Regiospecific photodimerization reactions of an unsymmetrical alkene in two coordination compounds†
Dong
Liu
ab and
Jian-Ping
Lang
*ac
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, PR China. E-mail: jplang@suda.edu.cn; Fax: +86 512 65880089
bCollege of Chemistry and Materials Science, Huaibei Normal University, Huaibei 235000, PR China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, PR China
First published on 13th September 2013
Abstract
Two coordination complexes [Cd2(CBA)4(4-spy)4] (1) and [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2(H2O)4]·(4-spy–H)2 (2) were prepared by the hydrothermal reactions of Cd(NO3)2·4H2O, 4-styrylpyridine (4-spy) and 4-chlorobenzoic acid (HCBA) or 5-nitrobenzene-1,3-dicarboxylic acid (5-NO2-1,3-H2BDC). Each pair of 4-spy molecules in [Cd2(CBA)4(4-spy)4] and [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2(H2O)4]·(4-spy–H)2 are arranged in a head-to-head (HH) or head-to-tail (HT) fashion, which, upon UV irradiation, dimerizes into 1,2-bis(4-pyridyl)-3,4-bis(phenyl)cyclobutane (HH-ppcb) or 1,3-bis(4-pyridyl)-2,4-bis(phenyl)cyclobutane (HT-ppcb) in 100% yield, respectively. Compared with liquid-phase organic reactions, these solid-state photochemical [2 + 2] cycloadditions of olefin molecules provide good opportunities to synthesize regiospecific cyclobutanes.
Introduction
The solid-state photochemical [2 + 2] cycloadditions of alkenes have attracted considerable attention in organic photochemistry because they can produce cyclobutanes or ladderanes that are less available or totally unavailable in solution.1,2 Compared with solution photodimerizations, one of the most intriguing aspects of such solid-state photoreactions is that the regio- or stereo-chemistry of the products is determined by the relative arrangement of the olefinic molecules in the crystals.1–3 For example, photodimerization of the unsymmetrical alkene 4-styrylpyridine (4-spy) in solution yields a regio- and stereo-isomeric mixture of cyclobutanes, whose ratio depends on the Woodward–Hoffmann selection rules, electronic and steric factors and stability of the diradical intermediates.3 Although the reaction selectivity can be enhanced by adding hydrochloric acid, such a solution reaction is still unable to afford a regiospecific product in a quantitative yield.3 Therefore, many attempts have been made to align olefinic molecules in an arrangement suitable for photodimerization in the solid state.4–10 The advance of crystal engineering has impelled the development of intriguing templates that adopt non-covalent interactions such as hydrogen bonds, coordination bonds, π–π interactions and halogen–halogen interactions to affect the relative orientation of olefinic molecules.4–10 This strategy generally exploits the excellent regularity and molecular orientation provided by a crystal lattice to afford products with specific morphology during reactions in high efficiency. However, at the current time it is still difficult to rationally design a molecule that crystallizes in an arrangement suitable for the desired photoreaction. Therefore, the work involved in creating photodimeric isomers from regiospecific photodimerizations has been rare until now.11In the solid state, each pair of the aforementioned 4-spy molecules may be arranged in a head-to-head (HH) or head-to-tail (HT) fashion with the help of the appropriate templates. Upon UV irradiation, the HH- or HT-arranged 4-spy molecules would dimerize to 1,2-bis(4-pyridyl)-3,4-bis(phenyl)cyclobutane (HH-ppcb) or 1,3-bis(4-pyridyl)-2,4-bis(phenyl)cyclobutane (HT-ppcb), respectively (Scheme 1). Recently, we found that photoreactive coordination polymers could be used to carry out regioselective photodimerization reactions to afford specific isomeric photodimers.11d In this paper, we report the preparation of two photoreactive carboxylate-mediated cadmium complexes with 4-spy molecules arranged in HH or HT configurations and two regiospecific isomers HH-ppcb and HT-ppcb formed through their photodimerization reactions.
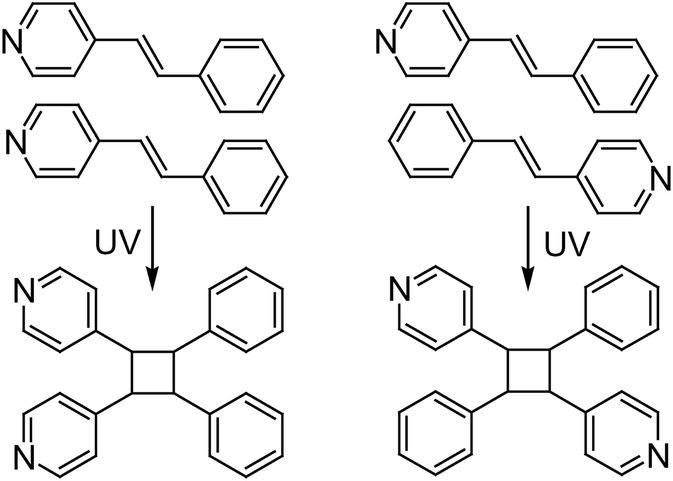 | ||
| Scheme 1 Possible photodimerization reactions based on the head-to-head (HH) and head-to-tail (HT) arranged 4-spy. | ||
Experimental
Materials and physical measurements
Reagents and solvents were purchased from TCI Co., Ltd., and Sigma-Aldrich Co. All the chemicals were used without any further purification. Powder XRD patterns were obtained using a PANalytical X’Pert PRO MPD system (PW3040/60). Thermal analysis was performed using a Perkin-Elmer TGA-7 thermogravimetric analyzer at a heating rate of 10 °C min−1 and a flow rate of 100 cm3 min−1 (N2). 1H and 13C NMR spectra were recorded at ambient temperature using a Bruker AVANCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) III (400 MHz) spectrometer. 1H and 13C NMR chemical shifts were referenced to the solvent signal in d6-DMSO. Infrared (IR) samples were prepared as KBr pellets, and spectra were obtained in the 4000–400 cm−1 range using a Nicolet Avatar 360 FT-IR spectrophotometer. The elemental analyses for C, H, and N were performed using an EA1110 CHNS elemental analyzer.
III (400 MHz) spectrometer. 1H and 13C NMR chemical shifts were referenced to the solvent signal in d6-DMSO. Infrared (IR) samples were prepared as KBr pellets, and spectra were obtained in the 4000–400 cm−1 range using a Nicolet Avatar 360 FT-IR spectrophotometer. The elemental analyses for C, H, and N were performed using an EA1110 CHNS elemental analyzer.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, 4-spy), 7.48 (d, 8H, Ph–H, CBA), 7.33 (m, 28H, Ph–H and Py–H, 4-spy).
CH, 4-spy).
CH, 4-spy), 7.48 (d, 8H, Ph–H, CBA), 7.33 (m, 28H, Ph–H and Py–H, 4-spy).
CH, 4-spy).
The structures were solved by direct methods, and non-hydrogen atoms were refined anisotropically by least-squares on F2 using the SHELXTL-97 program.12 All non-hydrogen atoms were refined anisotropically. The protonated H atom of the 4-spy–H molecule in 2 was located from the Fourier map with the N–H distances being fixed at 0.86 Å. The H atoms of the coordinated water molecules in 2 were located from the Fourier map and included in the final refinement by use of geometrical restraints with the O–H distances being fixed at 0.85 Å and Uiso(H) equivalent to 1.5 times Ueq(O). All other H atoms were introduced at the calculated positions and included in the structure-factor calculations. A summary of key crystallographic information for 1 and 2 is given in Table 1. Selected bond lengths and angles for 1 and 2 are listed in Table 2.
|
a
R = Σ||Fo|−|Fc||/Σ|Fo|.
b
wR = [Σw(Fo2−Fc2)2/Σw(Fo2)2]1/2.
c GOF = {Σ[w((Fo2−Fc2)2)/(n−p)]}1/2, where n = number of reflections and p = total number of parameters refined.
|
||
|---|---|---|
| Compound | 1 | 2 |
| Chemical formula | C80H60Cd2Cl4N4O8 | C58H46Cd2N6O28 |
| Formula weight | 1571.94 | 1499.83 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 10.584(2) | 8.7925(18) |
| b/Å | 11.362(2) | 11.978(2) |
| c/Å | 15.896(3) | 14.391(3) |
| α/° | 107.23(3) | 89.67(3) |
| β/° | 92.05(3) | 74.91(3) |
| γ/° | 109.70(3) | 81.01(3) |
| V/Å3 | 1699.3(8) | 1444.4(5) |
| Temperature/K | 223(2) | 223(2) |
| D c/g cm−3 | 1.536 | 1.724 |
| Z | 1 | 1 |
| μ(Mo-Kα)/mm−1 | 0.846 | 0.837 |
| F(000) | 796 | 756 |
| Total reflections | 13708 |
11913 |
| Unique reflections | 5970 | 5087 |
| No. observations | 4979 | 4595 |
| No. parameters | 460 | 429 |
| R a | 0.0371 | 0.0281 |
| wR b | 0.0868 | 0.0658 |
| GOFc | 0.974 | 0.990 |
| Δρmax/Δρmin (e Å−3) | 0.903/−0.725 | 0.945/−0.525 |
| a Symmetry codes for 1: A: −x + 1, −y + 1, −z + 1; for 2: A: −x, −y + 1, −z + 1. | |||
|---|---|---|---|
| Complex 1 | |||
| Cd(1)–N(1) | 2.322(3) | Cd(1)–O(4) | 2.324(2) |
| Cd(1)–N(2) | 2.335(3) | Cd(1)–O(3A) | 2.349(2) |
| Cd(1)–O(2) | 2.389(2) | Cd(1)–O(1) | 2.412(2) |
| Cd(1)–O(3) | 2.595(2) | ||
| N(1)–Cd(1)–N(2) | 174.68(9) | ||
| N(1)–Cd(1)–O(4) | 92.45(9) | N(1)–Cd(1)–O(3A) | 87.90(8) |
| O(4)–Cd(1)–N(2) | 91.12(9) | N(2)–Cd(1)–O(3A) | 86.79(9) |
| O(4)–Cd(1)–O(3A) | 128.22(8) | O(4)–Cd(1)–O(2) | 138.96(8) |
| N(1)–Cd(1)–O(2) | 85.15(9) | O(3A)–Cd(1)–O(2) | 92.69(8) |
| N(2)–Cd(1)–O(2) | 94.73(9) | O(4)–Cd(1)–O(1) | 84.65(8) |
| N(1)–Cd(1)–O(1) | 92.51(9) | O(3A)–Cd(1)–O(1) | 147.10(7) |
| N(2)–Cd(1)–O(1) | 91.75(9) | N(1)–Cd(1)–O(3) | 86.07(8) |
| O(2)–Cd(1)–O(1) | 54.64(8) | N(2)–Cd(1)–O(3) | 92.99(8) |
| O(4)–Cd(1)–O(3) | 52.57(8) | O(2)–Cd(1)–O(3) | 165.80(7) |
| O(3A)–Cd(1)–O(3) | 75.85(8) | ||
| O(1)–Cd(1)–O(3) | 137.02(7) | ||
| Complex 2 | |||
| Cd(1)–O(8) | 2.2683(18) | Cd(1)–O(13) | 2.274(2) |
| Cd(1)–O(14) | 2.2820(19) | Cd(1)–O(2) | 2.2897(18) |
| Cd(1)–O(7A) | 2.2983(17) | Cd(1)–O(1) | 2.5316(18) |
| Cd(1)–O(7) | 2.6469(18) | ||
| O(8)–Cd(1)–O(14) | 87.73(7) | ||
| O(8)–Cd(1)–O(13) | 88.68(7) | O(8)–Cd(1)–O(2) | 136.36(6) |
| O(13)–Cd(1)–O(14) | 172.63(7) | O(14)–Cd(1)–O(2) | 100.63(7) |
| O(13)–Cd(1)–O(2) | 86.40(8) | O(13)–Cd(1)–O(7A) | 88.01(8) |
| O(8)–Cd(1)–O(7A) | 132.29(6) | O(2)–Cd(1)–O(7A) | 90.87(7) |
| O(14)–Cd(1)–O(7A) | 89.65(7) | O(13)–Cd(1)–O(1) | 94.58(8) |
| O(8)–Cd(1)–O(1) | 83.37(6) | O(2)–Cd(1)–O(1) | 53.95(6) |
| O(14)–Cd(1)–O(1) | 91.40(6) | O(8)–Cd(1)–O(7) | 52.44(6) |
| O(7A)–Cd(1)–O(1) | 144.33(6) | O(14)–Cd(1)–O(7) | 82.40(7) |
| O(13)–Cd(1)–O(7) | 90.31(8) | O(7A)–Cd(1)–O(7) | 79.99(7) |
| O(2)–Cd(1)–O(7) | 170.39(6) | ||
| O(1)–Cd(1)–O(7) | 135.44(6) | ||
Results and discussion
Synthetic and spectral aspects
:2:2 at 150 °C. The powder X-ray diffraction (PXRD) patterns of the bulk products are identical to those simulated from the data of the single crystals (Fig. S1, ESI†). Hydrothermal treatment of a mixture of Cd(NO3)2·4H2O, 4-spy, HCBA and 5-NO2-1,3-H2BDC in a molar ratio of 1:1:2:2 at 150 °C afforded yellow blocks. The PXRD pattern of the product is similar to that of 2 which indicated that the product is 2 (Fig. S1, ESI†). Such a phenomenon may be determined by the structures of the two Cd complexes because 1 is a discrete coordination compound while 2 is a three-dimensional supramolecular compound that is linked by hydrogen-bonding interactions. Generally speaking, a complex with high-dimensional supramolecular structure is more insoluble in solvent. For competitive reactions in this system, the mixed reactants are prone to form complex 2.
UV irradiation of crystals of 1 for 24 h induced its internal cleavage associated with the formation of the irradiated product 1a. The PXRD patterns of 1a are similar to those of the simulated and observed ones of 1 (Fig. S1, ESI†), which implied that the main structure of 1 was retained in 1a after UV irradiation. The 1H NMR spectrum of 1a reveals a quantitative conversion (100%), showing a shift in the resonance of the pyridyl protons from δ 8.56 ppm to δ 8.33 ppm, and the total disappearance of the sharp signal of olefinic protons of 1 at δ 7.58 ppm and the appearance of the characteristic signal of cyclobutane protons at 4.57 ppm (Fig. S2, ESI†). Thus, the formula of 1a may be assumed to be [Cd2(CBA)4(HH-ppcb)2]. It is noted that the proton signals of HH-ppcb in 1a are similar to those observed in [Ag2(4-pyr-ph-cb)2][(CO2CF3)2] (4-pyr-ph-cb is equal to HH-ppcb), indicating that the stereoconfiguration of HH-ppcb in 1a is also the same as that in [Ag2(4-pyr-ph-cb)2][(CO2CF3)2].13a Therefore, the HH-ppcb in 1a can be identified to be rctt-1,2-bis(4-pyridyl)-3,4-bis(phenyl)cyclobutane.
UV irradiation of crystals of 2 for 24 h led to the photodimerization between the HT-4-spy–H cations to give the HT-ppcb–H2 dications. Attempts to accomplish the reaction through single-crystal-to-single-crystal transformation failed because it always resulted in the cracking of the crystals. However, the PXRD patterns of 2a indicated that its framework was retained after the UV irradiation (Fig. S1, ESI†). The 1H NMR spectrum of the photoproduct 2a showed the disappearance of the signal of the olefinic protons at δ 7.58 ppm and the appearance of a signal of cyclobutane protons at 4.57 ppm. Furthermore, the shift of the signal of the pyridyl protons from 8.56 to 8.32 ppm confirmed 100% conversion of 4-spy–H into ppcb–H2 (Fig. S2, ESI†). Based on the arrangement of the 4-spy–H anions in 2, the chemical formula of 2a was tentatively assumed to be [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2(H2O)4]·HT-ppcb–H2. The H-bonded metal complexes have rarely been used to align the CC bonds for photodimerizations.8d Utilization of the coordination anions to arrange photoreactive olefinic cations for photodimerization reactions has not been reported previously. When a mixture of 1a, Na2(H2edta), NaOH, H2O and CH2Cl2 were placed in a flask and stirred for one day, HH-ppcb would transfer to the organic phase. The organic phase was then separated from the reaction mixture and concentrated to dryness in vacuo to form a yellow powder of HH-ppcb. HT-ppcb could be readily isolated from 2a using the same method. The identities of HH-ppcb and HT-ppcb have been further confirmed by 1H and 13C NMR spectra (Fig. S2–S3, ESI†). In our previous work, we found a single-crystal-to-single-crystal transformation from 4-spy into rctt-HT-ppcb in [Cd(1,3-bdc)(4-spy)(HT-ppcb)0.5]n.13c Considering the similar proton signals of HT-ppcb that was isolated from 2a (Fig. S2, ESI†) and this known complex,13c the stereoconfiguration of HT-ppcb in 2a can be described as rctt. In order to investigate the crystal structures of 1a and 2a, we tried to re-crystallize them from various organic solvents but all attempts failed.
The maximum absorbance for 1 appears at 310 nm whereas that of 2 is at 301 nm (Fig. S4, ESI†). In order to achieve the single-crystal-to-single-crystal (SCSC) photoreactions of both 1 and 2, we attempted to use different lamps that emitted UV light with different wavelengths such as 365 nm, 302 nm, 285 nm or 254 nm. However, all the attempts were failed since the single crystals always cracked after UV irradiation.
, and its asymmetric unit contains half of a discrete [Cd2(CBA)4(4-spy)4] dimeric molecule. As shown in Fig. 1, each Cd center adopts a pentagonal bipyramidal geometry, coordinated by five O atoms from three different CBA ligands and two N atoms from two 4-spy ligands. Each pair of Cd atoms are combined by four CBA ligands to form one [Cd2(CBA)4] unit with a Cd⋯Cd contact of 3.903 Å. Such a relatively short distance makes the [Cd2(CBA)4] unit become a potential metal–organic template to arrange 4-spy molecules in close contact for photodimerization. In fact, the non-bonding C⋯C contacts of the olefinic C atoms between the two adjacent parallel 4-spy ligands in such a dimer are 3.880 Å (C6⋯C19A) and 3.896 Å (C7⋯C20A).
 | ||
| Fig. 1 View of the structure of 1. Symmetry code: (A) −x + 2, −y + 3, −z + 2. The cyan, green, red, blue and gray nodes represent Cd, Cl, O, N and C atoms, respectively. | ||
, and its asymmetric unit has half of a [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2(H2O)4] dianion and one protonated 4-spy–H cation. Each Cd atom takes a pentagonal bipyramidal coordination geometry with two O atoms from one 5-NO2-1,3-BDC dianion, three O atoms from two 5-NO2-1,3-HBDC anions and two O atoms from two H2O molecules (Fig. 2a). Each pair of Cd atoms are connected by two 5-NO2-1,3-BDC dianions and two 5-NO2-1,3-HBDC anions to form a dianion of [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2(H2O)4]. Such a coordination dianion is further stabilized by intramolecular O11–H11⋯O3A and O11A–H11A⋯O3 hydrogen bonding interactions (symmetry code, A: −x, −y + 1, −z + 1). In 2, the 4-spy molecules do not coordinate with Cd atoms but accept protons to form 4-spy–H cations to balance the charge of the coordination dianions. The 4-spy–H cations are anchored by [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2(H2O)4] dianions via N3–H3⋯O1 hydrogen bonds (Fig. 2a). Between the two neighbouring H-bonded [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2(H2O)4]·(4-spy–H)2 molecules, each pair of 4-spy–H cations are stacked in the HT fashion with π–π interactions (3.736 Å) between the adjacent pyridyl and phenyl groups. The olefinic groups (C22C23B and C22BC23) adopt a parallel arrangement and the separation between them is 3.905 Å (Fig. 2b). Such a distance is similar to those found in the photoreactive coordination complexes [Zn2(cca)2(4-spy)2] (cca = 4-carboxycinnamate) and [Zn2(ndc)2(4-spy)2] (2,6-naphthalenedicarboxylate).13b This structural feature provides us with a good chance to explore a photochemical [2 + 2] cycloaddition reaction.
 | ||
| Fig. 2 (a) View of the hydrogen-bonded molecule of 2. (b) View of the 1D supramolecular chain of 2. The gray dashed lines represent the hydrogen bonds. Symmetry codes: (A) −x, −y + 1, −z + 1; (B) x + 1, y + 1, z + 2. The cyan, red, blue, gray and green nodes represent Cd, O, N, C and H atoms, respectively. | ||
Conclusions
In summary, the present work demonstrated that two regiospecific photodimers HH-ppcb and HT-ppcb were isolated from the photoproducts of two carboxylate-mediated cadmium complexes, respectively. The arrangements of the 4-spy molecules in 1 and 2 are mainly determined by the metal–carboxylate structures. In 1, each pair of Cd atoms is combined by four CBA ligands to form an electronically neutral [Cd2(CBA)4] unit at the basal positions. Two pairs of 4-spy were further linked with two Cd atoms at the apical positions to saturate the coordination sites of the Cd atoms. Therefore, the [Cd2(CBA)4] unit may work as a template to arrange 4-spy molecules in the HH fashion. In 2, each pair of Cd atoms is combined by four carboxylates to form a negatively charged [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2] unit at the basal positions. In order to balance the electronic charge of the [Cd2(5-NO2-1,3-BDC)2(5-NO2-1,3-HBDC)2] unit, each neutral 4-spy molecule accepts a proton in 2. As a result, the 4-spy–H cations cannot coordinate with Cd atoms. Each pair of 4-spy–H cations are stacked in the HT fashion with π–π interactions between the adjacent pyridyl and phenyl groups. We believe that such a synthetic methodology may be applied to other unsymmetrical alkenes to yield specific isomeric photodimers through regioselective photoreactions.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21171124 and 21201068), the State Key Laboratory of Organometallic Chemistry of Shanghai Institute of Organic Chemistry (201201006), the support for the Qin-Lan Project and the “333” Project of Jiangsu Province, the Priority Academic Program Development of Jiangsu Higher Education Institutions, the “SooChow Scholar” Program of Soochow University, the Natural Science Foundation of Anhui Province (1208085QB26). The authors also greatly thanked the helpful comments from the editor and the reviewers.Notes and references
- (a) G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CrossRef CAS; (b) I. G. Georgiev and L. R. MacGillivray, Chem. Soc. Rev., 2007, 36, 1239 RSC; (c) L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D. K. Bučar, Q. L. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS PubMed; (d) J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781 CrossRef CAS PubMed; (e) M. Nagarathinam, A. M. P. Peedikakkal and J. J. Vittal, Chem. Commun., 2008, 5277 RSC.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS PubMed.
- (a) S. Yamada, N. Uematsu and K. Yamashita, J. Am. Chem. Soc., 2007, 129, 12100 CrossRef CAS PubMed; (b) A. Nakamura, H. Irie, S. Hara, M. Sugawara and S. Yamada, Photochem. Photobiol. Sci., 2011, 10, 1496 RSC.
- (a) L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817 CrossRef CAS; (b) G. S. Papaefstathiou, A. J. Kipp and L. R. MacGillivray, Chem. Commun., 2001, 2462 RSC; (c) T. D. Hamilton, G. S. Papaefstathiou and L. R. MacGillivray, J. Am. Chem. Soc., 2002, 124, 11606 CrossRef CAS PubMed; (d) X. Gao, T. Friščić and L. R. MacGillivray, Angew. Chem., Int. Ed., 2004, 43, 232 CrossRef CAS PubMed; (e) A. N. Sokolov, D. C. Swenson and L. R. MacGillivray, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1794 CrossRef CAS PubMed; (f) B. R. Bhogala, B. Captain, A. Parthasarathy and V. Ramamurthy, J. Am. Chem. Soc., 2010, 132, 13434 CrossRef CAS PubMed; (g) S. Yamada and Y. Nojiri, Chem. Commun., 2011, 47, 9143 RSC; (h) C. Karunatilaka, D. K. Bučar, L. R. Ditzler, T. Friščić, D. C. Swenson, L. R. MacGillivray and A. V. Tivanski, Angew. Chem., Int. Ed., 2011, 50, 8642 CrossRef CAS PubMed; (i) S. Dutta, D. K. Bučar and L. R. MacGillivray, Org. Lett., 2011, 13, 2260 CrossRef CAS PubMed; (j) S. Bhattacharya, J. Stojaković, B. K. Saha and L. R. MacGillivray, Org. Lett., 2013, 15, 744 CrossRef CAS PubMed.
- (a) G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641 CrossRef CAS; (b) S. Yamada and Y. Tokugawa, J. Am. Chem. Soc., 2009, 131, 2098 CrossRef CAS PubMed.
- (a) T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500 CrossRef CAS PubMed; (b) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114 CrossRef CAS PubMed.
- (a) G. S. Papaefstathiou, Z. M. Zhong, L. Geng and L. R. MacGillivray, J. Am. Chem. Soc., 2004, 126, 9158 CrossRef CAS PubMed; (b) G. S. Papaefstathiou, I. G. Georgiev, T. Friščić and L. R. MacGillivray, Chem. Commun., 2005, 3974 RSC; (c) I. G. Georgiev, D. K. Bučar and L. R. MacGillivray, Chem. Commun., 2010, 46, 4956 RSC; (d) J. F. Eubank, V. C. Kravtsov and M. Eddaoudi, J. Am. Chem. Soc., 2007, 129, 5820 CrossRef CAS PubMed.
- (a) N. L. Toh, M. Nagarathinam and J. J. Vittal, Angew. Chem., Int. Ed., 2005, 44, 2237 CrossRef CAS PubMed; (b) M. Nagarathinam and J. J. Vittal, Angew. Chem., Int. Ed., 2006, 45, 4337 CrossRef CAS PubMed; (c) M. Nagarathinam and J. J. Vittal, Chem. Commun., 2008, 438 RSC; (d) A. M. P. Peedikakkal, L. L. Koh and J. J. Vittal, Chem. Commun., 2008, 441 RSC; (e) A. M. P. Peedikakkal and J. J. Vittal, Inorg. Chem., 2010, 49, 10 CrossRef CAS PubMed; (f) M. H. Mir, L. L. Koh, G. K. Tan and J. J. Vittal, Angew. Chem., Int. Ed., 2010, 49, 390 CrossRef CAS PubMed; (g) M. H. Mir, J. X. Ong, G. K. Kole, G. K. Tan, M. J. McGlinchey, Y. Wu and J. J. Vittal, Chem. Commun., 2011, 47, 11633 RSC.
- (a) W. L. Nie, G. Erker, G. Kehr and R. Fröhlich, Angew. Chem., Int. Ed., 2004, 43, 310 CrossRef CAS PubMed; (b) J. Paradies, I. Greger, G. Kehr, G. Erker, K. Bergander and R. Fröhlich, Angew. Chem., Int. Ed., 2006, 45, 7630 CrossRef CAS PubMed; (c) A. J. Blake, N. R. Champness, S. S. M. Chung, W. S. Li and M. Schröder, Chem. Commun., 1997, 1675 RSC; (d) A. Michaelides, S. Skoulika and M. Siskos, Chem. Commun., 2004, 2418 RSC; (e) A. Briceño, D. Leal, R. Atencio and G. Díaz de Delgado, Chem. Commun., 2006, 3534 RSC; (f) Y. Hill and A. Briceňo, Chem. Commun., 2007, 3930 RSC; (g) D. Roiban, E. Serrano, T. Soler, I. Grosu, C. Cativielad and E. P. Urriolabeitia, Chem. Commun., 2009, 4681 RSC; (h) G. K. Kole, R. Medishetty, L. L. Koh and J. J. Vittal, Chem. Commun., 2013, 49, 6298 RSC.
- (a) X. Y. Wang, Z. M. Wang and S. Gao, Chem. Commun., 2007, 1127 RSC; (b) Y. F. Han, Y. J. Lin, W. G. Jia, G. L. Wang and G. X. Jin, Chem. Commun., 2008, 1807 RSC; (c) W. Z. Zhang, Y. F. Han, Y. J. Lin and G. X. Jin, Organometallics, 2010, 29, 2842 CrossRef CAS; (d) W.-B. Yu, Y.-F. Han, Y. J. Lin and G. X. Jin, Chem.–Eur. J., 2011, 17, 1863 CrossRef CAS PubMed; (e) Y. C. Ou, W. T. Liu, J. Y. Li, G. G. Zhang, J. Wang and M. L. Tong, Chem. Commun., 2011, 47, 9384 RSC; (f) M. H. Xie, X. L. Yang and C. D. Wu, Chem.–Eur. J., 2011, 17, 11424 CrossRef CAS PubMed; (g) T. Wu, L. H. Weng and G. X. Jin, Chem. Commun., 2012, 48, 4435 RSC; (h) Y. C. Ou, D. S. Zhi, W. T. Liu, Z. P. Ni and M. L. Tong, Chem.–Eur. J., 2012, 18, 7357 CrossRef CAS PubMed.
- (a) M. Khan, G. Brunklaus, V. Enkelmann and H. W. Spiess, J. Am. Chem. Soc., 2008, 130, 1741 CrossRef CAS PubMed; (b) S. Y. Yang, P. Naumov and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 7247 CrossRef CAS PubMed; (c) G. K. Kole, G. K. Tan and J. J. Vittal, Org. Lett., 2010, 12, 128 CrossRef CAS PubMed; (d) D. Liu, Z.-G. Ren, H. X. Li, J. P. Lang, N. Y. Li and B. F. Abrahams, Angew. Chem., Int. Ed., 2010, 49, 4767 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, SHELXS-97, Program for the refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL-97, Program for Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- (a) Q. Chu, D. C. Swenson and L. R. MacGillivray, Angew. Chem., Int. Ed., 2005, 44, 3569 CrossRef CAS PubMed; (b) R. Medishetty, L. L. Koh, G. K. Kole and J. J. Vittal, Angew. Chem., Int. Ed., 2011, 50, 10949 CrossRef CAS PubMed; (c) D. Liu, J. P. Lang and B. F. Abrahams, Chem. Commun., 2013, 49, 2682 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures. CCDC 860515–860516. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce41609g |
| This journal is © The Royal Society of Chemistry 2014 |