Temperature-dependent guest reorientation: a reversible order–disorder transformation in a single crystal†
Matteo
Lusi
and
Leonard J.
Barbour
*
Department of Chemistry and Polymer Science, University of Stellenbosch, Stellenbosch 7600, South Africa. E-mail: ljb@sun.ac.za
First published on 10th October 2013
Abstract
A Werner clathrate undergoes a reversible, temperature-induced, single-crystal to single-crystal polymorphic transformation. Such transformation involves a realignment of the Werner complex, which generates a twofold axis and increases the space group symmetry from P21/n to C2/c. An increase of the crystal's dynamic disorder is also associated with the phase transition. Crystallographic and thermal behaviours are discussed.
The occurrence of multiple phases in the solid state includes polymorphism, allotropism, tautomerism, etc. No word correctly identifies the phenomenon as a whole and the term polymorphism is often used in sensu lato to (incorrectly) refer to what would be better defined as “polyformism” or “multiformism”.1 Whatever the term used, the phenomenon originates from the presence of more than one kinetically accessible phase.2 The kinetic product would spontaneously convert into a more stable form with time, although the process might be extremely slow. Sometimes a thermodynamic equilibrium exists between different phases, in which case the transformation is observed only upon modification of temperature and pressure, and can be reversed when the original conditions are restored.
The occurrence of multiple crystal forms is of primary importance in both theoretical and applied crystallography.3,4 It represents one of the most intriguing phenomena in solid-state chemistry for two reasons: i) from a practical point of view crystallizations do not always occur as designed and the formation of the desired (“engineered”) form and its property might be precluded by the existence of a more (or similarly) stable phase; ii) by reversing the paradigm, multiple crystal forms might represent a resource for crystalline materials that could enable tuning the compound's physical properties by changing its organization in the solid state.5 Despite recent progresses in the field, to date it is still not possible to predict all the variables that influence the formation of a crystal product,6,7 which limits the success of designed crystal synthesis. Controlling the occurrence of multiple forms is a step towards proper “crystal engineering”.
Investigation of the phenomenon is difficult because little structural information is available during the solid to solid transformation. In fact general experience suggests that microscopic transformations at the molecular (and supramolecular) level may often result in the destruction of the macroscopic single crystallinity. Exceptions are particularly common when the microscopic transformations have only limited effect on the crystal symmetry and internal stress.8,9 Reported examples include tautomerism,10 order–disorder11,12 and conformational transformation,13 as well as the more extreme examples such as guest exchange14,15 or photo-induced chemical reactions.16 These cases represent an opportunity to understand the reaction mechanisms in the solid state.
While investigating the separation properties of a Werner complex17–19 we found that one of its clathrates undergoes a single-crystal to single-crystal transformation in which the crystal symmetry is increased while the unit cell metrics are preserved. The transformation is associated with an order–disorder phase transition involving the guest. Remarkably, despite their structural similarity, the same behaviour was not observed in other clathrates of the same host.17–19
Crystals of [Ni(NCS)2ppp4]·2m-xyl (1) (ppp = para-phenylpyridine, m-xyl = meta-xylene) were obtained following a previously reported procedure. A single crystal was selected and its crystal structure determined with the diffraction data collected at 100 K (1100).‡ Four metal complexes and eight xylene molecules arrange in a monoclinic P21/n system. The packing of the complex generates two non-equivalent pockets, each filled with two guest molecules (Fig. 1). In one of them the two xylene molecules pack in a head-to-head fashion (with the methyl groups pointing towards each other) while in the other they orient tail-to-tail (methyl groups pointing away from each other). This structure is different from the one previously reported, which was collected at room temperature (1RT).20 In the original structure a monoclinic C centered cell with similar dimensions was observed and four metal complexes pack in the same fashion to host eight xylene guests, but in this case the complex axis is slightly tilted, forming an extra two fold axis along the crystallographic b direction. The pockets are equivalent and all the m-xylene molecules are disordered over the two positions (Fig. 1).
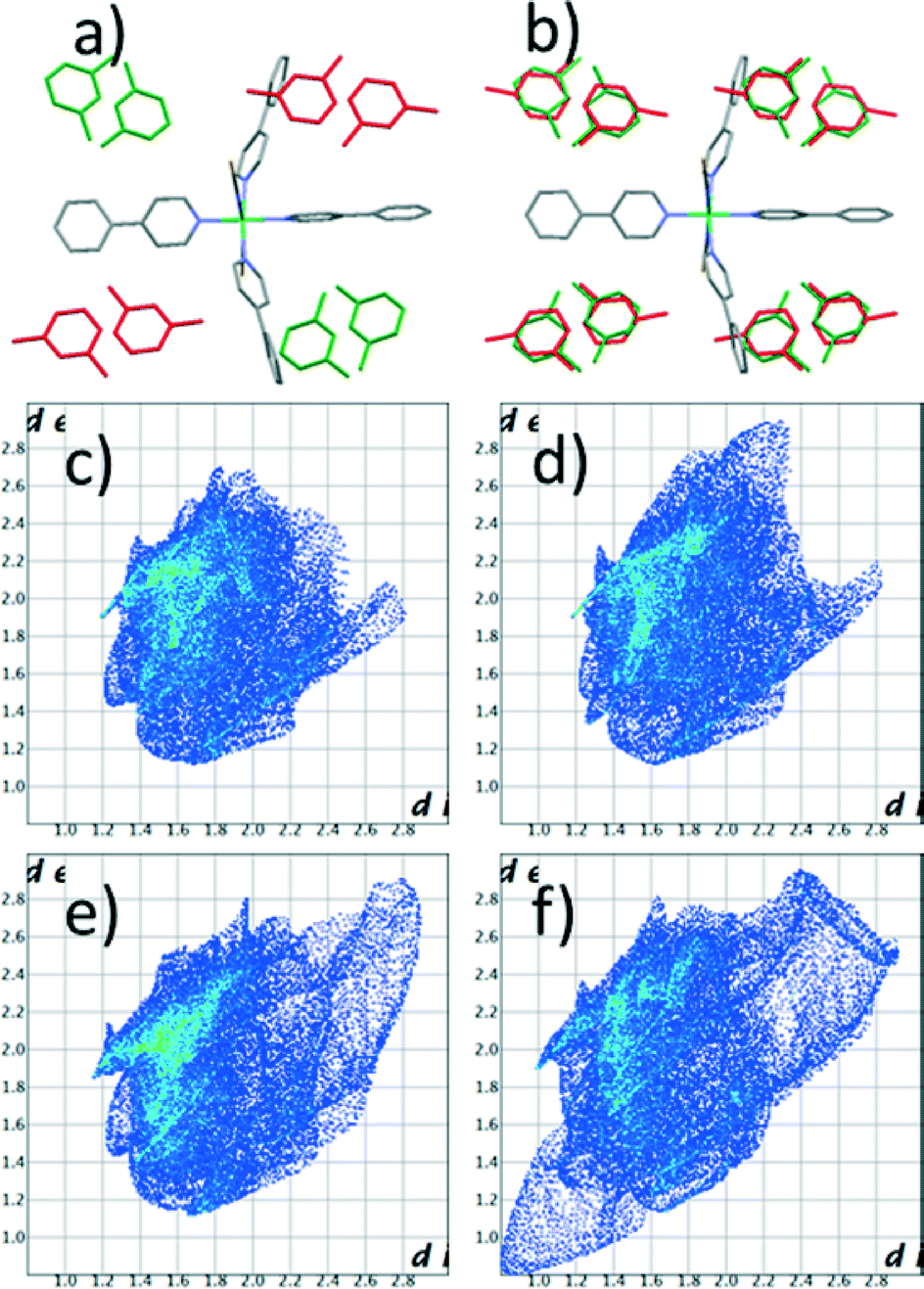 | ||
| Fig. 1 Crystal structure diagrams for 1100 (a) and 1RT (b). The structures are displayed along the a axis. The guests are highlighted in green (head-to-head) and red (tail-to-tail). Fingerprint plots of the Hirshfeld surfaces for the guests: head-to-head (c and d) and tail-to-tail (e and f). | ||
The two structures are so similar that the phase transition would probably be overlooked if analyzed by powder X-Ray diffraction (PXRD) at varied temperature. In fact the main difference between the two simulated patterns lies in two small peaks at around 6° and 13° in 2θ, which could be inadvertently ascribed to preferential orientation or experimental errors (Fig. 2). Nonetheless differential scanning calorimetry (DSC) confirms that, on heating, an endothermic phase transition of about 0.5 kcal mol−1 occurs, which is centered at approximately 225 K (see Fig. 3 and S2†). A second endothermic event occurs between 270 and 310 K. Both thermal events are reversible at around 200 and 270 K, respectively.
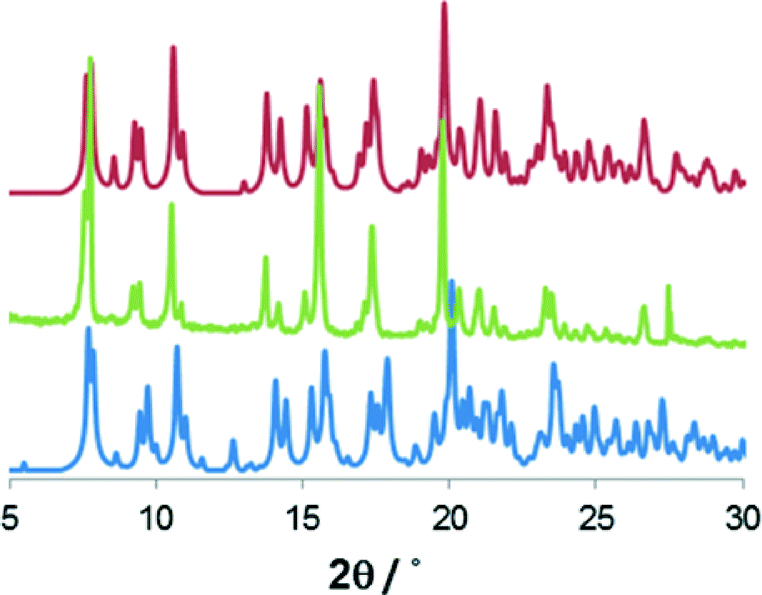 | ||
| Fig. 2 Comparison of powder X-ray diffraction measured for 1 (green) with the patterns simulated* for the structure determined at 100 K (blue) and at 313 K (red). | ||
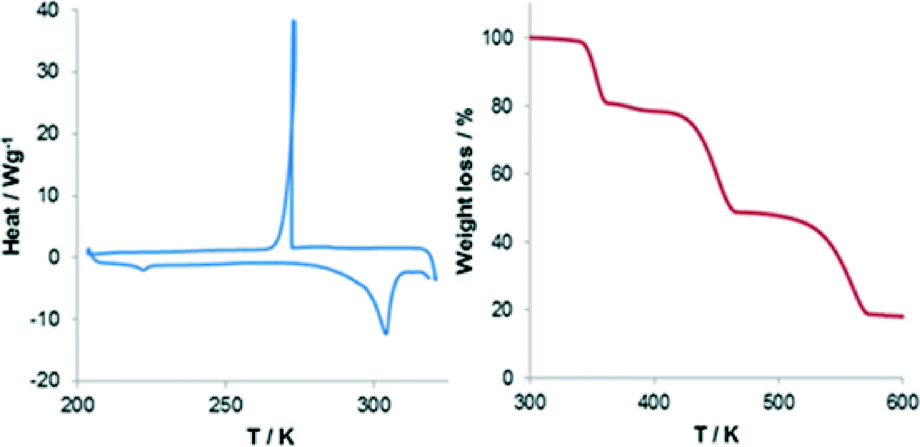 | ||
| Fig. 3 Differential scanning calorimetry (left) and thermogravimetric analysis (right) for 1. | ||
Fingerprint plot of the Hirshfeld surface for the guests: the head-to-head (c and d) and tail-to-tail (e and f).
We note that the low temperature transformation occurs near the operating limits of our calorimeter. The high temperature event it is not associated to any structural change. In our view this could be due to moisture adsorbed onto the surface of the sample during analysis, and which could not be dried without losing the organic guest. Thermogravimetric analysis (TGA) supports this hypothesis revealing that, beside the large 20% loss at around 350 K, polycrystalline 1 loses about 2% of mass from room temperature to about 380 K (Fig. 3 and S3†).
In order to understand the mechanism of the transformation, diffraction data of the same single crystal used for 1100 were collected at 173 (1173), 243 (1243) and 313 (1313) K. The structures were solved paying particular attention to the disorder of the xylene molecules. Unfortunately, structure refinement did not allow a precise anisotropic treatment of the disordered guest. Nonetheless some information could be retrieved from the analysis on the relative occupancy of the molecule in the two possible orientations (Table 1).
| Structure | 1100 | 1173 | 1243 | 1313 |
|---|---|---|---|---|
| Temperature (K) | 100 | 173 | 243 | 313 |
| R factor (%) | 4.66 | 5.72 | 6.01 | 6.02 |
| Space group | P21/n | P21/n | C2/c | C2/c |
| Rel, occupancy m-xyl 1 | 1 | 1 | 0.68 0.32 | 0.67 0.33 |
| 0 | 0 | |||
| Rel. occupancy m-xyl 2 | 1 | 0.79 | 0.69 | 0.67 |
| 0 | 0.21 | 0.31 | 0.33 | |
| Highest diff. peak (e Å−3) | 0.44 | 0.43 | 0.44 | 0.36 |
At 100 K the P21/n structure is practically ordered, with the xylene molecules frozen in their positions. The crystal disorder is modulated as a function of temperature.21 At 173 K one of the xylene molecules is still frozen while the disorder of the second guest increases, with the least favorable position observed 21% of the time. At 243 K the metal complex is tilted, aligning itself along the b axis and the space group symmetry is increased to C2/c. The xylene molecules become equivalent and the least favorable position is observed 33% of the time. No other change is observed at 313 K. The phase transformation is reversible upon cooling.
In all the structures the guests are kept in position by only weak interactions. Hirshfeld surface analysis22 reveals that about 80% of the guest surface is involved in interactions with hydrogen atoms. At 100 K the two xylene molecules have very similar fingerprints (Fig. 1c and 1e). The surface area for the guests in tail-to-tail orientation is only slightly larger, confirming that this molecule is less tightly bonded. In contrast, at high temperature a substantial difference exists between the two orientations (Fig. 1d and 1f). In particular it is clear that the guest in the tail-to-tail arrangement has extremely short H–H contact distances with symmetry-related parts. Of course these values are too short to be real and are the result of disorder. In our view this suggests that the guest rotation is only possible as a concerted process involving all the xylene molecules at once.
Conclusions
We have described an example of thermally induced polymorphism associated with an order–disorder phase transition of the guest in a Werner clathrate. The polymorphic transition is due to the in alignment of one of the complex's axes along [010]. Notably, the order–disorder transition involves only one of the guest molecules, whereas the disorder of the second is changes without discontinuity throughout the thermal range investigated.Acknowledgements
The National Research Foundation (South Africa) financed the research and the Claude Leon Foundation sponsored ML.Notes and references
- J. Bernstein, Cryst. Growth Des., 2011, 11, 632–650 CAS.
- T. Threlfall, Org. Process Res. Dev., 2003, 7, 1017–1027 CrossRef CAS.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford, 1st edn, 2007 Search PubMed.
- D. Braga, F. Grepioni, L. Maini and M. Polito, in Molecular Networks, ed. M. W. Hosseini, Springer, Berlin Heidelberg, 2009, vol. 132, pp. 25–50 Search PubMed.
- C. J. Adams, A. L. Gillon, M. Lusi and A. G. Orpen, CrystEngComm, 2010, 12, 4403–4409 RSC.
- T. Beyer, T. Lewis and S. L. Price, CrystEngComm, 2001, 3, 178–212 RSC.
- A. Gavezzotti, CrystEngComm, 2002, 4, 343–347 RSC.
- G. Kaupp, CrystEngComm, 2003, 5, 117–133 RSC.
- A. N. Sokolov, D. C. Swenson and L. R. MacGillivray, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1794–1797 CrossRef CAS PubMed.
- N. Zencirci, E. Gstrein, C. Langes and U. J. Griesser, Thermochim. Acta, 2009, 485, 33–42 CrossRef CAS PubMed.
- S. M. Haile, D. A. Boysen, C. R. I. Chisholm and R. B. Merle, Nature, 2001, 410, 910–913 CrossRef CAS PubMed.
- M. A. Garcia-Garibay, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10771–10776 CrossRef CAS PubMed.
- M. A. Malwitz, S. H. Lim, R. L. White-Morris, D. M. Pham, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2012, 134, 10885–10893 CrossRef CAS PubMed.
- L. Dobrzańska, G. O. Lloyd, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2006, 45, 5856–5859 CrossRef PubMed.
- T. Jacobs, J.-A. Gertenbach, D. Das and L. J. Barbour, Aust. J. Chem., 2010, 63, 573–577 CrossRef CAS.
- T. Friščić and L. R. MacGillivray, Z. Kristallogr., 2005, 220, 351–363 CrossRef.
- M. Lusi and L. J. Barbour, Angew. Chem., Int. Ed. Engl., 2012, 51, 3928–3931 CrossRef CAS PubMed.
- E. Batisai, M. Lusi, T. Jacobs and L. J. Barbour, Chem. Commun., 2012, 48, 12171–12173 RSC.
- M. Lusi and L. J. Barbour, Chem. Commun., 2013, 49, 2634–2636 RSC.
- M. H. Moore, L. R. Nassimbeni and M. L. Niven, Inorg. Chim. Acta, 1987, 131, 45–52 CrossRef CAS.
- A. Gavezzotti, J. Pharm. Sci., 2007, 96, 2232–2241 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic and computational details; DSC and TGA analysis. CCDC 933918–933921. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce41572d |
‡ Crystal data for 1100: C62H56N6NiS2, M = 1007.95, lilac plate, 0.30 × 0.20 × 0.05 mm3, monoclinic, space group P21/n (no. 14), a = 10.4770(4), b = 23.0584(10), c = 22.7761(9) Å, β = 99.028(2)°, V = 5434.1(4) Å3, Z = 4, Dc = 1.232 g cm−3, F000 = 2120, MoKα radiation, λ = 0.71073 Å, T = 100(2) K, 2θmax = 60.9°, 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 528 reflections collected, 14861 unique (Rint = 0.0537). Final GooF = 1.030, R1 = 0.0466, wR2 = 0.1020, R indices based on 10178 reflections with I > 2σ(I) (refinement on F2), 644 parameters, 0 restraints. 528 reflections collected, 14861 unique (Rint = 0.0537). Final GooF = 1.030, R1 = 0.0466, wR2 = 0.1020, R indices based on 10178 reflections with I > 2σ(I) (refinement on F2), 644 parameters, 0 restraints.Crystal data for 1313: C62H56N6NiS2, M = 1007.95, lilac plate, 0.30 × 0.25 × 0.05 mm3, monoclinic, space group C2/c (no. 15), a = 10.6221(7), b = 23.1917(16), c = 23.0275(15) Å, β = 99.649(4)°, V = 5592.4(6) Å3, Z = 4, Dc = 1.197 g cm−3, F000 = 2120, MoKα radiation, λ = 0.71073 Å, T = 313(2) K, 2θmax = 60.9°, 27 |
| This journal is © The Royal Society of Chemistry 2014 |