Oxalyl dihydrazide polymorphism: a periodic dispersion-corrected DFT and MP2 investigation†
Davide Prestia,
Alfonso Pedone*a,
Maria Cristina Menziania,
Bartolomeo Civallerib and
Lorenzo Maschio*b
aDipartimento di Scienze Chimiche e Geologiche and INSTM research unit, Università di Modena e Reggio Emilia, Via G. Campi 183, I-41125 Modena, Italy. E-mail: alfonso.pedone@unimore.it; Fax: +39 059 373543; Tel: +39 059 2055043
bDipartimento di Chimica, Università di Torino and NIS Centre of Excellence, Via P. Giuria 7, I-10129 Torino, Italy. E-mail: lorenzo.maschio@unito.it; Fax: +39 011 670 7855; Tel: +39 011 6707564
First published on 18th October 2013
Abstract
The molecular crystal of oxalyl dihydrazide differentiates into five polymorphs that are governed by inter- and intramolecular hydrogen bonds. The complex mixture of such interactions with long range dispersive forces makes its computational characterization very challenging; thus it represents an ideal benchmark for ab initio methods when striving for a description of polymorphism in molecular crystals. Indeed, a complete experimental energetic profile of this system is still lacking, and it is here investigated by means of periodic dispersion-corrected DFT and Local second order Møller–Plesset Perturbation theory (LMP2) calculations. In this work, the empirical dispersion correction schemes proposed by Tkatchenko and Scheffler (TS) [Tkatchenko et al., Phys. Rev. Lett., 2009, 102, 073005] and Grimme (D2) [Grimme, J. Comput. Chem., 2006, 27, 1787] have been used in combination with the PBE semilocal functional for geometry optimizations. We observed that PBE-TS provides a remarkable improvement in predicting the crystal structure of oxalyl dihydrazide polymorphs with respect to commonly used DFT-D functionals. The relative stabilities of the five forms have then been computed at the PBE-TS/D2, PBE0-D2, B3LYP-D2 and B3LYP-D3(BJ)+gCP level on the PBE-TS hydrogen-optimized geometries and benchmarked against high level periodic LMP2 calculations. PBE-TS, B3LYP-D2 and B3LYP-D3(BJ)+E(3) (that is including three-body corrections) achieve good predictions of the stability ordering, though the broadness of the energy range is slightly larger than in the case of LMP2.
1. Introduction
The theoretical modelling of molecular crystals plays a leading role in the design of new materials and polymorphs, making a significant contribution to crystal engineering. The ability to explore the polymorphic landscape for a molecule is of major interest in electronic, pigment, explosive industries1–3 and, in particular, in pharmaceutical research, were new crystalline active pharmaceutical ingredients (APIs) with improved properties such as solubility, biocompatibility, chemical stability, melting point, taste, etc. are increasingly requested from the market.Ab initio modelling of molecular crystals is challenging since the crystal packing is governed by intermolecular interactions, such as long range dispersive forces (vdW) or hydrogen bonding. Dispersive interactions are quantum mechanical in nature and correspond to the multipole moments induced in response to instantaneous fluctuations in the electron density. These long range correlation effects are naturally accounted for by correlated wave-function4–6 and Quantum Monte Carlo (QMC)7 methods. In recent years, post-Hartree–Fock methods have become available also for crystalline systems,5,8–11 and in this context molecular crystals have been specially used as a cross-benchmark of different proposals.12–18 On the other hand, Density Functional Theory (DFT)19,20 has become the most widely used method for evaluating energies and electronic properties of molecules and condensed phases since it generally provides accurate results in a reasonable computational time. However, most of the exchange-correlation functionals (including hybrid functionals) do not include the long range electron correlation effects responsible for van der Waals forces, and thus usually fail to predict structures and energetics of molecular crystals. To overcome this problem, several dispersion-correction schemes have been developed from London-type pairwise corrections21–27 based on either empirical parameters (like the well known DFT-D2 scheme21 and its enhanced D3 version)28 or non-empirical parameters (like the exchange-dipole moment model,29 the Tkatchenko–Scheffler (TS) scheme26 or the TS scheme with the inclusion of many-body dispersion (MBD) effects30) and non-local DFT functionals (e.g. vdw-DF31 and VV10).32
Among these, the Tkatchenko–Scheffler model26 is a simple approach that partly recovers a non-empirical character. It has been already successfully applied to investigate various systems by different research groups,33–40 and will be adopted in the present work in the computational study of the oxalyl dihydrazide (ODH) molecular crystals. ODH exploits a peculiar kind of conformational polymorphism with five known polymorphs (i.e. α, β, γ, δ and ε) whose stabilities are determined by the competition between different types of hydrogen bond (HB) and vdW forces between π-stacked molecules. The α phase is experimentally the denser one (1.76 g cm−3) and involves only the intermolecular hydrogen bonds. The other phases (β, γ, δ, ε), instead, present mixtures of inter- and intramolecular hydrogen bonds and are less dense than the α one. According to density measurements, the α phase was proposed to be the most stable even if there are still no certain proofs, due to the incompleteness of the experimental data.41 The authors, in fact, only point out that the stability ordering is α, δ, ε > γ > β (without any specification about the relative stability between α, δ and ε). Such ordering is justified by the endothermic phase transition of α, δ, ε into γ that occurs at different temperatures under 250 °C, and the great difficulty to obtain the receding metastable β phase.
A few theoretical works have been carried out to shed light on the ODH polymorphs. Karamertzanis et al.42 used a GGA dispersion-corrected D2-PW91 functional to predict the relative stability of the ODH, with reasonably good results. The energies span in a range of about 15 kJ mol−1, that is slightly wider than the usually accepted one (10 kJ mol−1) for experimentally observed polymorphs.43 Recently, Wen et al.44 showed that the energy ranking of the five polymorphs is remarkably sensitive to diverse functionals and basis sets,44 and compared B3LYP-D* results with their fragment-based Hybrid Many-Body Interaction method (HMBI)11,16,44,45 – that combines MP2 level short-range one- and two-body terms with MM two- and many-body long-range terms. They obtained structures in good agreement with the experimental ones, but underlined the inefficacy of DFT-D in predicting the energetics, specially if compared to HMBI. In fact, HMBI single-point energies (SPEs), performed on the B3LYP-D* optimized structures, achieved the correct stability ordering of the five ODH polymorphs within an energy span of 5 kJ mol−1.
Here, we then combine the well-known PBE46 functional with the TS dispersion correction and compare it with the PBE, PBE047 and B3LYP48 functionals, augmented with the DFT-D2 approach21 and the B3LYP functional augmented with the D3(BJ) correction.28 We investigate also the combination of B3LYP-D3(BJ) with the geometrical-CounterPoise (gCP) correction.49 The latter theoretical approach has recently been successfully applied to study the cohesive energy of molecular crystals.50 As a further assessment of the TS scheme and to compare with HMBI results, full periodic Local second order Møller–Plesset perturbation theory (LMP2) calculations of ODH polymorphs are also presented for the first time.
2. Computational details
Ab initio periodic PBE-TS and PBE-D2 computational simulations were carried out by using the CASTEP51,52 package, that employs plane-waves (PWs) as a basis-set in conjunction with norm-conserving pseudo-potentials (NCPP).In the Tkatchenko–Scheffler correction (TS),26 at difference with the DFT-D2 scheme,21 the electron density is used to calculate the Hirshfeld volumes of an atom in isolation and in the bonding environment. It takes advantage of the relationship between polarizability and volume to compute the effective dispersion coefficient for an atom in a molecule, thus accounting for the relative variation in dispersion coefficients of differently bonded atoms. Computed coefficients are then used in a London-type pairwise correction term. This approach is more rigorous from a physical point of view, and the determination of the dispersion coefficients could be considered nearly nonempirical.
An energy cut-off of 800 eV and a 7 × 4 × 3 k-points mesh (distance between k-points: 0.04 Å−1) have been employed in the calculations. They have been carefully individuated as a good compromise between computational cost and accuracy (see ESI†). In geometry optimizations, tolerances have been set on 5 × 10−6 eV per atom, 5 × 10−4 Å, 1 × 10−2 eV Å−1 and 2 × 10−2 GPa for the total energy convergence, the maximum ionic displacement, the maximum force and the maximum stress, respectively.
The starting experimental structures of ODH were taken from the original work of Ahn et al.41 (available in the Cambridge Crystal Structure Database with reference codes VIPKIO01–VIPKIO05). The polymorphs, all belonging to the P21/c symmetry, contain two molecules (28 atoms) per unit cell in the α, β, γ, δ, ε phases and four molecules (56 atoms) in the γ phase, respectively. Since three-dimensional structures from databases were obtained from X-ray diffraction (XRD) techniques, we carried out geometry optimizations of the hydrogen atoms with the PBE-TS method to obtain refined experimental structures. Hereafter, we will refer to them as revised experimental geometries.
Full relaxations of cell parameters and atomic positions have then also been performed at both PBE-TS and PBE-D2 levels of theory. The fully optimized structures have been compared with the experimental ones in order to assess the accuracy of these dispersion corrected functionals in the prediction of the crystal structure of the five oxalyl dihydrazide polymorphs. The last SCF ground-state energies have also been collected to calculate the relative stability of the five polymorphs at the fully relaxed geometry.
Single-point energy calculations on the revised experimental structures have also been carried out with the PBE0-D2, B3LYP-D2 and B3LYP-D3(BJ) functionals by using a development version of the CRYSTAL code.53,54 The B3LYP-D3(BJ) calculations were performed with and without the gCP correction and including both two- and three-body corrections. All electron basis sets have been employed. The large QZVP55 basis set has been considered for PBE0-D2, B3LYP-D2 and B3LYP-D3(BJ) calculations, while the 6-31G(d) basis set has been used when the gCP correction is included.
Periodic Local-MP2 energies have been computed by means of the CRYSCOR program6,8,56 on the revised experimental structures. A p-aug-6-31G(d,p) Gaussian basis-set has been chosen. This has already proven to be a good compromise between computational cost and accuracy of the results in the study of molecular crystals14 and graphene-based nanostructures.57 The local correlation approach58 allows one to exploit the fundamentally local character of dynamic electron correlation, leading to quantum chemical methods that scale linearly with the size of the system. CRYSCOR implements this kind of approach together with fast integral evaluation techniques59 for correlated calculations (MP2) in periodic systems. It employs the periodic Hartree–Fock solution in the basis of Gaussian-type orbitals (GTO) provided by the CRYSTAL code.53,54
All calculations (DFT and LMP2) have been performed at 0 K temperature. Zero-point energy (ZPE) effects have been evaluated through vibrational frequencies calculations with CASTEP – employing the linear-response phonon DOS (Density Of States)60 approach on the fully optimized PBE-TS structures that are, however, very close to the revised experimental ones. As is shown below, ZPE effects have in this case a negligible impact on relative stabilities.
3. Results and discussion
3.1 Crystal structure optimizations
Table 1 reports the relative deviation % of the optimized cell parameters and volume computed at 0 K, with respect to the experimental ones. B3LYP-D* results from the recent paper of Wen et al.44 and D2-PW91 plane-waves calculation42 are also reported for comparison.
| Method/basis-set | a (Å) | b (Å) | c (Å) | γ (°) | Volume (Å3) |
|---|---|---|---|---|---|
| α polymorph | |||||
| Experimenta | 3.622 | 6.832 | 9.129 | 99.30 | 223.0 |
| B3LYP-D*/6-31G(d,p)b | −3.66% | −1.24% | −1.44% | −1.81% | −5.80% |
| B3LYP-D*/TZPb | −1.33% | −0.26% | −1.73% | −1.58% | −2.90% |
| D2-PW91/PWc | −0.80% | −2.28% | −1.14% | −0.61% | −4.04% |
| PBE-D2/NCPP | −4.14% | −1.86% | −0.54% | −1.44% | −6.11% |
| PBE-TS/NCPP | 0.67% | −1.24% | 0.53% | −0.32% | 0.01% |
| ε polymorph | |||||
|---|---|---|---|---|---|
| a Experiment from ref. 41. b Ref. 44. c Ref. 42. | |||||
| Experimenta | 5.364 | 3.841 | 12.319 | 109.00 | 240.0 |
| B3LYP-D*/6-31G(d,p)b | 2.52% | −4.91% | 0.23% | 3.19% | −4.52% |
| B3LYP-D*/TZPb | 3.86% | −3.56% | 1.36% | 3.76% | −1.25% |
| D2-PW91/PWc | −0.39% | 0.57% | −1.44% | 3.01% | −3.43% |
| PBE-D2/NCPP | 2.78% | −2.02% | −0.68% | 3.83% | −2.76% |
| PBE-TS/NCPP | −0.62% | 1.96% | −0.82% | 1.59% | −0.60% |
The overall accuracy given by PBE-TS is remarkable: the error in predicting cell volumes is notably smaller with respect to the other methods and less than 0.6% for all phases but β whose deviation is around 4%. Moreover, in most cases, PBE-TS functional yields very small deviations on cell parameters.
On the other hand, the Grimme dispersion (D2) scheme tends to overestimate the long range interaction and consequently to give rise to more compact cells.
For the β polymorph – which is the less stable phase, and was experimentally obtained and characterized only once41 – the agreement of computational methods with experiments is rather poor. Although the a parameter is considerably overestimated by the PBE-TS (~7%), b and c compensate with an opposite trend, giving the smaller volume deviation (about 4.5%). Conversely, the PBE-D2 leads to a better agreement with experiment of single cell parameters but a larger discrepancy in the cell volume.
The PBE-TS fully optimized structures of the five polymorphs have been reported in Fig. 1 which also shows the complex intermolecular hydrogen bond network that constitutes the three dimensional structure.
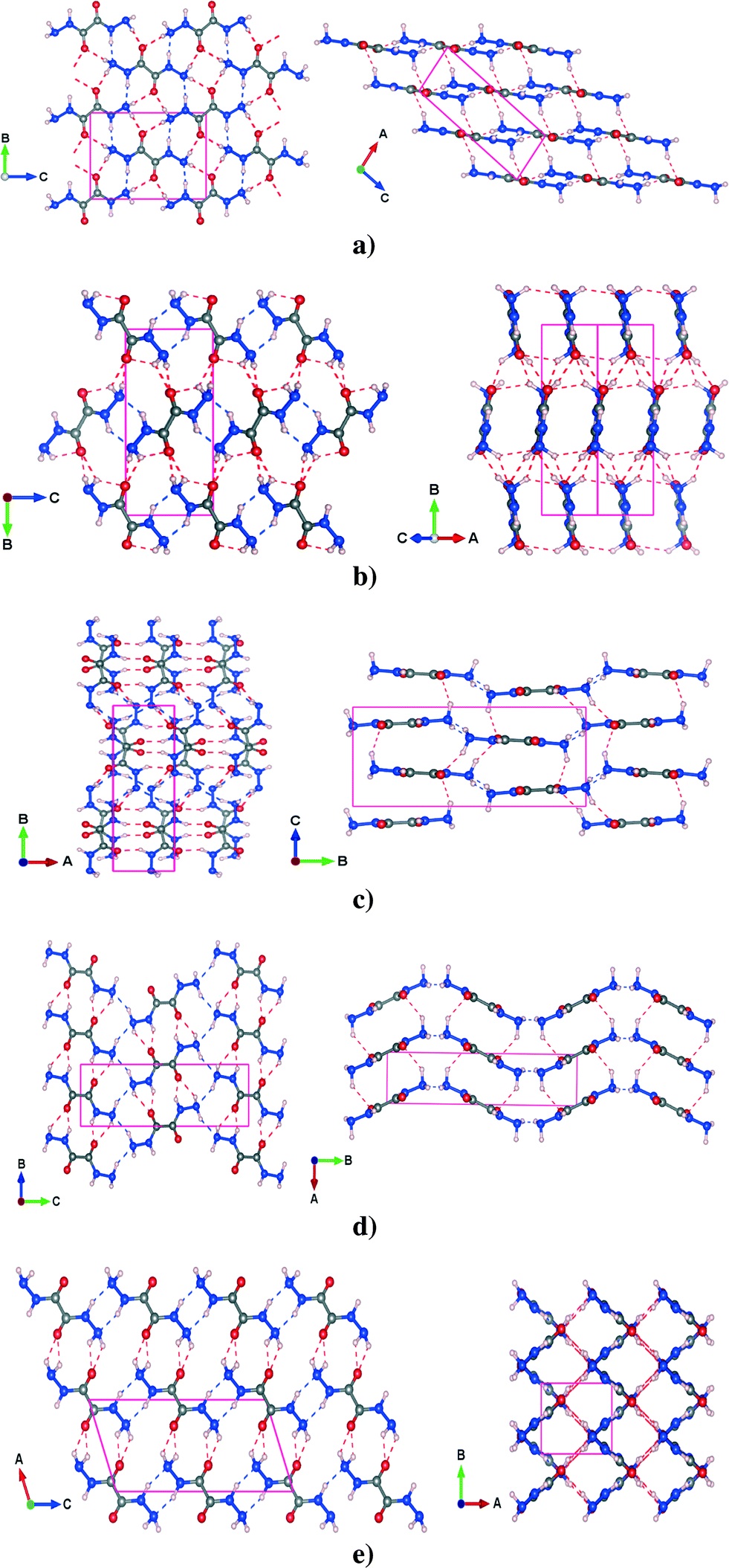 | ||
| Fig. 1 (a) α, (b) β (c) γ, (d) δ and (e) ε polymorphs of crystalline oxalyl dihydrazide, as predicted by PBE-TS/NCPP full structure relaxations. Only intermolecular hydrogen bonds are displayed. The red dashed lines indicate N–H⋯O hydrogen bonds, while the blue dashed lines indicate N–H⋯N hydrogen bonds. | ||
| Hydrogen bond | Type | PBE-TS (Å) | Deviation (Å) | PBE-D2 (Å) | Deviation (Å) | Exp.a (Å) |
|---|---|---|---|---|---|---|
| α polymorph | ||||||
| In-plane | N–H⋯N | 2.882 | −0.037 | 2.833 | −0.086 | 2.919 |
| N–H⋯O | 2.953 | −0.059 | 2.937 | −0.075 | 3.012 | |
| Out-of-plane | N–H⋯O | 3.094 | −0.082 | 3.037 | −0.139 | 3.176 |
| γ polymorph | ||||||
|---|---|---|---|---|---|---|
| In-plane | N–H⋯O | 2.851 | −0.002 | 2.798 | −0.055 | 2.853 |
| N–H⋯O | 2.834 | −0.036 | 2.807 | −0.063 | 2.870 | |
| N–H⋯N | 3.141 | −0.078 | 3.121 | −0.098 | 3.219 | |
| N–H⋯N | 3.105 | −0.043 | 3.092 | −0.063 | 3.184 | |
| Out-of-plane | N–H⋯O | 3.045 | −0.074 | 3.019 | −0.100 | 3.119 |
| N–H⋯O | 3.029 | −0.079 | 2.986 | −0.122 | 3.108 | |
| δ polymorph | ||||||
|---|---|---|---|---|---|---|
| In-plane | N–H⋯O | 2.821 | −0.027 | 2.801 | −0.047 | 2.848 |
| N–H⋯N | 3.086 | −0.076 | 3.060 | −0.102 | 3.162 | |
| Out-of-plane | N–H⋯O | 3.072 | −0.075 | 3.046 | −0.101 | 3.147 |
3.1.2.1 α polymorph. In the α phase, the molecules of ODH lie in planes stacked along the [1 0 −2] direction (see Fig. 1a). The molecules are linked by two types of HBs: N–H⋯N and N–H⋯O on the plane. The out-of-plane HB, which is another N–H⋯O, lies between planes and is a little longer than the other two. The PBE-TS is in good agreement with the experimental data. Distances are slightly underestimated (absolute deviation less than 0.08 Å) and the cell is more compact along the stacking direction. In this case, the PBE-TS slightly overestimates both the vdW π-stacking interactions and the N–H⋯O between planes (this is clearly observable in Table 2), that dominate over the in-plane HBs.
3.1.2.2 β polymorph. The β polymorph is characterized by long and nonplanar hydrogen bonds (see Fig. 1b), that probably are the main cause of its low stability. The only planar HBs are the N–H⋯N from the -NH groups, that link the synthons to form molecular ‘ribbons’ while two types of N–H⋯O HBs lie out-of-plane, between adjacent ribbons. As shown for cell parameters, the structure prediction of PBE-TS is not completely satisfactory: the N–H⋯O formed by –NH groups splits into two different HBs, in disagreement with experimental measurements that provide two equal HBs. However, the PBE-TS absolute deviations are always less than 0.08 Å.
3.1.2.3 γ polymorph. γ is characterized by four different HBs inside the ab molecular plane, two N–H⋯N and two N–H⋯O (see Fig. 1c) whereas two N–H⋯O are located out of the molecular plane. Even in this case the distances are slightly underestimated (max. abs. dev. < 0.08 Å), the cell parameters show such behavior only along the a and the b axis, probably because the in-plane interactions (intramolecular HBs are also present here) dominate the crystal packing over the out-of-plane interaction. This could be the result of a marked rearrangement of molecular synthons from the most stable phases (α, δ and ε). In fact, the γ phase contains four molecules per unit cell – whereas the other phases contain only two.
3.1.2.4 δ polymorph. The δ polymorph is structurally similar to γ, but it contains only two (N–H⋯N and the N–H⋯O) HBs in the ab molecular plane and an N–H⋯O bond out-of-the ab plane (see Fig. 1d). All these distances are slightly underestimated, especially the in-plane N–H⋯N and the out-of-plane N–H⋯O whose deviation with respect to the experimental measurements are around 0.075 Å. The distances between the –NH2 groups found experimentally are shorter, but less directional than in γ and consequently the a parameter and the γ
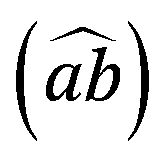 angle are slightly overestimated (see Table 1). Such molecular arrangement – the planes are not perfectly flat – leads also to an overestimation of c, though the N–H⋯O is underestimated.
angle are slightly overestimated (see Table 1). Such molecular arrangement – the planes are not perfectly flat – leads also to an overestimation of c, though the N–H⋯O is underestimated.
3.1.2.5 ε polymorph. This phase is quite different from the others: it shows a ‘grid-like’ shape, in the ab plane, that arises from the alternate arrangement of molecular ribbons perpendicular to each other (see Fig. 1e).
For each ribbon there are two different intermolecular N–H⋯O HBs – one in-plane and one out-of-plane (the latter between parallel ribbons). The N–H⋯N hydrogen bonds from the –NH groups and from the nitrogen of –NH2 groups link adjacent ribbons along c. As always, the PBE-TS gives shorter HB distances that are in agreement with the cell compacting along a and c, thus indicating an overestimation of the interactions between adjacent perpendicular ribbons.
In conclusion, the overall accuracy of PBE-TS in predicting cell parameters and hydrogen bond distances (mean abs. dev. = 0.06 Å and max. abs. dev. = 0.08 Å) of oxalyl dihydrazide appears higher than that obtained with the PBE-D2 functional (mean abs. dev. = 0.11 Å and max abs. dev. = 0.14 Å).
3.2. Relative stability
In this section, we discuss the relative stabilities of the five polymorphs of ODH with respect to the phase α considered as the reference. These have been calculated at the PBE-TS and PBE-D2 level on the hydrogen- (revised experimental) and fully-optimized structures at the same level of theory (PBE-TS/PBE-TS and PBE-D2/PBE-D2) as the energy difference of each polymorph with respect to the α phase.The PBE-TS and PBE-D2 energy rankings, reported in Fig. 2, are consistent with those obtained by D2-PW91 calculations.42 The difference between the most and the less stable phases corresponds to 12–15 kJ mol−1 per molecule, in accordance with the commonly accepted value (10–15 kJ mol−1 per molecule).43,44 PBE-TS leads to the expected experimental stability:41 α, δ, ε > γ > β, which is in agreement with the density ordering of the five polymorphs (α is the most dense – 1.76 g cm−3, β is the less dense – 1.59/1.66 g cm−3) and the experimentally observed endothermic transformations of α, δ, ε into γ while heated below 250 °C – temperature at which ODH decomposes to N,N′-dioxalylhydrazidylhydrazine (DOHH) and gaseous hydrazine through a solid state chemical reaction.
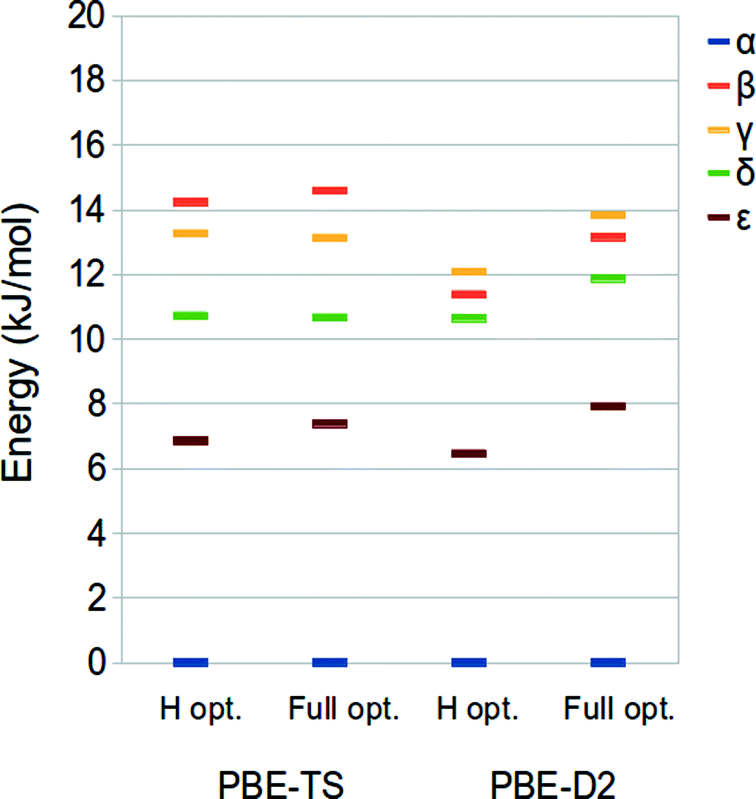 | ||
| Fig. 2 PBE-TS (left) and PBE-D2 (right) relative stability of crystalline oxalyl dihydrazide: for each method both the results from the optimizations of H atoms and full optimizations are shown. The α phase was taken as reference. | ||
The PBE-D2 method, instead, reverses the stability of the two less stable phases, giving β > γ.
The relative stability of the ODH polymorphs as computed for the revised experimental structure (i.e. optimized hydrogen atoms) and the fully relaxed ones is only slightly different, thus indicating that apart from the H atoms positions the potential energy surface of this system is very flat.
We have finally evaluated the zero-point energy (ZPE) contribution as described in the “computational details” section. Results are shown in Fig. 3. It is possible to observe that the inclusion of the ZPE correction does not change the energy ranking, and the energy range is only slightly broadened (different from that observed by Wen et al.).44
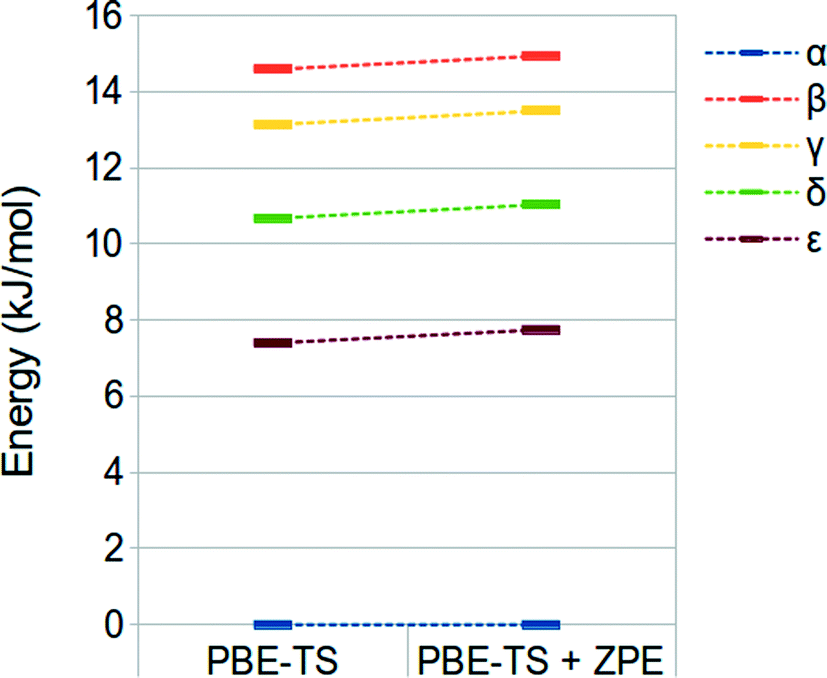 | ||
| Fig. 3 PBE-TS and ZPE corrected PBE-TS relative stabilities of oxalyl dihydrazide at the fully optimized geometry. | ||
A summary of the relative stabilities given by the aforementioned approaches is reported in Table 3 and shown in Fig. 4, together with LMP2 results.
| Relative stability, ΔE (kJ mol−1 per molecule) | ||||
|---|---|---|---|---|
| Method | β | γ | δ | ε |
| a Basis set: QZVP. b Basis set: 6-31G(d). c Basis set: p-aug-6-31G(d.p). | ||||
| PBE-TS | 14.23 | 13.26 | 10.72 | 6.85 |
| PBE-D2 | 11.38 | 12.08 | 10.65 | 6.46 |
| PBE0a-D2 | 26.21 | 8.86 | 9.35 | 6.21 |
| B3LYPa-D2 | 19.45 | 9.85 | 10.36 | 7.93 |
| B3LYPa-D3(BJ) | 20.35 | 7.42 | 7.86 | 4.25 |
| B3LYPa-D3(BJ)+E(3) | 19.22 | 6.63 | 7.20 | 3.37 |
| B3LYPb-D3(BJ)+gCP | 7.30 | 2.18 | 3.95 | −4.07 |
| LMP2c | 9.84 | 6.60 | 7.80 | 5.28 |
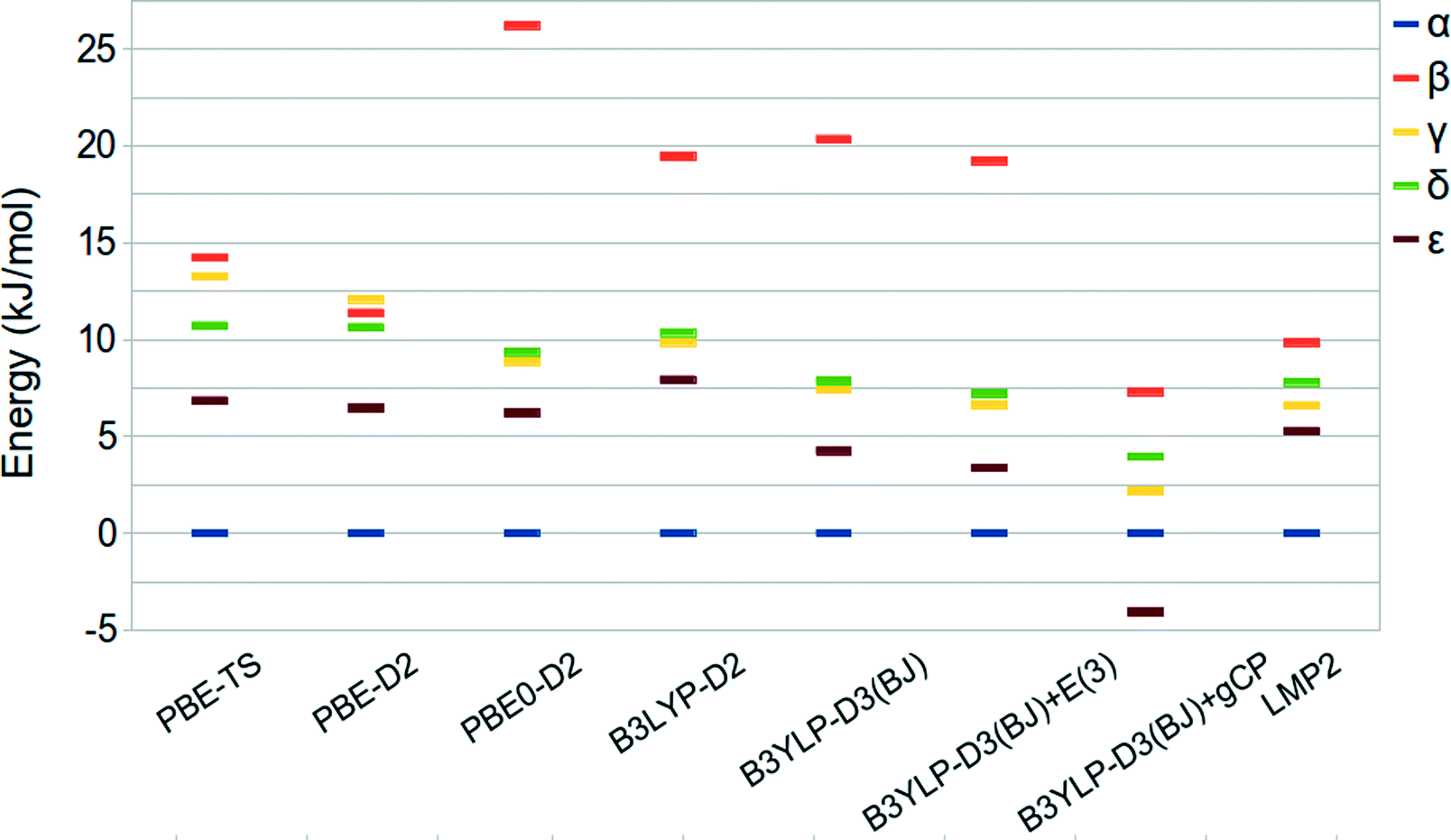 | ||
| Fig. 4 Relative stabilities of oxalyl dihydrazide at the PBE-TS revised experimental (hydrogen optimized) geometry. | ||
The predicted energy ranking obtained by LMP2 calculations is α > ε > γ > δ > β and an energy range below 10 kJ mol−1. These results are slightly different with respect to those reported by Wen et al.44 In fact, their HMBI calculations without including the three body correction provide the δ phase to be more stable than the γ one whereas the inclusion of the three-body dispersion terms reverses the stability order of the α and ε polymorphs. In both cases, the energy range among the five polymorphs is below 5 kJ mol−1. The difference between this value and our LMP2 result can be ascribed to the different approximations inherent to the HBMI and LMP2 approaches, different basis set adopted, and treatment of BSSE effects.
The energy differences obtained at the DFT level of theory underline a marked dependency from the exchange-correlation functional form. In particular, the PBE-TS functional provides a stability ordering more similar to the one obtained by Wen44 since the γ and δ stability is reversed with respect to the LMP2 calculations but the difference between the two phases is very small. Moreover, the energy range among the phases is wider and more similar to that computed at the LMP2 level. On the other hand, PBE0-D2 and B3LYP-D2 provide a stability ordering similar to LMP2 but with a much wider stability range of about 26 and 20 kJ mol−1, respectively.
B3LYP-D3(BJ)/QZVP gives relative stabilities similar to B3LYP-D2 whereas the inclusion of the three-body contribution (E(3)) stabilizes all the other phases with respect to α without changing the stability order. In fact, the E(3) contribution is very small, being about 1.0 kJ mol−1 in agreement with the results reported by Wen et al. in ref. 44.
However, it has to be noted that the Axilrod–Teller–Muto term61,62 is not the most important many-body contribution to dispersion interactions in solids. The polarizability of molecules in solids is significantly influenced by the environment through collective electrodynamic response.63 Very recently, Tkatchenko et al.63–65 have demonstrated that these many-body effects cannot be neglected if one aims at a more accurate prediction of solid-state properties and thus further investigations including such terms should be carried out in the future.
Since Wen et al.44 computed the relative stability of the five polymorphs by employing BSSE corrected total energies we decided to investigate the recently proposed gCP correction,49 which mimics the Boys–Bernardi66 counterpoise correction, in the prediction of the energy ranking of ODH. The gCP correction, as fitted for the 6-31G(d) basis set, is added to the B3LYP-D3(BJ) results. Oddly, the ε phase becomes the most stable one while all other polymorphs are stabilized with respect to α. When compared with other methods, this suggests that the gCP is probably not well balanced.
In spite of the larger stability range, the PBE-TS and B3LYP-D2 functionals seem to be, amongst the considered functionals the most compatible with LMP2 and HMBI results. Interestingly, the B3LYP-D3(BJ)+E(3)/QZVP method gives results very similar to LMP2 for all polymorphs but the β phase.
Conclusions
The polymorphism of oxalyl dihydrazide has been investigated by employing periodic dispersion-corrected DFT and accurate periodic local MP2 calculations. The semiempirical correction schemes proposed by Grimme (DFT-D2) and Tkatchenko and Scheffler (DFT-TS) have been combined with the PBE functional and used to predict the crystal structure of the five crystal forms of oxalyl dihydrazide molecule. Among the two schemes, PBE-TS provides better results on the crystal structure of the five forms of oxalyl dihydrazide and on the relative stability prediction, with respect to the PBE-D2 functional. The comparison with previous HMBI calculations and with fully periodic local MP2 calculations reported in this work for the first time, shows that a stability ordering can be defined, even though it spans a larger range of energies. Structure-dependent dispersion-correction schemes – such as the TS and D3 – lead to results that are reliable, even if relative stabilities of the polymorphs are slightly overestimated with respect to LMP2 results. The B3LYP-D3(BJ)+E(3)/QZVP methods is promising even if the β polymorph is predicted to be too unstable compared with the LMP2 relative stability. On the contrary, the gCP correction, designed to remove the BSSE and devised to compute BSSE-corrected interaction energies, appears to be unsuitable for the evaluation of the relative stability of ODH polymorphs. In spite of that, both D3 and gCP deserve to be further investigated in the prediction of molecular crystals polymorphism.The predicted energy ranking that can be deduced from the different methods adopted in the present work appears to be: α > ε > γ ~ δ > β. Overall, computed results motivate present and future efforts to assess the applicability of dispersion correction schemes to molecular crystal polymorphism.
Acknowledgements
D. P. thanks Regione Emilia-Romagna for financial support through the SPINNER 2013 program entitled ‘Ottimizzazione delle forme molecolari e cristalline di farmaci, fitofarmaci, pesticidi in relazione ad attività, biodisponibilità aspetti brevettuali, e alla produzione di polimorfi, solvati e co-cristalli con metodi a basso impatto ambientale’.References
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889–1896 RSC.
- D. Braga, F. Grepioni, G. I. Lampronti, L. Maini and A. Turrina, Cryst. Growth Des., 2011, 11, 5621–5627 CAS.
- F. R. Manby, D. Alfé and M. J. Gillan, Phys. Chem. Chem. Phys., 2006, 8, 5178–5180 RSC.
- M. Marsman, A. Grüneis, J. Paier and G. Kresse, J. Chem. Phys., 2009, 130, 184103 CrossRef CAS PubMed.
- C. Pisani, L. Maschio, S. Casassa, M. Halo, M. Schütz and D. Usvyat, J. Comput. Chem., 2008, 29, 2113 CrossRef CAS PubMed http://www.cryscor.unito.it .
- W. M. C. Foulkes, L. Mitas, R. J. Needs and G. Rajagopal, Rev. Mod. Phys., 2001, 73, 33–83 CrossRef CAS.
- C. Pisani, M. Schütz, S. Casassa, D. Usvyat, L. Maschio, M. Lorenz and A. Erba, Phys. Chem. Chem. Phys., 2012, 14, 7615–7628 RSC.
- G. H. Booth, A. Grüneis, G. Kresse and A. Alavi, Nature, 2013, 493, 365–370 CrossRef CAS PubMed.
- C. Müller and B. Paulus, Phys. Chem. Chem. Phys., 2012, 14, 7605–7614 RSC.
- G. J. O. Beran and K. Nanda, J. Phys. Chem. Lett., 2010, 1, 3480–3487 CrossRef CAS.
- L. Maschio, D. Usvyat, M. Schütz and B. Civalleri, J. Chem. Phys., 2010, 132, 134706 CrossRef PubMed.
- L. Maschio, D. Usvyat and B. Civalleri, CrystEngComm, 2010, 12, 2429 RSC.
- L. Maschio, B. Civalleri, P. Ugliengo and A. Gavezzotti, J. Phys. Chem. A, 2011, 115, 11179–11186 CrossRef CAS PubMed.
- A. Erba, L. Maschio, S. Salustro and S. Casassa, J. Chem. Phys., 2011, 134, 074502 CrossRef CAS PubMed.
- S. Wen and G. J. O. Beran, J. Chem. Theory Comput., 2011, 7, 3733–3742 CrossRef CAS.
- P. J. Bygrave, N. L. Allan and F. R. Manby, J. Chem. Phys., 2012, 137, 164102 CrossRef CAS PubMed.
- M. D. Ben, J. Hutter and J. VandeVondele, J. Chem. Theory Comput., 2012, 8(11), 4177–4188 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- K. Lee, E. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- P. Jurečka, J. Černý, P. Hobza and D. Salahub, J. Comput. Chem., 2007, 28, 555–569 CrossRef PubMed.
- F. Ortmann, F. Bechstedt and W. Schmidt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205101–205110 CrossRef.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. O. de-la Roza and E. R. Johnson, J. Chem. Phys., 2012, 137, 054103 CrossRef PubMed.
- B. Schatschneider, J. Liang, A. M. Reilly, N. Marom, G. X. Zhang and A. Tkatchenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 060104 CrossRef.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- O. A. Vydrov and T. Van Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- N. Marom, A. Tkatchenko, M. Rossi, V. V. Gobre, O. Hod, M. Scheffler and L. Kronik, J. Chem. Theory Comput., 2011, 7, 3944–3951 CrossRef CAS.
- G.-X. Zhang, A. Tkatchenko, J. Paier, H. Appel and M. Scheffler, Phys. Rev. Lett., 2011, 107, 245501 CrossRef.
- B. Santra, J. c. v. Klimeš, D. Alfè, A. Tkatchenko, B. Slater, A. Michaelides, R. Car and M. Scheffler, Phys. Rev. Lett., 2011, 107, 185701 CrossRef.
- N. Marom, A. Tkatchenko, S. Kapishnikov, L. Kronik and L. Leiserowitz, Cryst. Growth Des., 2011, 11, 3332–3341 CAS.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
- W. Al-Saidi, V. Voora and K. Jordan, J. Chem. Theory Comput., 2012, 8, 1503–1513 CrossRef CAS.
- G. Mercurio, E. R. McNellis, I. Martin, S. Hagen, F. Leyssner, S. Soubatch, J. Meyer, M. Wolf, P. Tegeder, F. S. Tautz and K. Reuter, Phys. Rev. Lett., 2010, 104, 036102 CrossRef CAS.
- A. Pedone, D. Presti and M. C. Menziani, Chem. Phys. Lett., 2012, 541, 12–15 CrossRef CAS PubMed.
- S. Ahn, F. Guo, B. M. Kariuki and K. D. M. Harris, J. Am. Chem. Soc., 2006, 128, 8441–8452 CrossRef CAS PubMed.
- P. G. Karamertzanis, G. M. Day, G. W. A. Welch, J. Kendrick, F. J. J. Leusen, M. A. Neumann and S. L. Price, J. Chem. Phys., 2008, 128, 244708 CrossRef PubMed.
- J. Bernstein, Polymorphism in molecular crystals, Oxford University Press, 2002, vol. 14 Search PubMed.
- S. Wen and G. J. O. Beran, J. Chem. Theory Comput., 2012, 8, 2698–2705 CrossRef CAS.
- G. J. O. Beran, J. Chem. Phys., 2009, 130, 164115 CrossRef PubMed.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
- J. G. Brandenburg, M. Alessio, B. Civalleri, M. F. Peintinger, T. Bredow and S. Grimme, J. Phys. Chem. A, 2013, 117, 9282–9292 CrossRef CAS PubMed.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- S. Clark, M. Segall, C. Pickard, P. Hasnip, M. Probert, K. Refson and M. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, K. Doll, N. M. Harrison, B. Civalleri, I. J. Bush, P. D'Arco and M. Llunell, CRYSTAL09 User's Manual, Università di Torino, Torino, 2010 Search PubMed.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. Saunders and C. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571–573 CrossRef CAS.
- A. J. Thakkar, T. Koga, M. Saito and R. E. Hoffmeyer, Int. J. Quantum Chem., 1993, 48, 343 CrossRef.
- L. Maschio, J. Chem. Theory Comput., 2011, 7, 2818–2830 CrossRef CAS.
- J. T. Tanskanen, L. Maschio, A. J. Karttunen, M. Linnolahti and T. A. Pakkanen, ChemPhysChem, 2012, 13, 2361–2367 CrossRef CAS PubMed.
- P. Pulay, Chem. Phys. Lett., 1983, 100, 151 CrossRef CAS.
- L. Maschio, D. Usvyat, F. Manby, S. Casassa, C. Pisani and M. Schütz, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 75101 CrossRef.
- S. Baroni, S. de Gironcoli, A. Dal Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515–560 CrossRef CAS.
- B. Axilrod and E. Teller, J. Chem. Phys., 1943, 11, 299 CrossRef CAS.
- Y. Muto, Proc. Phys.-Math. Soc. Jpn., 1943, 17, 629 CAS.
- R. A. DiStasio, O. A. von Lilienfeld and A. Tkatchenko, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14791–14795 CrossRef CAS PubMed.
- N. Marom, R. A. DiStasio, V. Atalla, S. Levchenko, A. M. Reilly, J. R. Chelikowsky, L. Leiserowitz and A. Tkatchenko, Angew. Chem., Int. Ed., 2013, 52, 6629–6632 CrossRef CAS PubMed.
- A. M. Reilly and A. Tkatchenko, J. Phys. Chem. Lett., 2013, 4, 1028–1033 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ce41758a |
| This journal is © The Royal Society of Chemistry 2014 |