Hydrogen bond-assisted solid-state formation of a salt-bridged calix[5]arene pseudo-dimer†
Giovanna
Brancatelli
a,
Sebastiano
Pappalardo
b,
Giuseppe
Gattuso
c,
Anna
Notti
c,
Ilenia
Pisagatti
c,
Melchiorre F.
Parisi
*c and
Silvano
Geremia
*a
aCentro di Eccellenza in Biocristallografia, Dipartimento di Scienze Chimiche e Farmaceutiche, Università di Trieste, via L. Giorgieri 1, 34127 Trieste, Italy. E-mail: sgeremia@units.it; Fax: +39 0405583903; Tel: +39 0405583936
bDipartimento di Scienze Chimiche, Università di Catania, viale A. Doria 6, 95125 Catania, Italy
cDipartimento di Scienze Chimiche, Università di Messina, viale F. Stagno d'Alcontres 31, 98166 Messina, Italy. E-mail: mparisi@unime.it; Fax: +39 0906765186; Tel: +39 0906765170
First published on 22nd October 2013
Abstract
Co-crystallization of a carboxylcalix[5]arene in the presence of n-butylamine afforded the corresponding host–guest carboxylate–butylammonium salt-bridged complex as a result of a proton transfer process assisted by a second non-ionized calixarene molecule. The crystals isolated consist of a supramolecular pseudo-dimer in which a host–guest inclusion complex is connected to a formally neutral receptor molecule – hosting a solvent molecule in the aromatic cavity – via a strong negative charge-assisted H-bond between the carboxyl/carboxylate substituents present at the narrow rim of the two interacting calixarene molecules.
Introduction
Salt bridges have important and often quite specific functions in protein chemistry (substrate binding, activity of catalytic triads, secondary-structure stabilization, stability of thermophilic proteins, etc.).1–5 The nature and strength of this interaction depend on the dielectric constant of the surrounding medium and the relative geometry of the pairs of interacting functional groups. For example, in the case of the methylammonium ion–acetate ion salt-bridge model, theoretical calculations show that the strength of the interaction is reduced over 50-fold on moving from the gas phase to water.6 Other theoretical studies suggest that in the gas phase the acid–base partners prefer to associate as a neutral hydrogen-bonded complex, whereas in aqueous solution, proton transfer from the acid to the base – leading to the formation of a solvated ion-pair complex – is energetically favoured.7 On the other hand, binding free energies in CHCl3 indicate a scarce tendency of low polarity solvents to promote proton transfer, thus preventing the formation of ionic species.1,8 In general, forcing an ion pair to form in very low dielectric media or within nonpolar environments (cavities and pockets) may result in proton transfer from the protonated base to the anion, with subsequent conversion of the ionic pair into a complex between neutral species.1,8In this respect, small artificial receptors possessing hydrophobic pockets, such as calixarenes,9 can be very useful models to study the role played by a nonpolar environment in the stability of salt bridges. In order to override the intrinsic ion-pair association between an ionic guest and its counterion, which is a phenomenon that generally reduces the binding efficiency of neutral receptors, the salt-bridge interaction has recently been exploited in selective amine recognition by ionizable calixarenes.10 To this end, calix[5]arenes 1a·H and 1b·H (Fig. 1) were endowed at the narrow rim with a carboxyl group and either four alkyl or four ester pendant chains, respectively, with the aim of triggering proton transfer from their acidic moiety to the basic nitrogen atom of amino substrates. These two calixarenes readily lend a proton to size/shape complementary amines, and the primary linear alkylammonium guests thus formed tightly bind inside the aromatic pocket of the receptor in a selective manner to counterbalance and ion pair the negatively charged carboxylate group generated at the bottom of the cavity.
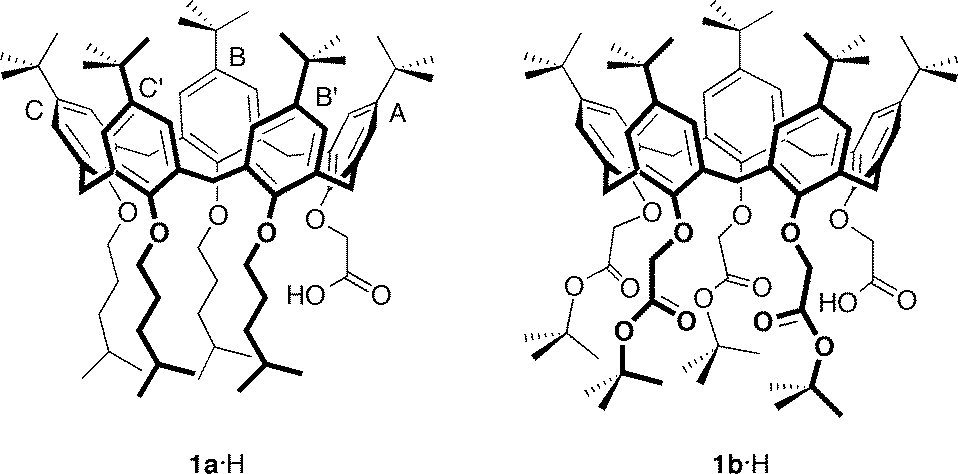 | ||
| Fig. 1 Molecular structures of ionizable carboxylcalix[5]arenes 1a·H and 1b·H with pertinent labelling of the aromatic rings. | ||
An X-ray crystallographic analysis of the inclusion complexes, obtained from co-crystallization of 1a·H or 1b·H and n-butylamine in the presence of chloroform and trifluoroethanol (TFE) as a protic polar solvent, has allowed us to pin down all the non-covalent forces contributing to the overall host–guest recognition and binding process.10 The calixarene receptor was found in the ionized form (i.e., as a carboxylate) in both complexes, hosting in an endo-cavity fashion the conjugated ammonium ion as a guest (n-BuNH3+⊂1a− and n-BuNH3+⊂1b−). In particular, in the case of the n-BuNH3+⊂1a− complex, the inwardly directed carboxylate moiety was seen to be involved in a salt-bridge interaction with the n-butylammonium ion, contributing to the overall stabilization of the complex. Conversely, no salt bridge was observed in the n-BuNH3+⊂1b− complex, with both oxygen atoms of the carboxylate moiety of 1b− being oriented outside the cavity and interacting via hydrogen bonds with TFE solvent molecules. It would then appear that the binding mode of ionizable host and guest species is determined by the presence of other coexisting interactions that ultimately contribute to the overall stabilization (or destabilization) of the complex.
All the above considerations prompted us to focus our attention on host–guest complexes obtained from ionizable calixarenes and amines in aprotic polar solvents. Herein, we report the crystal structure of a host–guest complex obtained by co-crystallization of 1a·H and n-butylamine in CH2Cl2–CH3CN and a parallel NMR investigation on the structural features of such a complex in solution (CD2Cl2).
Experimental
General
Calix[5]arene 1a·H was prepared according to a literature procedure.10Crystal structure determination
Block-shaped crystals of (n-BuNH3+⊂1a−·1a·H⊃CH3CN) were obtained by slow evaporation of a CH2Cl2–CH3CN 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture containing 1a·H and n-butylamine in a 1:1 molar ratio. Data collection was carried out using synchrotron radiation at the X-ray diffraction beam-line of Elettra Synchrotron, Trieste (Italy), using a MarCCD detector, with the rotating-crystal method and a monochromatic wavelength. The crystal dipped in Paratone®, used as cryo-protectant, was mounted in a loop and flash-frozen to 100 K with liquid nitrogen. Diffraction data were indexed and integrated using MOSFLM11 and scaled using SCALA.12 The structure was solved by direct methods using SIR2004,13 and non-hydrogen atoms were anisotropically refined (H atoms at the calculated positions) with bond length and angle restraints by full-matrix least-squares methods on F2 using SHELXL-97.14 Crystal data and refinement details are reported in Table S1, see ESI† (CCDC no. 853683).
:1 mixture containing 1a·H and n-butylamine in a 1:1 molar ratio. Data collection was carried out using synchrotron radiation at the X-ray diffraction beam-line of Elettra Synchrotron, Trieste (Italy), using a MarCCD detector, with the rotating-crystal method and a monochromatic wavelength. The crystal dipped in Paratone®, used as cryo-protectant, was mounted in a loop and flash-frozen to 100 K with liquid nitrogen. Diffraction data were indexed and integrated using MOSFLM11 and scaled using SCALA.12 The structure was solved by direct methods using SIR2004,13 and non-hydrogen atoms were anisotropically refined (H atoms at the calculated positions) with bond length and angle restraints by full-matrix least-squares methods on F2 using SHELXL-97.14 Crystal data and refinement details are reported in Table S1, see ESI† (CCDC no. 853683).
The electron density maps and the refinement showed that a tert-butyl group and a 4-methylpentyl chain were disordered over two sites. Furthermore, restraints were introduced to account for the anisotropic thermal parameters of carbon atoms in the pendant 4-methylpentyl chains using the card SIMU. Solvent molecules (CH2Cl2 and CH3CN) with partial occupancy were observed to reside in the interstitial voids generated between neighbouring calixarene molecules belonging to n-BuNH3+⊂1a− and CH3CN⊂1a·H. To investigate the solvent-containing cavities, the SQUEEZE function of the PLATON program15 was used. A residual density (480 electrons per cell) found in the voids of (n-BuNH3+⊂1a−·1a·H⊃CH3CN) (~14% of the total cell volume) was ascribed to approximately 6 CH2Cl2 and 10 CH3CN disordered solvent molecules. A refinement using reflections modified by the SQUEEZE procedure behaved well, and the R-factor was reduced from 19.1 to 14.2%.
Results and discussion
Crystals suitable for X-ray analysis were obtained by slow evaporation of a CH2Cl2–CH3CN 2:1 solution containing a 1:1 mixture‡ of n-BuNH2 and 1a·H. The asymmetric unit of the triclinic crystals contains four calixarene molecules arranged in independent pairs of complexes forming two structurally equivalent supramolecular pseudo-dimers (designated hereafter as I and II, Fig. 2; see also Fig. S1†), both displaying very similar geometries. The two calixarene molecules of I and II are nearly orthogonally oriented with respect to each other. In the two crystallographically independent pseudo-dimers, the dihedral angles between the calixarene reference planes, defined by the five methylene bridging carbon atoms, are 77.2° and 76.7° for I and II, respectively.
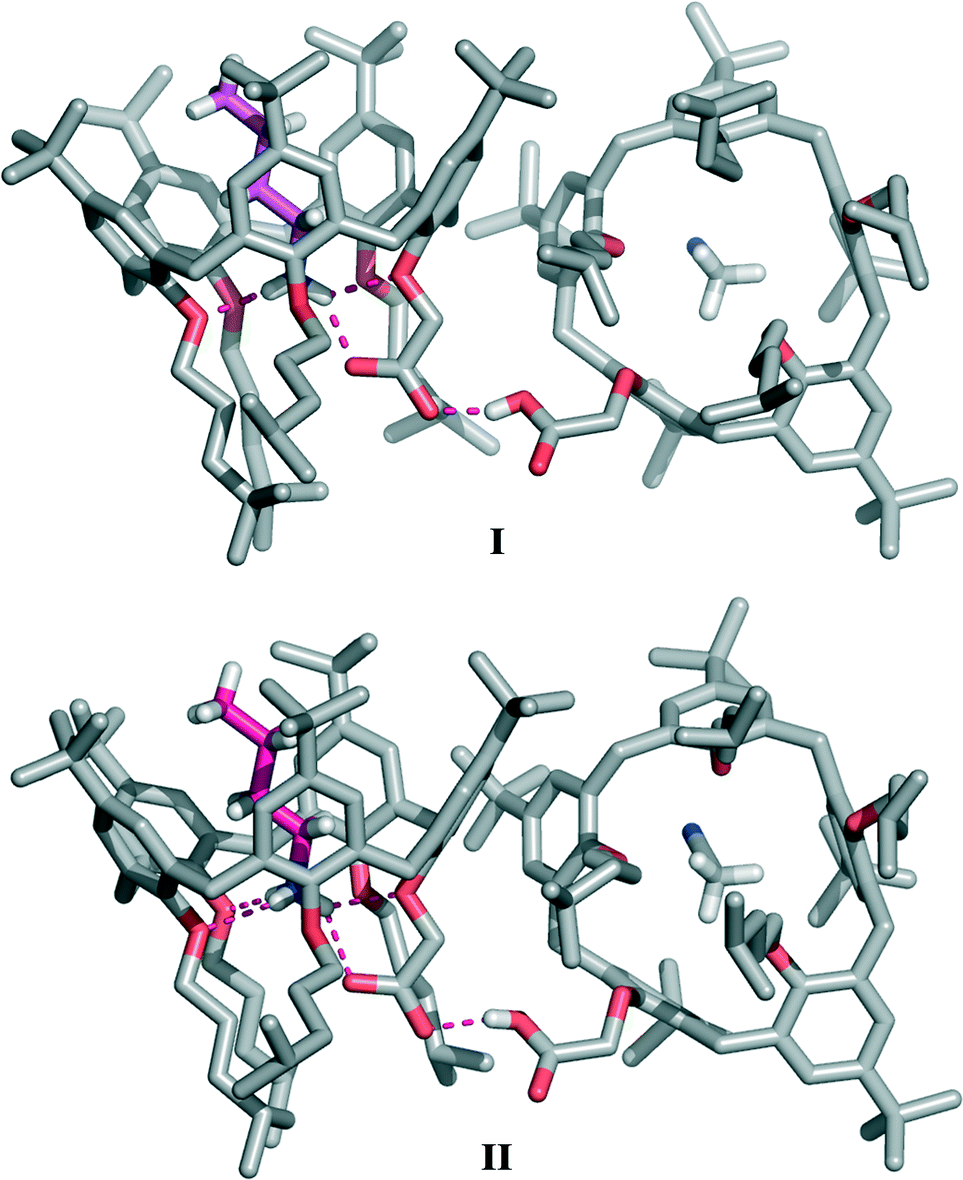 | ||
| Fig. 2 The crystallographic structure of the supramolecular pseudo-dimers I and II (n-BuNH3+⊂1a−·1a·H⊃CH3CN) obtained from a CH2Cl2–CH3CN solution of 1a·H and n-BuNH2. The pseudo-dimer consists of an ‘internal’ ion-pair complex (n-BuNH3+⊂1a−) stabilized by a salt-bridge interaction and a cavity-solvated neutral calixarene molecule (CH3CN⊂1a·H). The two calixarene units are connected by a strong carboxyl–carboxylate negative charge-assisted H-bond. The n-BuNH3+ guest included inside the calixarene cavity is depicted in magenta. | ||
In the pseudo-dimer, one calixarene molecule hosts inside its cavity a n-butylammonium ion in a fully extended conformation held in place by a network of four intermolecular hydrogen bonds connecting the NH3+ group of the guest to the phenolic oxygen atoms of rings A, C and C′ (d(N⋯O) in the 2.815(7)–2.857(7) Å and 2.783(6)–2.828(7) Å ranges (Table 1)), as well as to the carboxylate moiety of the host (d(N⋯O) of 2.767(7) and 2.772(7) Å for I and II, respectively). Guest inclusion is additionally secured by CH–π interactions between the α- and β-CH2 groups of the n-BuNH3+ ion and rings B, B′ and C, C′ of the host, respectively (Table S2†). The second adjacent calix[5]arene molecule accommodates inside its cavity one acetonitrile molecule forming the CH3CN⊂1a·H complex.
| D–H⋯A | d(D⋯A) | |
|---|---|---|
| n-BuNH3+⊂1a− (I) | N(1)–H(1b)⋯O(1l) | 2.767(7) |
| N(1)–H(1b)⋯O(1g) | 2.820(7) | |
| N(1)–H(1a)⋯O(4g) | 2.815(7) | |
| N(1)–H(1c)⋯O(5g) | 2.857(7) | |
| n-BuNH3+⊂1a− (II) | N(9)–H(9b)⋯O(11l) | 2.772(7) |
| N(9)–H(9b)⋯O(11g) | 2.791(7) | |
| N(9)–H(9c)⋯O(13g) | 2.783(6) | |
| N(9)–H(9a)⋯O(14g) | 2.828(7) | |
| Pseudo-dimer I | O(6l)–H(6l)⋯O(1m) | 2.445(8) |
| Pseudo-dimer II | O(16l)–H(16l)⋯O(11m) | 2.462(7) |
Diffraction data were of sufficient quality to allow a precise location of the hydrogen atoms involved in H-bonds. A careful analysis of the Fourier difference map (Fig. 3) reveals the presence of three residual electron density peaks around the nitrogen atom of the guest (mean value: 0.70 e Å−3), recognized as the three hydrogen atoms of the ammonium group.
 | ||
| Fig. 3 Sections of the electron density map (2Fo − Fc, contour level 1.4 σ, purple) and the residual electron density map (mFo − Fuc, contour level 3 σ, red) observed for pseudo-dimer I showing the position of the hydrogen atoms involved in hydrogen bonds (green dashed line). | ||
This piece of experimental evidence confirms that a proton transfer has occurred from the acidic host to the basic guest, leading to the formation of an ‘internal’ ion-paired complex stabilized by a salt-bridge interaction. The presence of a similar salt-bridge interaction had previously been detected in the n-BuNH3+⊂1a− complex crystallized in the presence of TFE, where the carboxylate group was found to be involved in additional ‘external’ hydrogen bonds with three solvent molecules.10
In the present case, the use of a mixture of aprotic solvents (CH2Cl2–CH3CN) was expected to prevent the formation of ‘external’ hydrogen bonds, and it was therefore predicted that no stabilization of the carboxylate–ammonium salt-bridged complex would take place. Surprisingly, however, in the absence of protic solvents, this overall ionization/complexation process is still assisted, in the solid state, by a different type of ‘external’ H-bond that now involves the carboxyl group of the CH3CN⊂1a·H complex acting as the hydrogen-bond donor (Fig. 2 and 3). According to the ‘pKa slide rule,’16 the interaction between homomolecular moieties with null ΔpKa by definition, such as the carboxylic/carboxylate fragments of 1a·H and 1a−, gives rise to a negative charge-assisted H-bond, (−)CAHB, characterized by a very short O⋯O distance (usually d(O⋯O) ≤ 2.5 Å).17 In extreme cases, a strong homonuclear (−)CAHB, with a symmetric location of the hydrogen atom, can indeed be considered as a three-centre four-electron bond17,18 with a highly covalent character, consistent with an equal contribution from two identical and therefore isoenergetic valence-bond resonance forms [–O–H⋯−O– ↔ –O−⋯H–O–].16,19–21 In the present structure, this H-bond is responsible for the self-assembly of the two calixarene complexes in both crystallographically independent pseudo-dimers (d(O⋯O) of 2.445(8) and 2.462(7)Å for I and II, respectively). Difference maps show that the peak position of the H atom is to some extent asymmetric (Fig. 3), with the hydrogen atom being slightly closer (relative distances of 0.1 and 0.2 Å) to the carboxylic oxygen atom of the CH3CN⊂1a·H complex in both pseudo-dimers.
The role played by this ‘external’ H-bond in the host–guest complex formation is likely that of increasing the acidity of the inwardly oriented carboxyl group, as well as stabilizing the charge separation. Proton transfer to the amine is then favoured (despite the hydrophobic environment), leading to the formation of an ‘internal’ salt bridge.22
It is worth mentioning that the close similarities between the salt-bridge geometry observed for the n-BuNH3+⊂1a− complex present in I and II (d(N⋯O) of 2.767(7) and 2.772(7) Å, respectively) and that for the ‘monomeric’ species studied earlier10 (d(N⋯O) of 2.779(5) Å) suggest that the effect of a single (−)CAHB on the ‘internal’ salt-bridge geometry is, in a way, comparable to that provided by the three H-bonds from TFE solvent molecules.
Furthermore, the strong hydrogen bond connecting the two subunits of this supramolecular aggregate is also likely responsible for the outward orientation (with respect to the cavity) of the CH2CO2H pendant group of CH3CN⊂1a·H (Fig. 2 and 4) and for forcing ring A to stand orthogonal to the calixarene reference plane (θA = 89.8(2) and 86.5(2)° for I and II, respectively). The superimposition of the calix[5]arene skeletons (devoid of p-tert-butyl and 4-methylpentyl substituents) of 1a·H and 1a−, which are present in aggregate I, shows an rmsd value of 3.7 Å (Fig. 4). This indicates that the formation of a (−)CAHB together with the interchange of an acetonitrile molecule for a n-butylammonium ion cause a severe conformational rearrangement of the host cavity. The pre-organized receptor cavities retain an approximate cone-out conformation23,24 in both complexes but, in the case of the ionized calix[5]arene 1a−, the dihedral angles between the aryl rings and the macrocycle reference plane were all found to have values above 96° (Table S3†), indicating a wider cavity that is better suited to enclose a bulkier n-butylammonium ion guest in place of an acetonitrile solvent molecule.
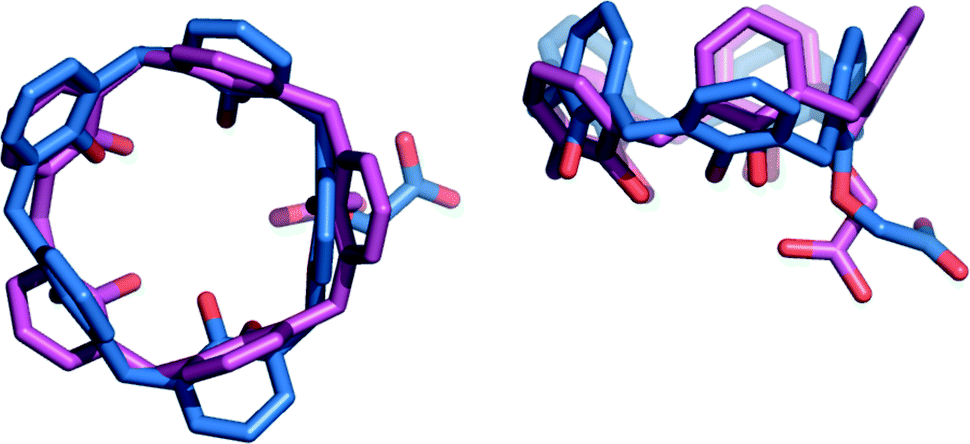 | ||
| Fig. 4 Top and side views of the superimposed X-ray molecular structures of calix[5]arenes 1a·H (blue) and 1a− (magenta) (rmsd = 3.7 Å) present in pseudo-dimer I. In both structures, p-tert-butyl and 4-methylpentyl substituents have been omitted for the sake of clarity. The CH3CN molecule and the n-BuNH3+ ion present inside the cavities of 1a·H and 1a−, respectively, are not shown for the same reason. | ||
In I and II, the positively-charged nitrogen atoms of the guest are located below the calixarene reference planes at a distance of −0.735(5) and −0.603(5) Å, respectively. These values are in keeping with those found in the previously reported solid-state structure of the n-BuNH3+⊂1a− ‘monomeric’ complex (−0.938(1) Å),10 but are significantly lower than that measured in the crystallographic structure of the penta-tert-butyl-pentakis(4-methylpentyloxy)calix[5]arene–n-butylammonium inclusion complex,25 where the nitrogen atom is 0.133(3) Å above the reference plane. This result suggests that salt-bridge interactions between the primary n-butylammonium ion and the carboxylate moiety positively cooperate together with tripodal hydrogen bonds and CH–π interactions to efficiently anchor a cationic guest at the bottom of a calix[5]arene cavity.
In an attempt to detect the formation of hydrogen-bonded pseudo-dimers in solution (anhydrous CD2Cl2), 1H NMR and diffusion-ordered spectroscopy (DOSY) experiments were carried out. The spectrum of a 2:1 mixture of 1a·H and n-BuNH2 showed – consistently with the host–guest ratio used – the presence of the BuNH3+⊂1a− complex, as well as that of uncomplexed 1a·H (see Fig. S2†). However, according to a comparison of the DOSY data (see Fig. S3 and Table S4†) collected on the above-mentioned mixture, a covalently-linked bis-calix[5]arene of known structure26 (2),§ as well as calixarene 1a·H on its own, no supramolecular pseudo-dimers similar to those observed in the solid-state (n-BuNH3+⊂1a−·1a·H⊃CH3CN) are formed. Specifically, the diffusion coefficients measured for n-BuNH3+⊂1a− and 1a·H (present in a 2:1 mixture of 1a·H and BuNH2) as well as that obtained for 1a·H (in the absence of BuNH2) were all found to have comparable values between themselves but were notably higher with respect to that detected for the model bis-calix[5]arene 2, indicating that in solution, a single carboxyl–carboxylate hydrogen-bond is not sufficiently strong to hold two calix[5]arenes together.27
Conclusions
In conclusion, acid–base proton transfer from carboxylcalix[5]arene 1a·H to n-butylamine triggers the formation of an endo-cavity complex (n-BuNH3+⊂1a−). This ‘internal’ complex takes advantage of a number of H-bonds and CH–π interactions as well as an ammonium–carboxylate salt bridge which is in turn stabilized by a very strong ‘external’ carboxylate–carboxyl negative charge-assisted hydrogen bond. The latter takes place between the ionized carboxylate moiety of 1a− and the carboxyl group of a second non-ionized calix[5]arene molecule, giving rise, in the solid state, to the (n-BuNH3+⊂1a−·1a·H⊃CH3CN) aggregate. The X-ray diffraction structure of this aggregate shows two different types of charged intermolecular hydrogen bonds, namely a (−)CAHB and a (+/−)CAHB (i.e. a double charge-assisted hydrogen bond or, in other words, a salt bridge). These co-existing H-bonds are in turn responsible for pseudo-dimer aggregation and guest inclusion.Our data demonstrate that ionizable macrocycles play, in the absence of protic solvents, a dual role to ensure optimal molecular recognition of neutral guests. Carboxylcalix[5]arene 1a·H, on one hand, ionizes and lends a proton to a target amine so that endo-cavity binding of the corresponding cationic guest occurs; on the other hand, it assists and stabilizes the formation of the resultant salt-bridged complex by acting, in the non-ionized form, as an H-bond donor.
Acknowledgements
MIUR (PRIN-2009A5Y3N9 project) is gratefully acknowledged for financial support of this research.Notes and references
- A. Warshel, P. K. Sharma, M. Kato and W. W. Parson, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 1647–1676 CrossRef CAS PubMed.
- S. Kumar and R. Nussinov, ChemBioChem, 2002, 3, 604–617 CrossRef CAS.
- P. Strop and S. L. Mayo, Biochemistry, 2000, 39, 1251–1255 CrossRef CAS PubMed.
- F. G. Pearce, R. C. J. Dobson, G. B. Jameson, M. A. Perugini and J. A. Gerrard, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1900–1909 CrossRef CAS PubMed.
- Q. Xu, H. Guo, A. Wlodawer and H. Guo, J. Am. Chem. Soc., 2006, 128, 5994–5995 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, J. Am. Chem. Soc., 2000, 122, 870–874 CrossRef CAS.
- S. Kong, I. G. Shenderovich and M. V. Vener, J. Phys. Chem. A, 2010, 114, 2393–2399 CrossRef CAS PubMed.
- X. Barril, C. Alemán, M. Orozco and F. J. Luque, Proteins: Struct., Funct., Genet., 1998, 32, 67–79 CrossRef CAS.
- C. D. Gutsche, Calixarenes – An Introduction, in Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 2nd edn, 2008, vol. 6 Search PubMed.
- C. Capici, G. Gattuso, A. Notti, M. F. Parisi, S. Pappalardo, G. Brancatelli and S. Geremia, J. Org. Chem., 2012, 77, 9668–9675 CrossRef CAS PubMed.
- A. G. W. Leslie and H. R. Powell, Processing diffraction data with mosflm, in Evolving Methods for Macromolecular Crystallography, ed. R. J. Read and J. L Sussman, NATO Science Series, Springer, 2007, vol. 245, pp. 41–51 Search PubMed.
- (a) P. R. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 72–82 CrossRef PubMed; (b) M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick and T. R. Schneider, SHELXL: High resolution refinement, Methods in Enzymology, Academic Press, Orlando, FL, 1997, vol. 277, pp. 319–343 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- P. Gilli, L. Pretto, V. Bertolasio and G. Gilli, Acc. Chem. Res., 2009, 42, 33–44 CrossRef CAS PubMed.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1994, 116, 909–915 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- P. Gilli, V. Bertolasi, L. Pretto, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2004, 126, 3845–3855 CrossRef CAS PubMed.
- G. Gilli and P. Gilli, J. Mol. Struct., 2000, 552, 1–15 CrossRef CAS.
- P. Gilli and J. Gilli, J. Mol. Struct., 2010, 972, 2–20 CrossRef CAS PubMed.
- P. L. Huyskens, J. Am. Chem. Soc., 1977, 99, 2578–2582 CrossRef CAS.
- G. Gattuso, A. Notti, S. Pappalardo, M. F. Parisi, T. Pilati and G. Terraneo, CrystEngComm, 2012, 14, 2621–2625 RSC.
- G. De Salvo, G. Gattuso, A. Notti, M. F. Parisi and S. Pappalardo, J. Org. Chem., 2002, 67, 684–692 CrossRef CAS PubMed.
- G. Gattuso, A. Notti, S. Pappalardo, M. F. Parisi, T. Pilati, G. Resnati and G. Terraneo, CrystEngComm, 2009, 11, 1204–1206 RSC.
- G. Gattuso, A. Notti, A. Pappalardo, M. F. Parisi, L. Pisagatti, S. Pappalardo, D. Garozzo, A. Messina, Y. Cohen and S. Slovak, J. Org. Chem., 2008, 73, 7280–7289 CrossRef CAS PubMed.
- A. Brück, L. L. McCoy and K. V. Kiway, Org. Lett., 2000, 2, 2007–2009 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal data for n-BuNH3+⊂1a−·1a·H⊃CH3CN and DOSY/1H NMR spectra on n-BuNH3+⊂1a−. CCDC reference number 853683. For ESI and crystallographic data in CIF format see DOI: 10.1039/c3ce41667d |
| ‡ Crystal growth in the presence of an excess of n-BuNH2 (2 equiv.), under analogous conditions, produced the same supramolecular pseudo-dimer. |
| § Bis-calix[5]arene 2 was chosen as a model compound for a species with a molecular weight roughly similar to that of the pseudo-dimer (n-BuNH3+⊂1a−·1a·H⊃CH3CN). |
| This journal is © The Royal Society of Chemistry 2014 |