 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-state coexistence of {Zr12} and {Zr6} zirconium oxocarboxylate clusters†
Iurie L.
Malaestean
a,
Meliha Kutluca
Alıcı
a,
Claire
Besson
a,
Arkady
Ellern
b and
Paul
Kögerler
*a
aInstitute of Inorganic Chemistry, RWTH Aachen University, 52074 Aachen, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de; Fax: +49 241 80 92642
bAmes Laboratory, Iowa State University, Ames, IA 50011, USA
First published on 30th October 2013
Abstract
Ligand metathesis, Co(II) coordination, and partial condensation reactions of an archetypal {Zr6} zirconium oxocarboxylate cluster result in the first example of the coexistence of the distinct zirconium oxide frameworks {Zr6O8} and {Zr12O22}. Even minor modifications to the reaction conditions push this apparent equilibrium towards the {Zr6O8}-based product.
Molecular high-nuclearity zirconium(IV) and cerium(III/IV) oxide structures have emerged as an extension to classical polyoxometalate chemistry with numerous structural analogies and similar formation paradigms.1 In this context, we have explored the magnetic functionalization of several zirconium(IV) oxide clusters based on highly condensed {Zr6O8}2 and {Zr12O22}3 frameworks. Whereas little is known about the equilibria of different structural zirconium oxide archetypes in their reaction solutions, their similarity to group-5/6 polyoxometalates, where numerous species or isomers commonly coexist, suggests that, under certain boundary reaction conditions, different zirconium oxide structures could coexist as well. We now scanned the reaction parameter space for Co(II)-substituted zirconium oxocarboxylate clusters, starting from a member of the well-established {Zr6O4(OH)4(carboxylate)12}2-type cluster family of clusters, namely [Zr6O4(OH)4(pr)12] (pr: propionate). Reactions were performed in the presence of potential bridging ligands in acetonitrile and small, well-defined amounts of water that facilitate further hydrolysis or condensation steps.‡‡ This approach yielded a crystalline phase of composition {H[Zr12Co2O8(OH)14(pr)22(MeCN)2(μ-pz)][Zr6Co6O8(pr)12(Hbda)6](NO3)3·8MeCN}n (1), in which clusters of both the {Co6Zr6} and the {Co2Zr12} types co-crystallize (H2bda: N-butyldiethanolamine). Here, pyrazine (pz) selectively coordinates to the Co centers of the {Co2Zr12} moieties and interlinks the latter into a one-dimensional coordination polymer; we note that while a few zirconium oxide-based MOFs characterized by single-crystal X-ray diffraction exist,41 represents the first structurally characterized heterometallic zirconium oxide-based coordination polymer. Importantly, replacing the pz linker with an alternative potential linker reagent, 4,4′-biphenol (H2bph), under otherwise comparable reaction conditions results in the formation of crystals of only the discrete {Co6Zr6}-type cluster, isolated as [Co6Zr6O8(pr)12(Hbda)6](Hbph)2·(H2bph) (2).
The crystal structure of compound 1 (space group C2/c) features two non-connected main constituents, the charge-neutral [Zr12Co2O8(OH)14(pr)22(MeCN)2(μ-pz)]n coordination polymer (A) and the discrete cluster cation [Zr6Co6O8(pr)12(Hbda)6]2+ (B), next to nitrate counteranions and acetonitrile solvate molecules (Fig. 1).
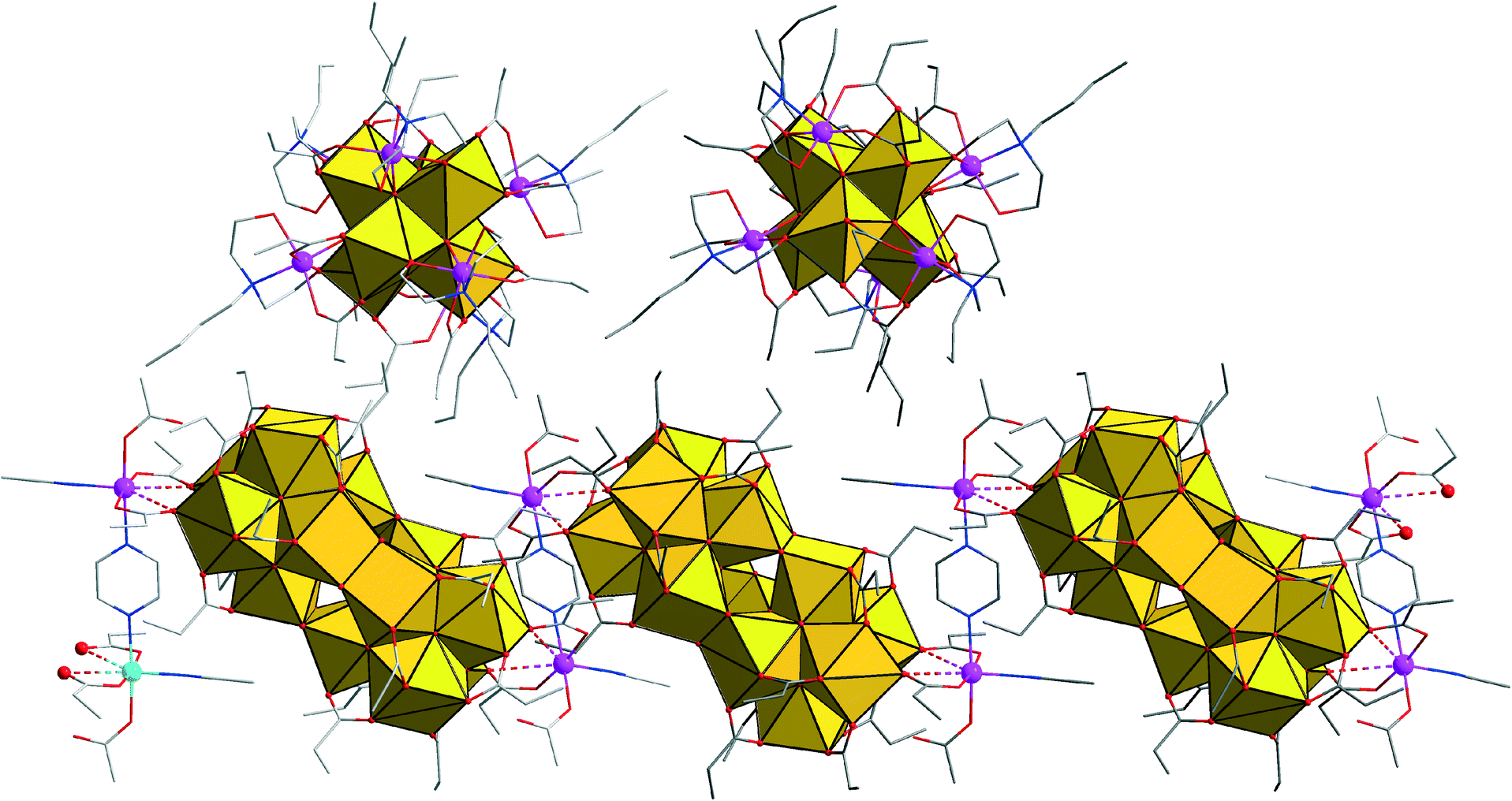 | ||
| Fig. 1 A segment of the solid-state structure of 1 illustrating the coexistence of one helical polymer strand A, consisting of three pyrazine-linked {Co2Zr12} groups, and two adjacent {Co6Zr6} (B) clusters. The zirconium oxide substructures are shown in an octahedral representation. Co: purple spheres, C: grey, N: blue, O: red. Nitrate groups, acetonitrile solvate molecules, and hydrogen positions are omitted for clarity. | ||
A comprises the rare {Zr12O22} cluster core, in which two octahedral {Zr6} building blocks are directly condensed via six μ2-hydroxo bridges, whereas most other known {Zr12}-type structures comprise {Zr6} halves interlinked by four bidentate carboxylate bridges.5 In A, all external coordination sites of the {Zr12} subunit are assumed by oxygen atoms of 20 propionate ligands, 16 of which bridge neighboring Zr sites, and four bridge Zr2/Zr3 and their symmetry equivalents to the Co(II) sites (Fig. 2). The Zr centers are 7- or 8-coordinated by four μ3-O(H) or μ2-OH atoms and three or four O donors from the carboxylate groups (Zr–O: 2.178(6)–2.28(1) Å). The protonation of the μ2-OH groups interlinking the two {Zr6} halves can be deduced from longer Zr–O bond distances ranging from 2.139(8) to 2.159(7) Å, as well as from the presence of weak hydrogen bonds (2.91(2)–3.00(2) Å) to the NO3− anions. The {Zr6(μ-O(H))8} subunits consist of four μ3-O, the atoms O4, O6, O8, and O10 (Zr–(μ3-O): 2.050(7)–2.195(8) Å), as well as the four protonated μ3-O atoms O5, O7, O9, and O11 (Zr–(μ3-O): 2.360(6)–2.236(7) Å). The protonation of these atoms is also evident from the presence of hydrogen bonds to the monodentate carboxylates (2.70(1) Å) and to the nitrate groups (2.81(2) Å and 2.89(2) Å).
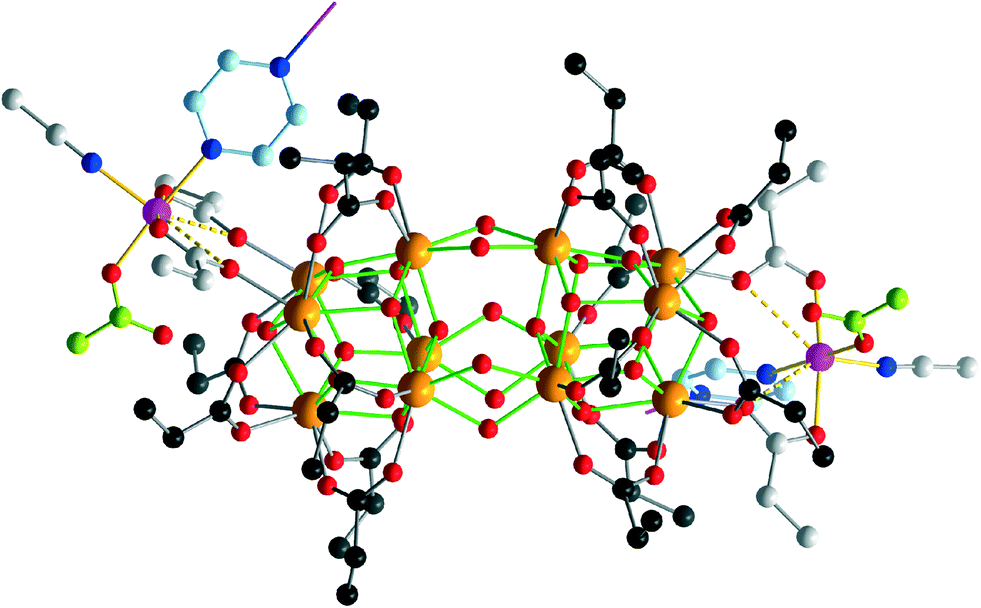 | ||
| Fig. 2 The {Co6Zr12} repetition unit in A. Green Zr–O bonds highlight the central zirconium oxide core. Zr: orange, Co: purple, O: red, N: dark blue spheres. C atoms, bidentate propionate groups: dark grey; pr groups bridging Zr and Co: light grey; monodentate pr: light green; pyrazine: light blue spheres. Solid yellow lines: Co–O/N coordinative bonds <2.1 Å; dashed yellow lines: weaker Co–O contacts (>2.7 Å). Hydrogen positions are omitted for clarity. | ||
The two (crystallographically equivalent) Co(II) sites each coordinate to two oxygen atoms of two propionate ligands, and a pyrazine ligand coordinates to two adjacent Co sites of neighboring {Co2Zr12} groups, resulting in an infinite helical chain (Fig. S1†). The square-prismatic CoN2O3 coordination environment includes one deprotonated terminal monodentate carboxylate displaying short C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O distances (1.20(2) Å), two bidentate carboxylates (Co–Ocarb: 1.98(2)–2.07(1) Å), one MeCN and one pyrazine (Co–N: 2.11(1)–2.20(1) Å). The O sites of the bidentate pr groups binding to Zr form weak contacts with Co (Co–O: 2.72(1)/2.87(1) Å).
O distances (1.20(2) Å), two bidentate carboxylates (Co–Ocarb: 1.98(2)–2.07(1) Å), one MeCN and one pyrazine (Co–N: 2.11(1)–2.20(1) Å). The O sites of the bidentate pr groups binding to Zr form weak contacts with Co (Co–O: 2.72(1)/2.87(1) Å).
B features a {Zr6(μ3/4-O)8} core to which six additional Co atoms are attached via Hbda− groups to the μ3-O sites. Chelating bda and carboxylate ligands connect the Co centers and adjacent Zr sites. The monodeprotonated Hbda− groups form intramolecular short hydrogen bonds (2.67(2)–2.68(1) Å) to neighboring carboxylate groups. Note that the {Zr6(μ3(4)-O)8} core in molecule B is not protonated as evident from the coordination of three μ4-O (and their symmetry equivalents) to Co atoms and of one remaining μ3-O (and its symmetry equivalent) to three zirconium atoms at distances of 2.081(8)–2.19(1) Å typical of Zr–(μ3-O) bonds. The carboxylate ligands coordinate in μ1.2-bridging mode. The Co sites adopt a distorted octahedral NO5 environment of two bidentate carboxylate ligands (Co–Ocarb: 2.054(9)–2.10(1) Å) and one N (Co–Nbda: 2.17(1)–2.19(3) Å) and two O donors of a bdaH− ligand (Co–Obda: 2.003(9)–2.09(1) Å), as well as one μ4-O (2.118(8)–2.17(1) Å). The Zr sites are 7- or 8-coordinated by four μ3(4)-O atoms and three or four O donors from carboxylate and alkoxide groups (Zr–O: 2.079(8)–2.237(9) Å) (Fig. 3).
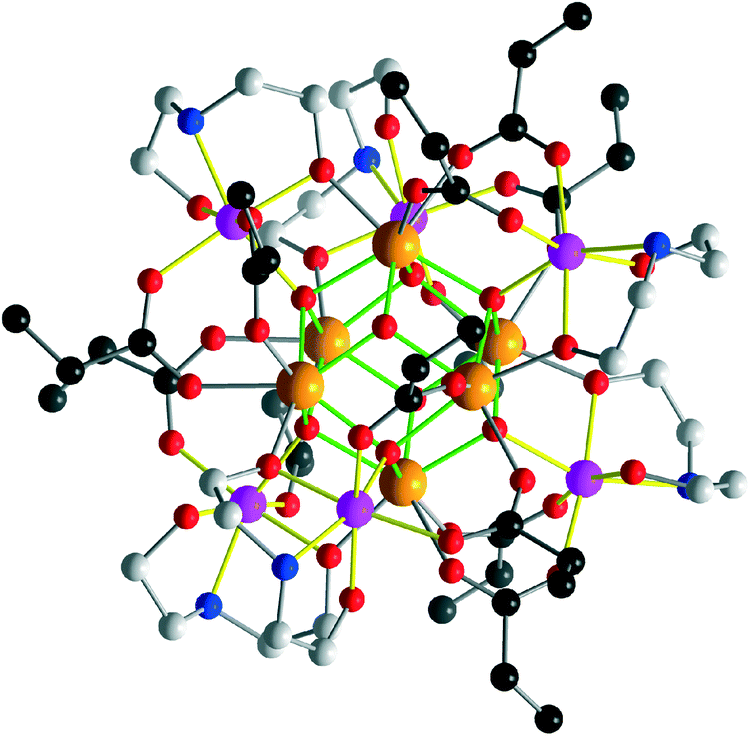 | ||
| Fig. 3 Molecular structure of B. Color codes as in Fig. 2. The {Co6Zr6} cluster in is virtually isostructural to B. Hydrogen and nBu groups belonging to bda ligands are omitted for clarity. | ||
The NO3− groups in the crystal lattice of 1 are arranged around the bridging μ2-OH groups that join the {Zr6(μ-O(H))8} subunits in A, thus forming a circular arrangement around the {Zr12} group (Fig. 4). Two of the three nitrates are symmetry equivalent with atom N10, while the third NO3− is crystallographically independent (N11), a potential indication for an interaction with an extra proton, which is required for the charge balance of complex 1.
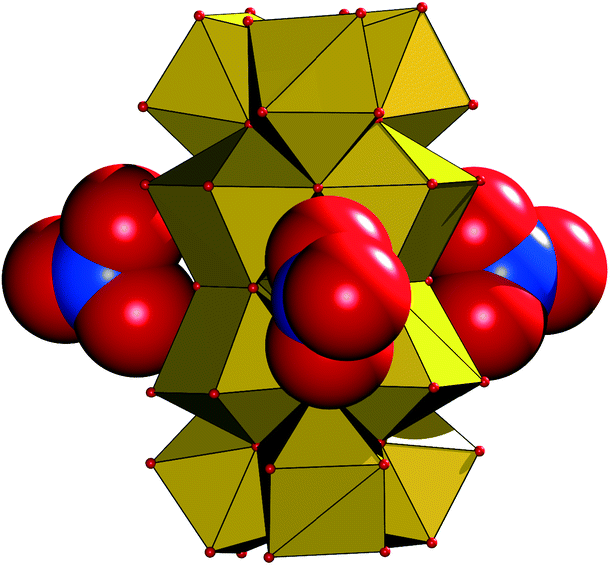 | ||
| Fig. 4 Arrangement of the three nitrate groups (shown in their van der Waals spheres) around the {Zr12} core (in a polyhedral representation) in 1. | ||
The crystal structure of compound 2 (space group P6322) comprises the cationic cluster [Zr6Co6O8(pr)12(Hbda)6]2+, which is nearly identical to molecule B of compound 1 (see Fig. S2† for a comparison of the core geometries), and three 4,4′-biphenol groups per formula unit. As there is only one crystallographically unique biphenol group in the crystal lattice of 2, charge balance requires it to formally be partly deprotonated (H4/3bph2/3−) on average. As in compound 1, chelating bda and carboxylate ligands bind the Co centers and adjacent Zr sites. The bda alkoxide groups are monoprotonated and form intramolecular short hydrogen bonds (2.75(2) Å) to the neighboring carboxylate groups. As for B, the {Zr6(μ3/4-O)8} core is also not protonated, and the indicative Zr–(μ3-O) bonds measure 2.105(5)–2.189(9) Å. The NO5-type Co coordination again shows typical geometrical parameters: Co–Ocarb: 2.08(1)–2.08(2) Å, Co–Nbda: 2.19(1) Å, Co–Obda: 2.02(1)–2.11(1) Å, and Co–(μ4-O): 2.189(9) Å; the Zr–O bonds range from 2.105(5) to 2.23(1) Å.
Conclusions
The close similarity between the reaction conditions yielding compounds 1 and 2 highlights the sensitive nature of the equilibrium conditions under which different {Zr6} and {Zr12} zirconium oxide cluster archetypes can co-crystallize. We note that we, however, cannot rule out the co-existence of these cluster types in the reaction solution that eventually produces crystals of 2: whereas the {Zr12} units in 1 are interlinked by pyrazine into a 1D coordination polymer, effectively reducing their solubility and facilitating precipitation, the 4,4′-biphenol linker employed in the reaction solution for compound 2 might simply be less affine to Co(II) coordination so as to adopt the same role. More systematic in situ studies of the reaction solutions are currently underway to determine how far the coexistence relations seen in the solid state relate to the conditions in the solution.Notes and references
- I. L. Malaestean, M. Speldrich, A. Ellern, S. G. Baca and P. Kögerler, Dalton Trans., 2011, 40, 331 RSC; I. L. Malaestean, A. Ellern, S. Baca and P. Kögerler, Chem. Commun., 2012, 48, 1499 RSC; I. L. Malaestean, A. Ellern and P. Kögerler, Eur. J. Inorg. Chem., 2013, 1635 CrossRef CAS.
- F. R. Kogler, M. Jupa, M. Puchberger and U. Schubert, J. Mater. Chem., 2004, 14, 3133 RSC; P. Piszczek, A. Radtke, A. Grodzicki, A. Wojtczak and J. Chojnacki, Polyhedron, 2007, 26, 679 CrossRef CAS.
- P. Piszczek, A. Radtke, A. Wojtczak, T. Muzioł and J. Chojnacki, Polyhedron, 2009, 28, 279 CrossRef CAS; I. L. Malaestean, M. Kutluca, M. Speldrich, A. Ellern and P. Kögerler, Inorg. Chim. Acta, 2012, 380, 72 CrossRef.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443 CrossRef CAS PubMed.
- M. Puchberger, F. E. Kogler, M. Jupa, S. Gross, H. Fric, G. Kickelbick and U. Schubert, Eur. J. Inorg. Chem., 2006, 3283 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic details, additional structural plots, thermal stability studies. CCDC 931733 and 931734. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce41829d |
‡ See ESI† for the synthesis of [Zr6O4(OH)4(pr)12]2·6Hpr precursors.Synthesis of {H[Zr12Co2O8(OH)14(pr)22(MeCN)2(pz)]·[Zr6Co6O8(pr)12(Hbda)6](NO3)3·8MeCN}n (1). A solution of 0.36 g (0.1 mmol) of [Zr6O4(OH)4(pr)12]2·6Hpr (derived from Zr(OBu)4),5N-butyldiethanolamine (0.085 mL, 0.51 mmol), 60 mg (0.2 mmol) of Co(NO3)2·6H2O, and pyrazine (40 mg, 0.5 mmol) in 5 mL acetonitrile was refluxed for 1 h. Upon cooling to room temperature, the solution was kept in a parafilm-covered vial. Pink crystals of 1 were obtained after several days, washed with acetonitrile and dried in air (yield: 0.027 g). Elemental analysis, calcd. (found) for C174H327Co8N21O119Zr18: Co, 7.0 (6.14); C, 31.2 (32.25); H, 4.9 (3.04); N, 4.4 (4.75); Zr, 24.4 (26.4)%. IR data (KBr, ν/cm−1): 3425 br/m, 2973 s, 2940 m, 1628 s, 1568 s, 1469 s, 1439 s, 1384 s, 1303 s, 1081 m, 1013 w, 906 w, 811 m, 632 s, 482 m, 414 w. Synthesis of [Co6Zr6O8(pr)12(Hbda)6](Hbph)2·(H2bph) (2). A solution of 0.19 g (0.053 mmol) of [Zr6O4(OH)4(pr)12]2·6Hpr (derived from ZrCl4, see ESI†), N-butyldiethanolamine (0.085 mL, 0.51 mmol), 60 mg (0.2 mmol) of Co(NO3)2·6H2O and 90 mg (0.48 mmol) of 4,4′-biphenol was refluxed for 1 h in 5 mL of acetonitrile. Upon cooling to room temperature, the solution was kept in a parafilm-covered vial. Pink crystals of 1 formed after several days and were separated and washed with acetonitrile and dried in air (yield: 0.018 g). Elemental analysis, calcd. (found) for C120H196Co6N6O50Zr6: Co, 10.32 (9.16); C, 42.09 (42.29); H, 5.77 (5.65); N, 2.45 (2.49); Zr, 15.98 (15.6). IR data (KBr, ν/cm−1): 3265 br/m, 2966 s, 1627 vs, 1502 s, 1467 m, 1438 m, 1302 mw, 1262 m, 1168 m, 1082 w, 904 m, 826 s, 619 w, 568 w, 542 w, 478 s. Single-crystal X-ray diffraction experiments were carried using a Bruker diffractometer with APEX II CCD detector with graphite monochromated MoKα radiation at a detector distance of 50.6 mm. Full-sphere data collection with exposures of 30 s per frame was made with ω scans in the range of 0–180° at φ = 0, 120, and 240°. A semi-empirical absorption correction was based on the fit of a spherical harmonic function to the empirical transmission surface as sampled by multiple equivalent measurements6 using SADABS software.7 Measurements were optimized to collect data to a resolution of 0.71 Å; however, the datasets have been truncated to obtain the statistically relevant resolution. The positions of metal atoms were found by direct methods. The remaining atoms were located in an alternating series of least-squares cycles and difference Fourier maps. All non-hydrogen atoms were refined in full-matrix anisotropic approximation. All hydrogen atoms were placed in the structure factor calculation at idealized positions and were allowed to ride on the neighboring atoms with relative isotropic displacement coefficients. 1: C174H287Co8N21O119Zr18, monoclinic, C2/c, Mr = 6690.63 g mol−1, T = 173(2) K, a = 36.819(4), b = 31.046(3), c = 28.384(5) Å, β = 123.688(1)°, V = 26996(6) Å3, Z = 4, 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 371 reflections, R1 = 0.0752, wR2 = 0.1916, R1 (all data) = 0.1198, wR2 (all data) = 0.2613; CCDC 931734. 2: C120H180Co6N6O50Zr6, hexagonal, P6322, Mr = 3407.6 g mol−1, T = 173(2) K, a = 18.423(5), c = 26.934(7) Å, V = 7917(4) Å3, Z = 2, 42012 reflections, R1 = 0.0850, wR2 = 0.2102, R1 (all data) = 0.0855, wR2 (all data) = 0.2106; CCDC 931733. 371 reflections, R1 = 0.0752, wR2 = 0.1916, R1 (all data) = 0.1198, wR2 (all data) = 0.2613; CCDC 931734. 2: C120H180Co6N6O50Zr6, hexagonal, P6322, Mr = 3407.6 g mol−1, T = 173(2) K, a = 18.423(5), c = 26.934(7) Å, V = 7917(4) Å3, Z = 2, 42012 reflections, R1 = 0.0850, wR2 = 0.2102, R1 (all data) = 0.0855, wR2 (all data) = 0.2106; CCDC 931733. |
| This journal is © The Royal Society of Chemistry 2014 |