai, Tetrahedron Lett., 1991, 32, 5111 CrossRef CAS.
- (a) P. Timmerman, W. Verboom, F. C. J. M. van Veggel, J. P. M. van Duynhoven and D. N. Reinhoudt, Angew. Chem., Int. Ed. Engl., 1994, 33, 2345 CrossRef; (b) A. M. A. van Wageningen, P. Timmerman, J. P. M. van Duynhoven, W. Verboom, F. C. J. M. van Veggel and D. N. Reinhoudt, Chem.–Eur. J., 1997, 3, 639 CrossRef CAS; (c) A. M. A. van Wageningen, J. P. M. van Duynhoven, W. Verboom and D. N. Reinhoudt, J. Chem. Soc., Chem. Commun., 1995, 1941 RSC.
- J. Rebek, Jr., Chem. Commun., 2000, 637 RSC.
- (a) J. D. van Loon, R. G. Janssen, W. Verboom and D. N. Reinhoudt, Tetrahedron Lett., 1992, 33, 5125 CrossRef; (b) K. Koh, K. Araki and S. Shinkai, Tetrahedron Lett., 1994, 35, 8255 CrossRef CAS.
- (a) K. D. Shimazu and J. Rebek, Jr., Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 12403 CrossRef; (b) J. Scheerder, R. H. Vreekamp, J. F. Engbersen, W. Verboom, J. P. M. Van Duyhoven and D. N. Reinhoudt, J. Org. Chem., 1996, 61, 3476 CrossRef CAS; (c) O. Mogck, V. Bohmer and W. Vogt, Tetrahedron, 1996, 52, 8489 CrossRef CAS; (d) R. S. Meissner, J. Rebek and J. Mendoza, Science, 1995, 270, 1485 CAS.
- O. Mogck, E. F. Paulus, V. Bohmer, I. Thondorf and W. Vogt, Chem. Commun., 1996, 2533 RSC.
- (a) R. K. Castellano, D. M. Rudkevich and J. Rebek, Jr., J. Am. Chem. Soc., 1996, 118, 10002 CrossRef CAS; (b) R. K. Castellano, B. H. Kim and J. Rebek, Jr., J. Am. Chem. Soc., 1997, 119, 12671 CrossRef CAS; (c) T. Martin, U. Obst and J. Rebek, Science, 1998, 281, 1842 CrossRef CAS; (d) Y. L. Cho, D. M. Rudkevich and J. Rebek, Jr., J. Am. Chem. Soc., 2000, 122, 9868 CrossRef CAS; (e) R. Zadmard, M. Junkers, T. Schrader, T. Grawe and A. Kraft, J. Org. Chem., 2003, 68, 6511 CrossRef CAS PubMed.
- (a) T. Heinz, D. M. Rudkevich and J. Rebek, Jr., Nature, 1998, 394, 764 CrossRef CAS PubMed; (b) K. N. Rose, L. J. Barbour, G. W. Orr and J. L. Atwood, Chem. Commun., 1998, 407 RSC; (c) S. Ma, D. M. Rudkevich and J. Rebek, Jr., J. Am. Chem. Soc., 1998, 120, 4977 CrossRef CAS; (d) A. Shivanyuk and J. Rebek, Jr., Chem. Commun., 2001, 2374 RSC; (e) M. H. K. Ebbing, M. J. Villa, J. M. Valpuesta, P. Prados and J. de Mendoza, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4962 CrossRef CAS PubMed; (f) S. J. Dalgarno, J. Antesberger, R. M. McKinlay and J. L. Atwood, Chem.–Eur. J., 2007, 13, 8248 CrossRef CAS PubMed.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 470 Search PubMed.
- (a) T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257 CrossRef CAS; (b) G. W. V. Cave, J. Antesberger, L. J. Barbour, R. M. McKinlay and J. L. Atwood, Angew. Chem., Int. Ed., 2004, 43, 5263 CrossRef CAS PubMed.
- (a) K. Kobayashi, T. Shirasaka, K. Yamaguchi, S. Sakamoto, E. Horn and N. Furukawa, Chem. Commun., 2000, 41 RSC; (b) K. Kobayashi, K. Ishii, S. Sakamoto, T. Shirasaka and K. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 10615 CrossRef CAS PubMed; (c) H. Kitagawa, M. Kawahata, R. Kitagawa, Y. Yamada, M. Yamanaka, K. Yamaguchi and K. Kobayashi, Tetrahedron, 2009, 65, 7234 CrossRef CAS PubMed; (d) Y. S. Park, S. Seo, E. H. Kim and K. Paek, Org. Lett., 2011, 13, 5904 CrossRef CAS PubMed; (e) D. Ajami and J. Rebek, Jr., J. Org. Chem., 2009, 74, 6584 CrossRef CAS PubMed; (f) W. Jiang and J. Rebel, Jr., J. Am. Chem. Soc., 2012, 134, 17498 CrossRef CAS PubMed; (g) O. Ugono and K. T. Holman, Chem. Commun., 2006, 2144 RSC; (h) T. Gerkensmeier, J. Mattay and C. Nätheri, Chem.–Eur. J., 2001, 7, 465 CrossRef CAS.
- (a) J. J. Gonzalez, R. Ferdani, E. Albertini, J. M. Blasco, A. Arduini, A. Pochini, P. Prados and J. de Mendoza, Chem.–Eur. J., 2000, 6, 73 CrossRef CAS; (b) A. Arduini, R. Ferdani, A. Pochini, A. Secchi, F. Ugozzoli, G. M. Sheldrick, P. Prados, J. J. Gonzalez and J. de Mendoza, J. Supramol. Chem., 2002, 2, 85 CrossRef CAS; (c) A. M. Rincon, P. Prados and J. de Mendoza, Eur. J. Org. Chem., 2002, 640 CrossRef CAS; (d) D. Garozzo, G. Gattuso, F. H. Kohnke, P. Malvagna, A. Notti, S. Occhipinti, S. Pappalardo, M. F. Parisib and I. Pisagatti, Tetrahedron Lett., 2002, 43, 7663 CrossRef CAS; (e) M. Makha, J. J. McKinnon, A. N. Sobolev, M. A. Spackman and C. L. Raston, Chem.–Eur. J., 2007, 13, 3907 CrossRef CAS PubMed.
- (a) P. Schmitt, P. B. Beer, M. G. Drew and P. D. Sheen, Angew. Chem., Int. Ed. Engl., 1997, 36, 1840 CAS; (b) A. P. Marchand, H. S. Chong, M. Takhi and T. D. Power, Tetrahedron, 2000, 56, 3121 CrossRef CAS; (c) B. S. Creavena, D. F. Donlona and J. McGinley, Coord. Chem. Rev., 2009, 253, 893 CrossRef PubMed; (d) The calixarene lower-rim has also recently been exploited in the construction of discrete polynuclear metal clusters that exhibiting promising magnetic properties, but this is also out with the scope of this article. For some recent examples please see: M. M. Olmstead, G. Sigel, H. Hope, X. Xu and P. P. Power, J. Am. Chem. Soc., 1985, 107, 8087 CrossRef CAS; (e) G. Karotsis, S. J. Teat, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 8285 CrossRef CAS PubMed; (f) G. Karotsis, S. Kennedy, S. J. Teat, C. M. Beavers, D. A. Fowler, J. J. Morales, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, J. Am. Chem. Soc., 2010, 132, 12983 CrossRef CAS PubMed.
- M. J. Hardie, Chem. Soc. Rev., 2010, 39, 516 RSC.
- Z. Zhong, A. Ikeda, S. Shinkai, S. Sakamoto and K. Yamaguchi, Org. Lett., 2001, 3, 1085 CrossRef CAS PubMed.
- (a) B. F. Abrahams, B. A. Boughton, N. J. FitzGerald, J. L. Holmes and R. Robson, Chem. Commun., 2011, 47, 7404 RSC; (b) B. F. Abrahams, N. J. FitzGerald and R. Robson, Angew. Chem., Int. Ed., 2010, 49, 2896 CrossRef CAS PubMed.
- R. M. McKinlay, G. W. V. Cave and J. L. Atwood, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5944 CrossRef CAS PubMed.
- R. M. McKinlay, P. K. Thallapally, G. W. V. Cave and J. L. Atwood, Angew. Chem., Int. Ed., 2005, 44, 5733 CrossRef CAS PubMed.
- N. P. Power, S. J. Dalgarno and J. L. Atwood, Angew. Chem., Int. Ed., 2007, 46, 860 Search PubMed.
- (a) D. A. Fowler, A. V. Mossine, C. M. Beavers, S. J. Teat, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2011, 133, 11069 CrossRef CAS PubMed; (b) J. L. Atwood, E. K. Brechin, S. J. Dalgarno, R. Inglis, L. F. Jones, A. Mossine, M. J. Paterson, N. P. Power and S. J. Teat, Chem. Commun., 2010, 46, 3484 RSC.
- (a) P. Jacopozzi and E. Dalcanale, Angew. Chem., Int. Ed. Engl., 1997, 36, 613 CrossRef CAS; (b) F. Fochi, P. Jacopozzi, E. Wegelius, K. Rissanen, P. Cozzini, E. Marastoni, E. Fisicaro, P. Manini, R. Fokkens and E. Dalcanale, J. Am. Chem. Soc., 2001, 123, 7539 CrossRef CAS PubMed; (c) D. Zuccaccia, L. Pirondini, R. Pinalli, E. Dalcanale and A. Macchion, J. Am. Chem. Soc., 2005, 127, 7025 CrossRef CAS PubMed.
- (a) L. Pirondini, F. Bertolini, B. Cantadori, F. Ugozzoli, C. Massera and E. Dalcanale, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4911 CrossRef CAS PubMed; (b) M. Yamanaka, Y. Yamada, Y. Sei, K. Yamaguchi and K. Kobayash, J. Am. Chem. Soc., 2006, 128, 1531 CrossRef CAS PubMed; (c) S. J. Park, D. M. Shin, S. Sakamoto, K. Yamaguchi, Y. K. Chung, M. S. Lah and J. I. Hong, Chem.–Eur. J., 2005, 11, 235 CrossRef PubMed.
- R. Pinalli, V. Cristini, V. Sottili, S. Geremia, M. Campagnolo, A. Caneschi and E. Dalcanale, J. Am. Chem. Soc., 2004, 126, 6516 CrossRef CAS PubMed.
- T. Haino, M. Kobayashi, M. Chikaraishi and Y. Fukazawa, Chem. Commun., 2005, 2321 RSC.
- (a) O. D. Fox, M. G. B. Drew and P. D. Beer, Angew. Chem., Int. Ed., 2000, 39, 135 CrossRef; (b) O. D. Fox, J. Cookson, E. J. S. Wilkinson, M. G. B. Drew, E. J. MacLean, S. J. Teat and P. D. Beer, J. Am. Chem. Soc., 2006, 128, 6990 CrossRef CAS PubMed.
- (a) O. D. Fox, N. K. Dalley and R. G. Harrison, J. Am. Chem. Soc., 1998, 120, 7111 CrossRef CAS; (b) D. Fox, N. K. Dalley and R. G. Harrison, Inorg. Chem., 1999, 38, 5860 CrossRef.
- F. A. Cotton, P. Lei, C. Lin, C. A. Murillo, X. Wang, S. Y. Yu and Z. X. Zhang, J. Am. Chem. Soc., 2004, 126, 1518 CrossRef CAS PubMed.
- O. Ugono, J. P. Moran and K. T. Holman, Chem. Commun., 2008, 1404 RSC.
- S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 785 CrossRef PubMed.
- P. P. Cholewa, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Chem. Commun., 2013, 49, 3203 RSC.
- P. P. Cholewa, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2013, 13, 5165 CAS.
| This journal is © The Royal Society of Chemistry 2014 |