DOI:
10.1039/C3CE41706A
(Paper)
CrystEngComm, 2014,
16, 82-88
Two octamolybdate-based complexes: hydrothermal synthesis, structural characterization and properties†
Received
26th August 2013
, Accepted 24th September 2013
First published on 10th October 2013
Abstract
Two octamolybdate-based inorganic–organic hybrid complexes constructed from rigid mmt ligand and flexible ttmb ligand respectively (mmt = 5-mercapto-1-methyltetrazole, ttmb = 1,3,5-tri(1,2,4-triazol-1-ylmethyl)-2,4,6-trimethylbenzene), namely, [Ag8(H2O)2(mmt)4(β-Mo8O26)] (1), Ag4(ttmb)2(β-Mo8O26) (2), have been successfully synthesized. Crystal structural analysis reveals that compound 1 is built up of [β-Mo8O26]4− clusters covalently linked by Ag–mmt metal organic layers, which possess interesting hexa-nuclear Ag clusters. In compound 2, the wave-like Ag–ttmb layers are linked by 1D Ag–[β-Mo8O26]4− chains to form a 3D framework. The electrochemical behavior of 1–2 and the luminescent property of 2 were investigated.
Introduction
Polyoxometalates (POMs), as an outstanding class of metal-oxide clusters, arouse chemists' attention not only because of their intriguing structural particularities such as controllable size, shape, high-negative charge, and nucleophilic oxygen-enriched surface, but also due to their potential applications in catalysis, biology, magnetism, materials science, optics, and medicine.1–6 Octamolybdate, as a notable subset of POM clusters, have been subjected to a vast number of studies because of their diverse isomers and attractive properties.7–11 According to the different numbers and arrangement of MoO4, MoO5, and MoO6, octamolybdate (Mo8) have eight isomers (α, β, γ, δ, ε, ζ, η, θ),12–16 which are easily transformed from one to the other under mild environment modifications. Furthermore, octamolybdate can be easily obtained under hydrothermal conditions by using (NH4)6Mo7O24 with controlled pH values.
The assembly of metal–organic frameworks (MOFs) constructed from metal ions as connectors and organic ligands as linkers have drawn more and more attention, owing to their interesting structural features and wide applications as functional materials.17 By rational design and assembly, it is appealing to introduce MOFs to POMs (especially Mo8) for constructing POMs supporting MOFs with diverse applications.18,19
In the synthesis of POMs supporting MOFs, the organic moieties have significant effects on the final structures. Usually, the rigid ligands have a few or even no conformational changes when they react with metal ions,20,21 while flexible ligands can adopt varied conformations and coordination modes according to the different geometric requirements of the metal ions.22,23 Additionally, transition-metal ions, especially the soft d10 Ag+ ions, are often utilized as versatile connectors in the construction of transition-metal complexes. Ag+ ions have a strong coordination ability with organic ligands with various coordination geometry and have been known to form many types of coordination polymers.
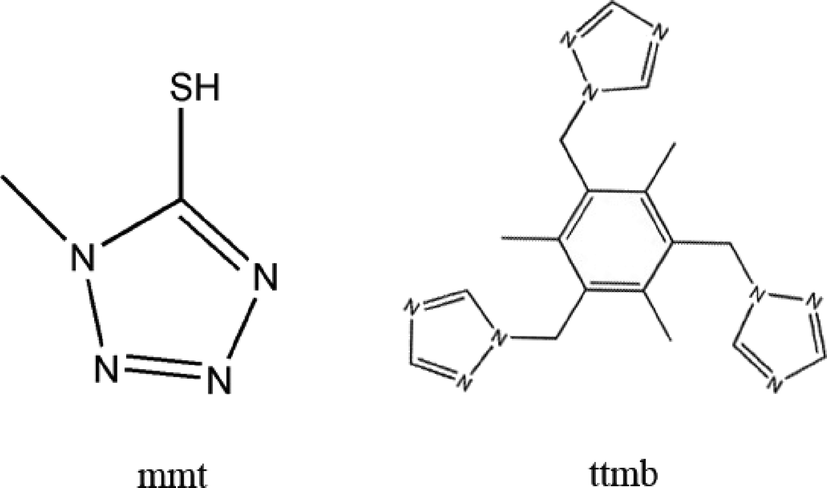
On the basis of the above-mentioned considerations, in this paper, by utilizing the structurally rigid 5-mercapto-1-methyltetrazole (mmt) ligand and flexible 1,3,5-tri(1,2,4-triazol-1-ylmethyl)-2,4,6-trimethylbenzene (ttmb) (Scheme 1), two novel compounds, namely [Ag8(H2O)2(mmt)4(β-Mo8O26)] (1), Ag4(ttmb)2(β-Mo8O26) (2), have been synthesized under hydrothermal conditions. Structural analysis indicates that 1 is built up of [β-Mo8O26]4− clusters covalently linked by Ag–mmt metal organic layers, which possess interesting hexa-nuclear Ag clusters. To the best of our knowledge, compound 1 presents the first example of octamolybdate containing mmt ligands and hexa-nuclear Ag clusters. In compound 2, the wave-like Ag–ttmb layers are linked by 1D Ag–[β-Mo8O26]4− chains to form a 3D framework.
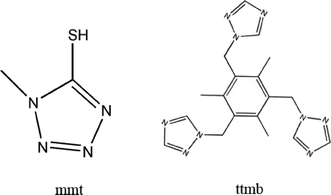 |
| | Scheme 1 The organic ligands used in this work (mmt for 1 and ttmb for 2). | |
Experimental
Materials and equipment
The ttmb ligands were prepared according to literature methods.24 While other chemicals purchased were of reagent grade and used without further purification. The elemental analysis (C, H, N) were performed on a Perkin-Elmer 2400 elemental analyzer. The IR spectra of compounds 1–2 were recorded with a Nicolet Impact 410 FTIR spectrometer with pressed KBr pellets in the 4000–500 cm−1 region. TG measurement was carried out on a Diamond thermogravimetric analyzer in flowing N2 atmosphere from 50 to 800 °C with a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) data were obtained using a Bruker D8 Advance diffractometer with Cu Kα radiation (λ = 1.54056 Å), with a step speed of 0.1° per second. A CHI 440 electrochemical quartz crystal microbalance connected to a Digital-586 personal computer was used for the electrochemical experiments. A conventional three-electrode cell was used at room temperature. A saturated calomel electrode (SCE) was used as the reference electrode and a platinum wire as the counter electrode. The compounds 1–2 bulk-modified carbon paste electrodes (1–CPE and 2–CPE) were used as the working electrodes. The solid-state emission/excitation spectra of compound 2 were measured on a FP-6500 spectrofluorimeter equipped with a 450 W xenon lamp as the excitation source.
Synthesis of [Ag8(H2O)2(mmt)4(β-Mo8O26)] (1)
A mixture of (NH4)6Mo7O24·4H2O (0.3219 g, 0.2606 mmol), AgNO3 (0.1835 g, 1.086 mmol), mmt (0.0599 g, 0.5 mmol), and deionized water (10 ml) was stirred for 20 min in open air. The pH was adjusted to 3.08 by NaOH (2 M) and HNO3 (2 M). Then the solution was stirred for another 20 minutes. At last the mixture was transferred to a 20 mL Teflon-lined autoclave and heated to 170 °C for 3 days. After being cooled to room temperature, the colourless block-like crystals were filtered, washed with deionized water and dried in air (0.1682 g, yield 32% based on Mo). Elemental analysis: found C 3.38%, H 0.59%, and N 8.18% (calc. C 3.78%, H 0.55%, and N 8.82%).
Synthesis of Ag4(ttmb)2(β-Mo8O26) (2)
A mixture of (NH4)6Mo7O24·4H2O (0.2396 g, 0.1940 mmol), AgNO3 (0.0692 g, 0.4094 mmol), ttmb (0.0715 g, 0.2 mmol), and deionized water (10 ml) was stirred for 20 min in open air. The pH was adjusted to 1.13 by HNO3 (1 M). Then the solution was stirred for another 20 minutes. At last the mixture was transferred to a 20 mL Teflon-lined autoclave and heated to 170 °C for 3 days. After being cooled to room temperature, the colourless block-like crystals were filtered, washed with deionized water and dried in air (0.1152 g, yield 29% based on Mo). Elemental analysis: found C 19.21%, H 1.61%, and N 10.41% (calc. C 18.47%, H 1.81%, and N 10.77%).
X-ray crystallography
The single crystals of compounds 1–2 were singled out by visual examination under the microscope and glued at the top of a thin glass fiber with epoxy glue in air for data collection, and the crystallographic data were collected on a Bruker Apex II CCD with Mo-Kα radiation (λ = 0.71073 Å) at 296 K using ω–2θ scan method. The crystal structures were solved by direct method and refined on F2 by full-matrix least-squares methods using the SHELX97 program package.25 [β-Mo8O26]4− in 2 is disordered, the occupied factors of Mo2, Mo3, Mo4, Mo5, Mo6, Mo7, O1, O2, O3, O4, O5, O6 and O7 are 0.5. The crystal data are presented in Table 1. The selected bond lengths for 1 and 2 are listed in Tables S1 and S2† respectively.
Table 1 Crystal data and structure refinement for 1 and 2
| Compound |
1
|
2
|
| Empirical formula |
C8H14Ag8Mo8N16O28S4 |
C18H21Ag2Mo4N9O13 |
| Formula weight |
2541.07 |
180.14 |
| Temperature (K) |
296(2) |
296(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
| Crystal system, space group |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
|
a (Å) |
10.5781(14) |
9.982(2) |
|
b (Å) |
11.0107(14) |
11.711(4) |
|
c (Å) |
11.389(3) |
13.987(2) |
|
α (°) |
101.089(2) |
72.133(3) |
|
β (°) |
109.288(2) |
74.511(2) |
|
γ (°) |
108.4730(10) |
78.879(2) |
| Volume (Å3) |
1120.3(3) |
1488.7(6) |
|
Z
|
1 |
13 |
| Calculated density (g cm−3) |
3.767 |
2.612 |
| Absorption coefficient (mm−1) |
5.850 |
3.001 |
|
F(000) |
1174 |
1116 |
| Crystal size (mm3) |
0.18 × 0.14 × 0.12 |
0.15 × 0.13 × 0.12 |
| Limiting indices |
−12 ≤ h ≤ 12 |
−12 ≤ h ≤ 11 |
| −12 ≤ k ≤ 13 |
−14 ≤ k ≤ 12 |
| −13 ≤ l ≤ 13 |
−16 ≤ l ≤ 16 |
| Reflections collected |
8032 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 869 869 |
| Independent reflection |
3906 [R(int) = 0.0222] |
5353 [R(int) = 0.0255] |
| Completeness |
98.80% |
98.20% |
| Max. and min. transmission |
0.5403 and 0.4190 |
0.7147 and 0.6617 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
3906/2/326 |
5353/37/542 |
| Goodness-of-fit on F2 |
1.09 |
1.089 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0323, wR2 = 0.0714 |
R
1 = 0.0503, wR2 = 0.1457 |
Results and discussion
Crystal structures
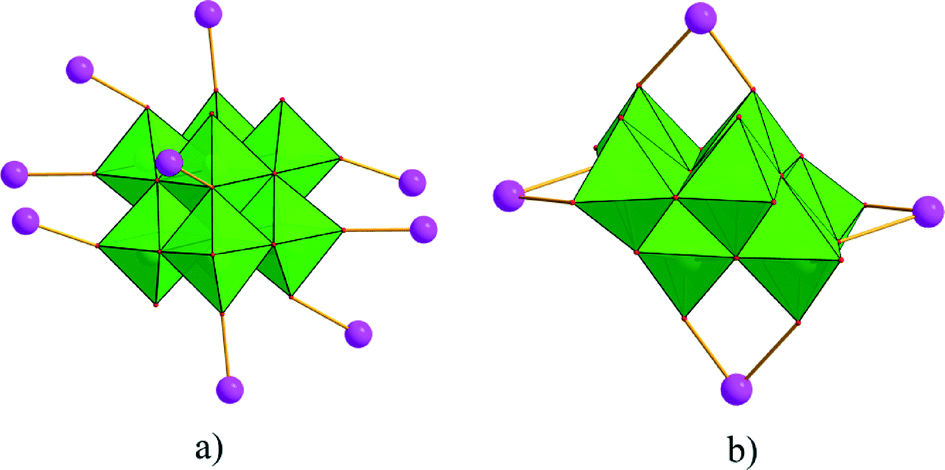
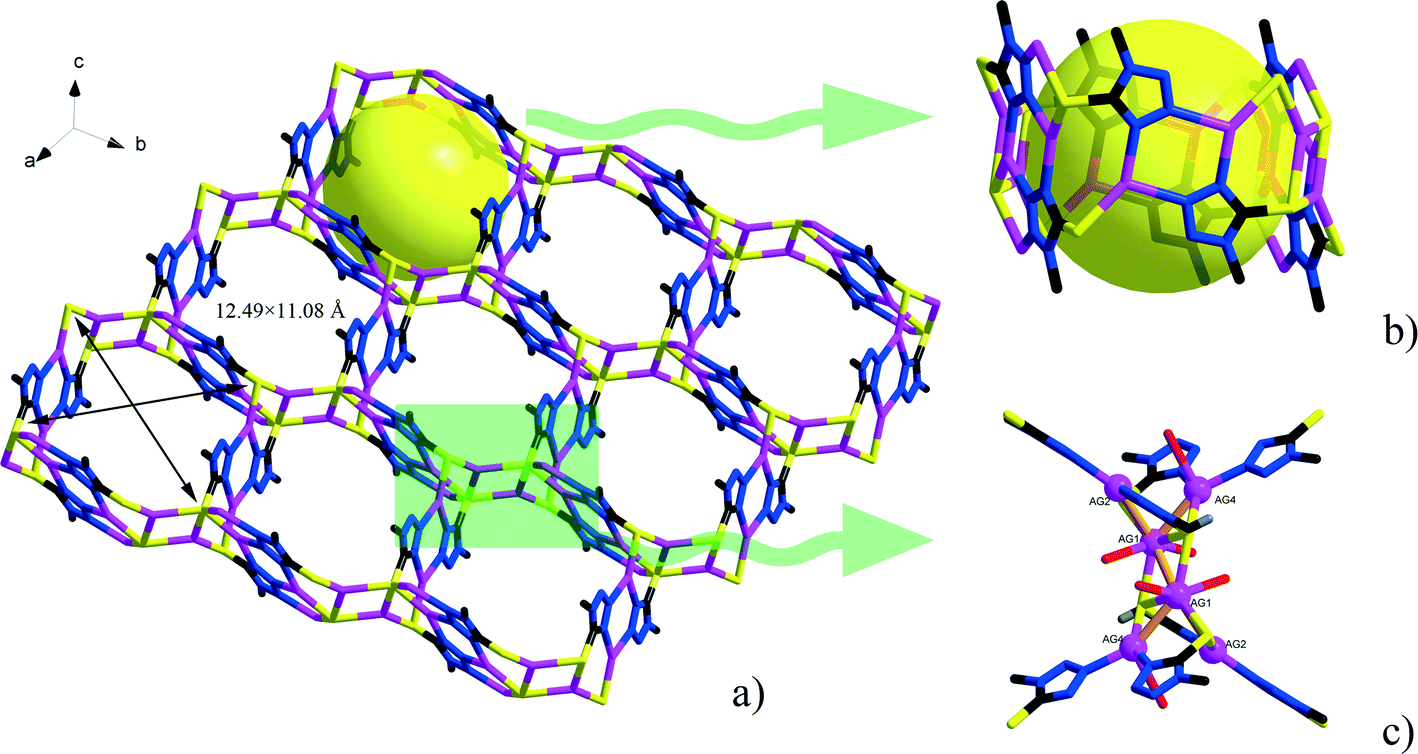
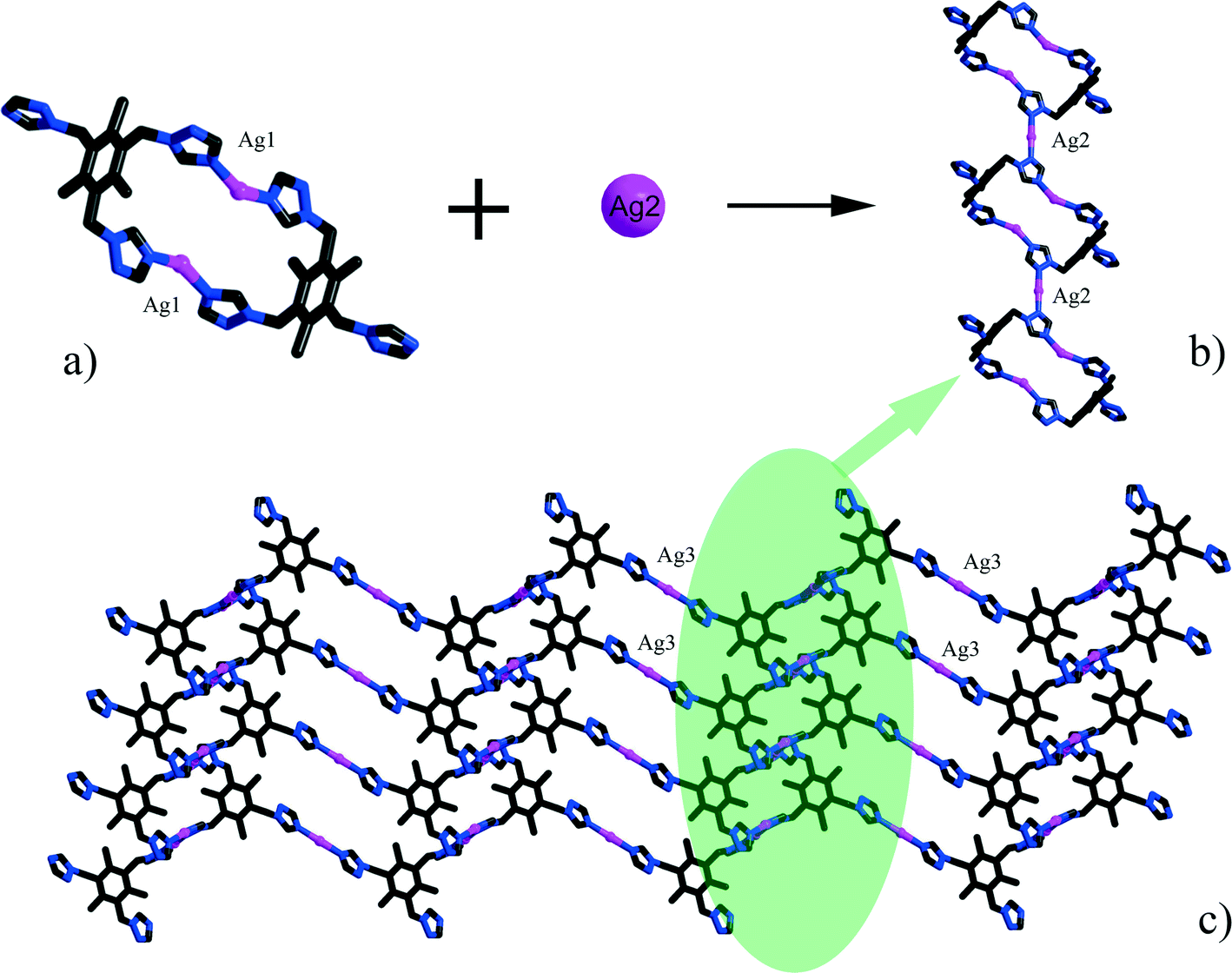
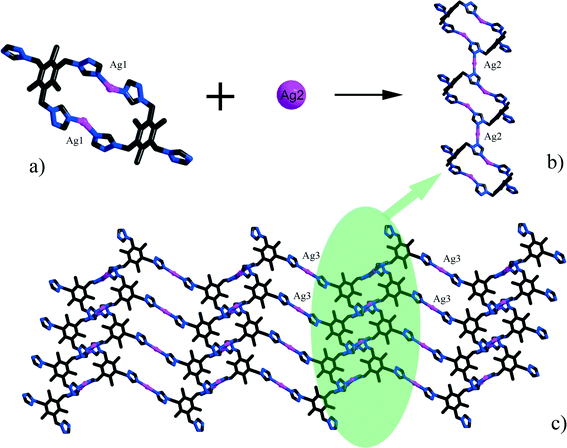
Single-crystal X-ray diffraction analyses of the two compounds reveal that they are both in the low-symmetry triclinic space group P. The asymmetric unit of 1 consists of four Ag+ ions, two mmt ligands, one coordinated water, and a half [β-Mo8O26]4− anion. The typical [β-Mo8O26]4− anion in 1 is composed of eight distorted {MoO6} octahedra with four kinds of O atoms, six μ2-O, four μ3-O, two μ5-O, and twelve Ot (Fig. 1). In the structure of compound 1, there are four coordination types of Ag. Ag1 adopts distorted trigonal bipyramid geometry, coordinated by two O atoms from [β-Mo8O26]4− anion and three S atoms from mmt ligands, with Ag–O bond distances of 2.4213(6)–2.5895(8) Å, Ag–S bond distances of 2.5885(7)–2.7711(11) Å. Ag2 shows a three-coordinated “Y-type” geometry, and is coordinated by two N atoms and one S atom from three mmt ligands. The bond distances around Ag2 are 2.5069(8) Å for Ag–S, 2.2207(9)–2.3622(10) Å for Ag–N. Ag3 exhibits distorted trigonal bipyramid geometry, coordinated by two O atoms from [β-Mo8O26]4− anion, one S atom and one N atom from mmt, and one O atom from water molecule. The bond distances are 2.4094(8) Å for Ag–N, 2.6089(7) Å for Ag–S, 2.4176(6)–2.6946(9) Å for Ag–O. Ag4 adopts distorted tetrahedral geometry, coordinated by one O atoms from [β-Mo8O26]4− anion, one S atom and two N atoms from mmt ligands, with Ag–O bond distance of 2.8313(8) Å, Ag–S bond distance of 2.4938(7) Å and Ag–N bond distances of 2.2200(8)–2.3467(7) Å. All the bond lengths and angles are in agreement with the reported compounds.26–29 In compound 1, the mmt ligands are connected by Ag+ ions to form a metal–organic layer, which possesses a cycle with the dimension of 12.49 × 11.08 Å (Fig. 2). Notably, the mmt ligands in compound 1 adopt two different coordination modes and conformations. They bridge adjacent Ag+ through N and S atoms as pentadentate and hexadentate linker respectively to form the grid-like metal–organic layers. The [β-Mo8O26]4− anion, acts as ten-connected inorganic ligand located in the cycle and further stabilizes the metal organic frameworks (Fig. 3b). Then, adjacent layers are linked through Ag3 to form the 3D POMs supporting MOFs (Fig. 3a). In the structure of 1, two notable features exist: (1) the [β-Mo8O26]4− anion, acting as multidentate inorganic ligand, is coordinated by ten Ag+ ion, which is rare in the octamolybdate-supporting MOFs (Fig. 1a). (2) An interesting hexanuclear Ag cluster is found in the 2D metal–organic layer (Fig. 2c), which represents the highest nuclear Ag cluster in octamolybdate-based inorganic–organic hybrid complexes.
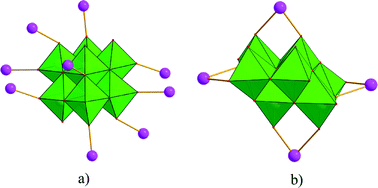 |
| | Fig. 1 Coordination types of [β-Mo8O26]4− anion in compounds 1 and 2. | |
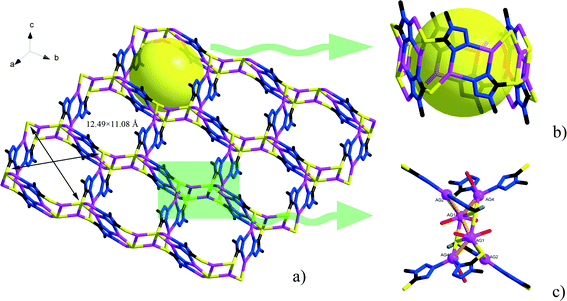 |
| | Fig. 2 a) The 2D metal–organic layer constructed from Ag+ and mmt ligands. b) The detailed representation of the channel in the 2D layer. c) The hexanuclear Ag cluster in the 2D layer. | |
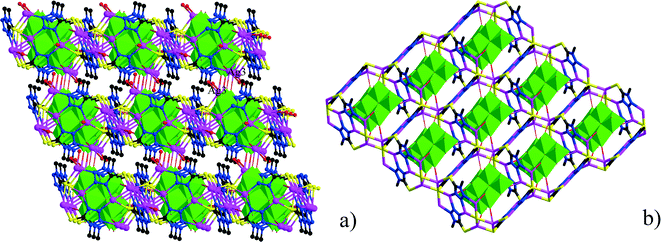 |
| | Fig. 3 The 3D frameworks in 1. | |
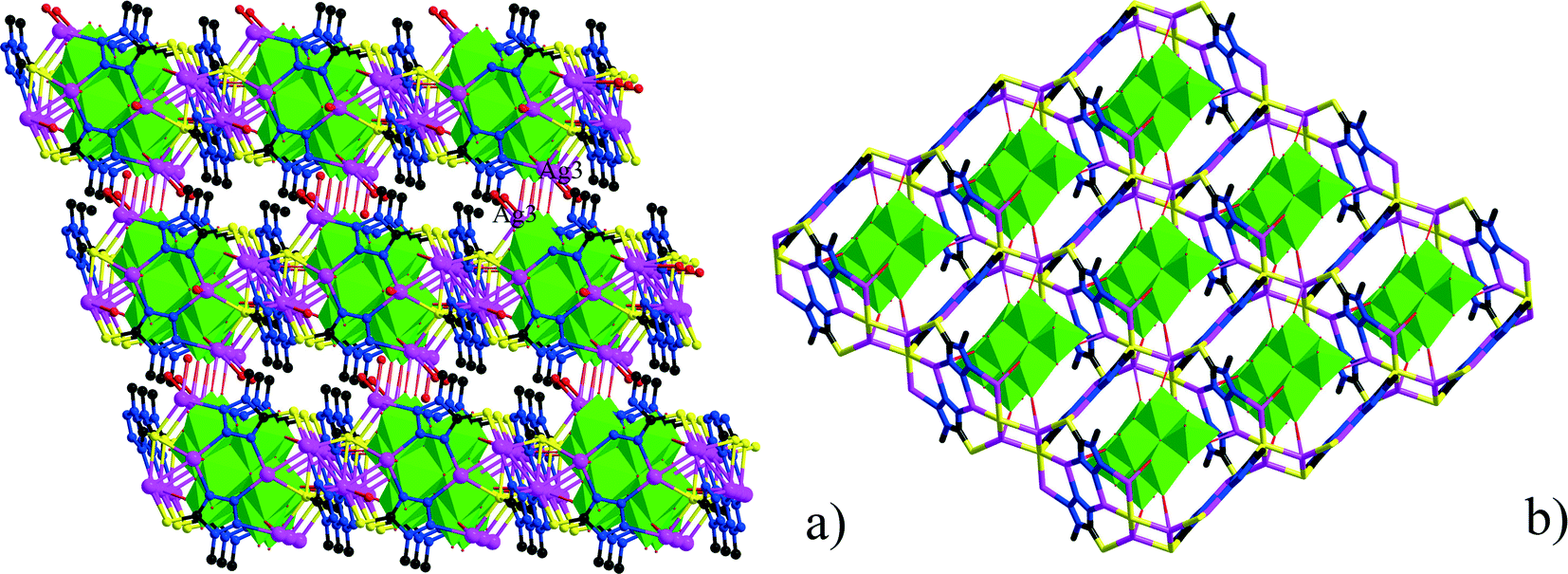
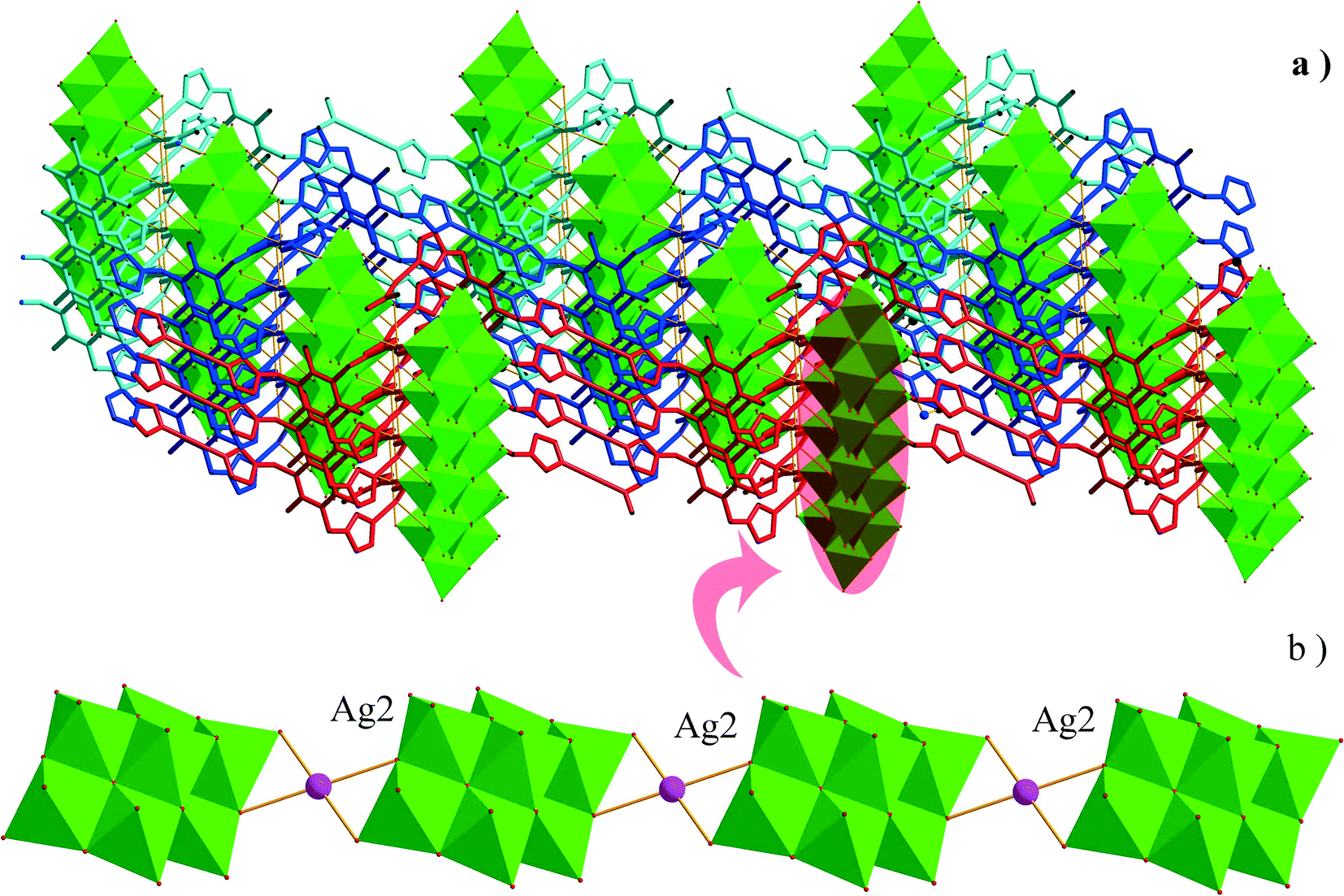
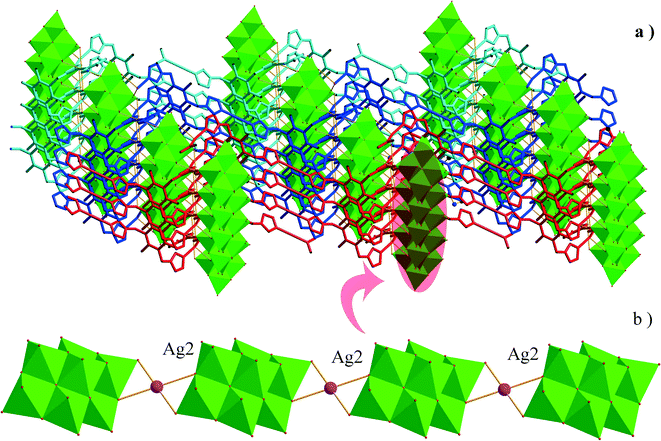
The asymmetric unit of 2 consists of three Ag+ ions, one ttmb ligand, and a half [β-Mo8O26]4− anion. In compound 2, [β-Mo8O26]4− anion is disordered. Mo2, Mo3, Mo4, Mo5, Mo6, Mo7, O1, O2, O3, O4, O5, O6, O7, Ag2 and Ag2A are with the occupied factors of 0.5. The Ag+ cations in compound 2 have three types of coordination environment. Ag1 adopts distorted tetrahedral geometry, coordinated by two O atoms from [β-Mo8O26]4− anion and two N atoms from ttmb, with Ag–O bond distances of 2.5116(4)–2.7655(5) Å, Ag–N bond distances of 2.1505(3)–2.7711(11) Å. Ag2 exhibits distorted octahedral geometry, coordinated by four O atoms from [β-Mo8O26]4− anion and two N atoms from ttmb. The bond distances are 2.1520(4) Å for Ag–N, 2.5844(4)–2.7628(8) Å for Ag–O. The Ag3 is two-coordinated by two N atoms from ttmb in a linear geometry. All the bond lengths and angles are in agreement with the reported compounds.30,31 In 2, Ag1 is linked by ttmb ligands to form a metal–organic ring (Fig. 4a), which is further linked through Ag2 to form an infinite chain (Fig. 4b). While adjacent chains are connected by Ag3 to form a wave-like metal–organic layer (Fig. 4c). The [β-Mo8O26]4− anion acts as an inorganic ligand bridging Ag2 to form a 1D inorganic chain (Fig. 5b). The connection of 2D metal–organic layer and 1D inorganic chain in vertical direction constructs the 3D framework of 2 by sharing Ag2 and Ag1 (Fig. 5a). Ma and coworkers had reported one 2D compound using the ttmb ligand.32 Nevertheless, the different conditions in this paper resulted in the formation of an interesting 3D conformation.
 |
| | Fig. 4 a) The metal–organic ring in 1. b) The metal–organic chain in 1. c) The 2D wave-like layer in 1. | |
 |
| | Fig. 5 a) The connection types of 1D inorganic chain and 2D metal–organic layers. b) The 1D inorganic chain in 2. | |
Influence of pH value in the synthesis
Parallel experiments indicate that compound 1 could be obtained in a wider pH range of about 3–4. However, the yield is rather low when the pH is adjusted to about 4. In this paper, the crystals of compound 1 were synthesized at pH 3.08. The synthesis of compound 2 was sensitive to the pH value, no crystals could be obtained if the pH was lower than 1 or higher than 1.5.
Influence of ligand configurations on the structure of POMs supporting MOFs
Generally speaking, many factors affect the construction of POMs supporting MOFs, such as the selection of metal cations, organic ligands and the charge of POMs, etc. In this paper, we focus on the influence of ligand configurations on the final structures of POMs supporting MOFs. As reported previously, rigid bridging ligands, such as piperazine, imidazole, and 1,2,4-triazole, which exhibit an extensively documented ability to bridge metal ions to afford polynuclear clusters, are regarded as an excellent candidate for the synthesis of coordination polymers. By using mmt ligands which has three adjacent N-donor atoms and one additional S-donor atom, it forms an hexa-nuclear Ag cluster in compound 1. Compared with the mmt ligands, the ttmb ligands used in the synthesis of compound 2 is more structurally flexible. The 1,2,4-triazole groups of ttmb ligands can freely twist around the –CH2– groups, which means that ttmb can adopt different coordination conformations to construct coordination polymers. On the other hand, the distance between adjacent coordination sites is rather long, which may restrain the formation of polynuclear clusters.
IR spectra
The IR spectra of 1–2 are presented in Fig. S3–S4.† The peaks in the range of 582–887 cm−1 for 1, 665–938 cm−1 for 2 are attributed to ν(Mo–O–Mo) and ν(Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibrations. While peaks in the range of 1166–1630 cm−1 for 1 and 1135–1645 cm−1 for 2 are due to the stretching vibrations of organic ligands. The bands around 3400 cm−1 are associated with the water molecules. The features are in agreement with the reported compounds.33–35
O) vibrations. While peaks in the range of 1166–1630 cm−1 for 1 and 1135–1645 cm−1 for 2 are due to the stretching vibrations of organic ligands. The bands around 3400 cm−1 are associated with the water molecules. The features are in agreement with the reported compounds.33–35
Thermal analysis
The thermogravimetric analysis (TGA) experiments were performed under N2 atmosphere in the range from room temperature to 800 °C. The TG curve of 1 indicates that weight loss can be divided into two distinct stages (Fig. S5†). The first weight loss of 1.32% (calc. 1.54%) at 50–300 °C corresponds to loss of water molecules. From 300 to 800 °C, the second weight loss is attributed to the decomposition of organic ligands (weight loss: exp. 18.78%, calc. 18.21%). As shown in Fig. S6,† compound 2 is thermally stable up to around 300 °C, and mass losses occur between 300–800 °C due to the decomposition of organic ligands.
Powder X-ray diffractions
Fig. S1–S2† presents the powder X-ray diffraction patterns for compounds 1–2. The diffraction peaks of both simulated and experimental patterns match well in relevant positions, thus indicating that the phase purities of the compounds 1–2 are good. The difference in reflection intensities between the simulated and the experimental patterns is due to the different orientation of the crystals in the powder samples.
Luminescent property
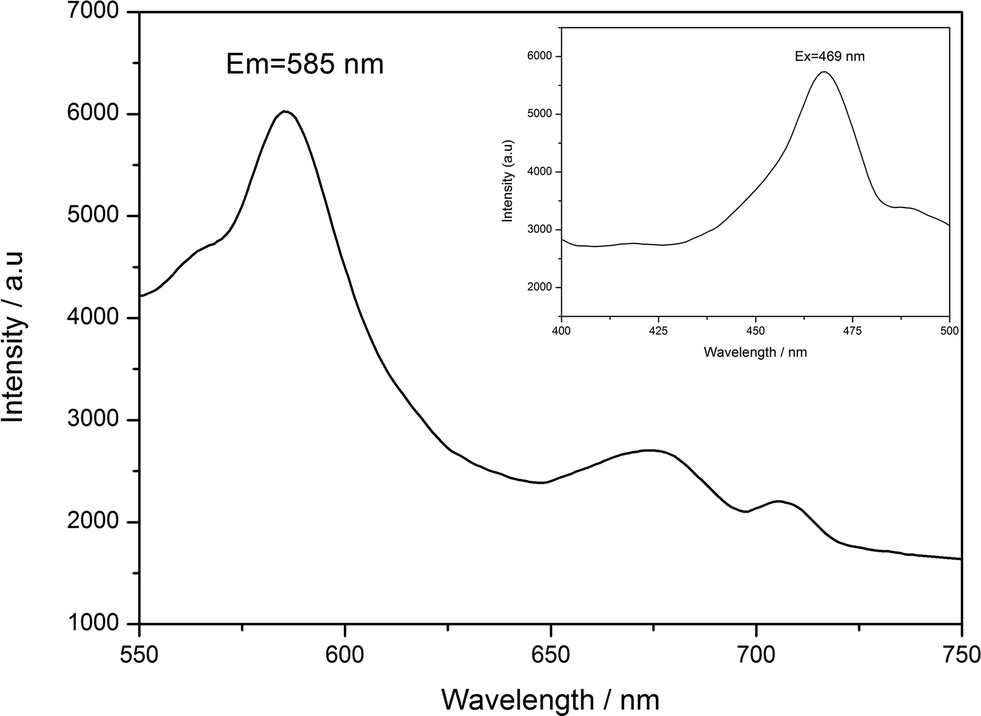
The fluorescence properties of POMs-based inorganic–organic hybrid compounds have been reported by many groups due to their potential applications as luminescent materials.36,37 According to the literature,38 free ttmb ligand shows feeble emission peak at 400 nm, while excited at 340 nm in the ultraviolet (UV) region. As shown in Fig. 6, compound 2 displays strong green fluorescence, the maximal emission and excitation wavelength are 585 nm and 469 nm, respectively. The origin of the emission can be tentatively attributed to ligand-to-metal charge transfer (LMCT) and the fluorescence property shows that compound 2 is a good candidate for fluorescent materials. The enhanced fluorescence efficiency of 2 is attributed to the coordination of the ttmb ligand to the Ag(I) ions that reduces the loss of energy by radiationless and increase the rigidity of the ligand, while the energy levels difference caused by the coordination environment makes the red shift of the emission from UV ttmb light to the green color region. However, the fluorescence property of compound 1 cannot be observed under experimental conditions.
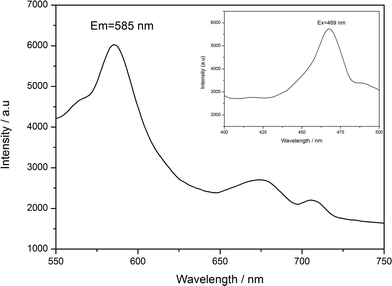 |
| | Fig. 6 Emission spectrum of 2 at room temperature. | |
Voltammetric behavior
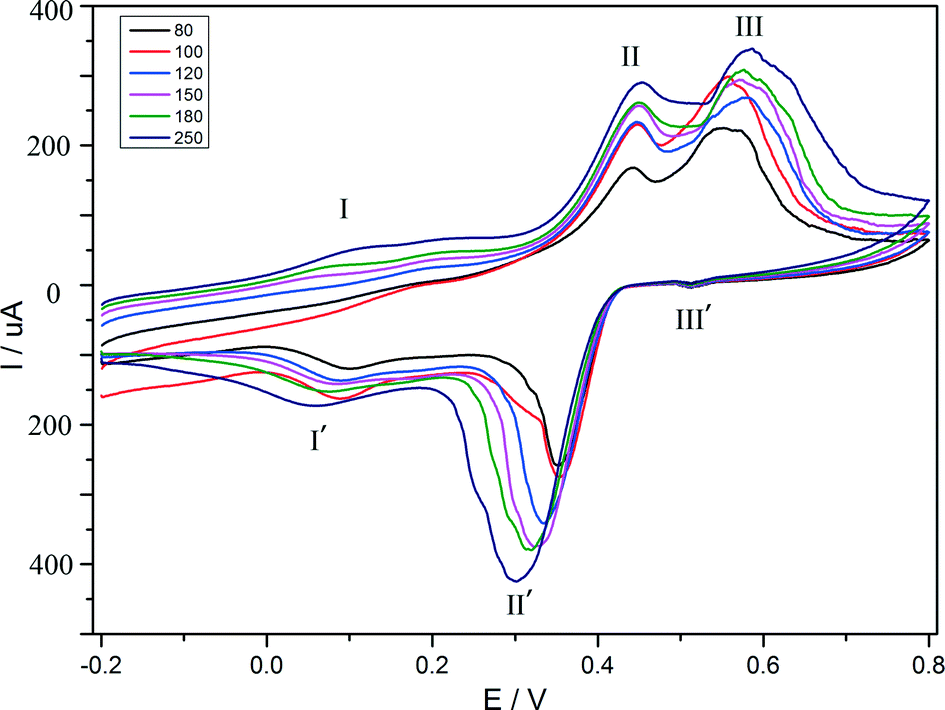
The electrochemical behaviour of compound 1 is investigated in 1 M H2SO4 aqueous solution at different scan rates. Because compound 1 is insoluble in water and common organic solvents, the bulk-modified CPE becomes the optimal choice to study the electrochemical properties, which is easy to prepare and handle. The CV plot for 1–CPE in 1 M H2SO4 aqueous solution at different scan rates is presented in Fig. 7. Three reversible redox peaks appear in the potential range of 800 to −200 mV. The mean peak potentials (E1/2 = (Epa + Epc)/2) are 521 mV (III–III′), 395 mV (II–II′), 111 mV (I–I′), respectively. THe redox peaks I–I′, II–II′, III–III′ can be ascribed to three consecutive two-electron processes.39,40 The peak potentials change gradually following the scan rates from 80 to 250 mV s−1: the cathodic peak potentials shift toward the negative direction and the corresponding anodic peak potentials to the positive direction with increasing scan rates. The electrochemical behaviour of compound 2 is similar to compound 1 and is presented in the supporting information (Fig. S7†).
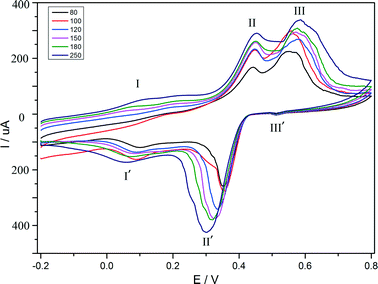 |
| | Fig. 7 Cyclic voltammograms of the 1–CPE in 1 M H2SO4 aqueous solution at different scan rates (from inner to outer: 80, 100, 120, 150, 180, 250 mV s−1). | |
Conclusions
Two octamolybdate-based inorganic–organic hybrid complexes were obtained under hydrothermal conditions. The 3D structure of compound 1 is built up of [β-Mo8O26]4− clusters covalently linked by Ag–mmt metal organic layers, which possess interesting hexa-nuclear Ag clusters. To the best of our knowledge, the Ag clusters in 1 represent the highest nuclear Ag cluster in octamolybdate-based inorganic–organic hybrid complex. In compound 2, the wave-like Ag–ttmb layers are linked by 1D Ag–[β-Mo8O26]4− chains to form a 3D framework. The different structures of compounds 1 and 2 demonstrate that the conformations of organic ligands have a great influence on the final structure.
Acknowledgements
We thank the Natural Science Foundation of Jiangsu Province (Grant BK2012823) and the Ph.D. Programs Foundation of Ministry of Education of China (20113221120002) for financial support.
References
- C. L. Hill and G. C. White, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed.
- D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef CAS PubMed.
- A. Dolbecq, E. Dumas, C. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed.
- X. L. Wang, C. Qin, E. B. Wang, Z. M. Su, Y. G. Li and L. Xu, Angew. Chem., Int. Ed., 2006, 45, 7411 CrossRef CAS PubMed.
- J. M. C. Juan, E. Coronado and A. G. Arino, Chem. Soc. Rev., 2012, 41, 7464 RSC.
- J. M. Poblet, X. Lopez and C. Bo, Chem. Soc. Rev., 2003, 32, 297 RSC.
- Z. Y. Shi, X. J. Gu, J. Peng and Z. F. Xin, Eur. J. Inorg. Chem., 2005, 2005, 3811 CrossRef.
- V. Coue, R. Dessapt, M. B. Doeuff, M. Evain and S. Jobic, J. Solid State Chem., 2006, 179, 3615 CrossRef CAS PubMed.
- Z. G. Han, Y. Z. Gao and C. W. Hu, Cryst. Growth Des., 2008, 8, 1261 CAS.
- J. X. Meng, Y. Lu, Y. G. Li, H. Fu and E. B. Wang, Cryst. Growth Des., 2009, 9, 4116 CAS.
- T. Arumuganathan, A. S. Rao and S. K. Das, Cryst. Growth Des., 2010, 10, 4272 CAS.
- H. Fu, C. Qin, Y. Lu, Z. M. Zhang, Y. G. Li, Z. M. Su, W. L. Li and E. B. Wang, Angew. Chem., Int. Ed., 2012, 51, 7985 CrossRef CAS PubMed.
- W. Q. Kan, J. F. Ma, Y. Y. Liu, H. Wu and J. Yang, CrystEngComm, 2011, 13, 7037 RSC.
- H. Y. Zang, Y. Q. Lan, S. L. Li, G. S. Yang, K. Z. Shao, X. L. Wang, L. K. Yan and Z. M. Su, Dalton Trans., 2011, 40, 3176 RSC.
- P. Roman, J. M. Gutierrezzorrilla, M. Martinezripoll and M. Garciablanco, Polyhedron, 1986, 5, 1799 CrossRef CAS.
- P. Roman, A. S. Jose, A. Luque and J. M. Gutierrezzorrilla, Z. Kristallogr., 1993, 204, 179 CrossRef CAS PubMed.
- C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883 CrossRef CAS PubMed.
- X. F. Kuang, X. Y. Wu, J. Zhang and C. Z. Lu, Chem. Commun., 2011, 47, 4150 RSC.
- X. L. Wang, J. Li, A. X. Tian, D. Zhao, G. C. Liu and H. Y. Lin, Cryst. Growth Des., 2011, 11, 3456 CAS.
- Y. N. Yang, J. J. Gong, W. S. Zhang, H. C. Zhang, L. L. Zhang, Y. Liu, H. Huang, H. L. Hu and Z. H. Kang, RSC Adv., 2012, 2, 6414 RSC.
- Q. G. Zhai, X. Y. Wu, S. M. Chen, Z. G. Zhao and C. Z. Lu, Inorg. Chem., 2007, 46, 5046 CrossRef CAS PubMed.
- J. Fu, H. X. Sun, Y. Xu, C. L. Wang, D. R. Zhu, Q. Sun and H. K. Liu, CrystEngComm, 2012, 14, 5148 RSC.
- H. Wu, J. Yang, Y. Y. Liu and J. F. Ma, Cryst. Growth Des., 2012, 12, 2272 CAS.
- A. W. Van der Made and R. H. Van der Made, J. Org. Chem., 1993, 58, 1262 CrossRef CAS.
-
G. M. Sheldrick, SHELXTL version 5.10, Bruker AXS Inc., Madison, WI, USA, 1997 Search PubMed.
- X. T. Zhang, P. H. Wei, D. F. Sun, Z. H. Ni, J. M. Dou, B. Li, C. W. Shi and B. Hu, Cryst. Growth Des., 2009, 9, 4424 CAS.
- H. Abbas, A. L. Pickering, D. L. Long, P. Kogerler and L. Cronin, Chem.–Eur. J., 2005, 11, 1071 CrossRef CAS PubMed.
- H. Fu, Y. Lu, Z. L. Wang, C. Liang, Z. M. Zhang and E. B. Wang, Dalton Trans., 2012, 41, 4084 RSC.
- D. L. Long, P. Kogerler and L. Cronin, Angew. Chem., Int. Ed., 2004, 43, 1817 CrossRef CAS PubMed.
- E. F. Wilson, H. Abbas, B. J. Duncombe, C. Streb, D. L. Long and L. Cronin, J. Am. Chem. Soc., 2008, 130, 13876 CrossRef CAS PubMed.
- H. Y. Zang, D. Y. Du, S. L. Li, Y. Q. Lan, G. S. Yang, L. K. Yan, K. Z. Shao and Z. M. Su, J. Solid State Chem., 2011, 184, 1141 CrossRef CAS PubMed.
- H. Y. Liu, H. Wu, J. F. Ma, Y. Y. Liu, J. Yang and J. C. Ma, Dalton Trans., 2011, 40, 602 RSC.
- D. R. Xiao, Y. Hou, E. B. Wang, S. T. Wang, Y. G. Li, L. Xu and C. W. Hu, Inorg. Chim. Acta, 2004, 357, 2525 CrossRef CAS PubMed.
- W. J. Chang, Y. C. Jiang, S. L. Wang and K. H. Li, Inorg. Chem., 2006, 45, 6586 CrossRef CAS PubMed.
- S. Reinoso, P. Vitoria, J. M. Zorrilla, L. Lezama, J. M. Madariaga, L. S. Felices and A. Iturrospe, Inorg. Chem., 2007, 46, 4010 CrossRef CAS PubMed.
- J. Q. Sha, J. W. Sun, C. Wang, G. M. Li, P. F. Yan and M. T. Li, Cryst. Growth Des., 2012, 12, 2242 CAS.
- J. Q. Sha, L. Y. Liang, J. W. Sun, A. X. Tian, P. F. Yan, G. M. Li and C. Wang, Cryst. Growth Des., 2012, 12, 894 CAS.
- X. Wang, W. Y. Wang, S. M. Liu, H. W. Hou and Y. T. Fan, J. Mol. Struct., 2009, 938, 185 CrossRef CAS PubMed.
- B. X. Wang and S. J. Dong, Electrochim. Acta, 1992, 37, 1859 CrossRef CAS.
- P. Wang, X. P. Wang and G. Y. Zhu, Electrochim. Acta, 2001, 46, 637 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 957456 and 957457 contain the supplementary crystallographic data for compound 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce41706a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.