Pseudopolymorphism leading and two different supramolecular aggregations in a phosphate monoester: role of a rare water-dimer†
Alok Ch.
Kalita‡
,
Kamna
Sharma‡
and
Ramaswamy
Murugavel
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: rmv@chem.iitb.ac.in; Fax: +91 22 2572 3480; Tel: +91 22 2576 7163
First published on 8th October 2013
Abstract
Whereas 2,6-diisopropyl- and 2,6-dimethylphenyl-phosphate forms a 1-D chain, 2,6-diisopropyl-4-amino-phenylphosphate crystallizes as two different 3-D supramolecular aggregates. The first pseudopolymorph 1a is a porous framework built around a rare water-dimer template that undergoes single-crystal to single-crystal transformations to yield desolvated form 1b. The second-pseudopolymorph 1c, obtained by re-crystallisation of 1a in methanol, is a dense 3-D framework.
Owing to the presence of many –OH groups on the central main group element, organosilanols and organophosphorus acids have been extensively studied as ligands in materials chemistry.1 An additional interesting aspect of this class of compounds is their solution and solid-state aggregation behaviour.2 The association behaviour of organosilanols have been studied as early in 1980s in view of the implication of hydrogen bonding interactions emanating from Si–OH groups in their use as silane coupling agents (Scheme 1).3
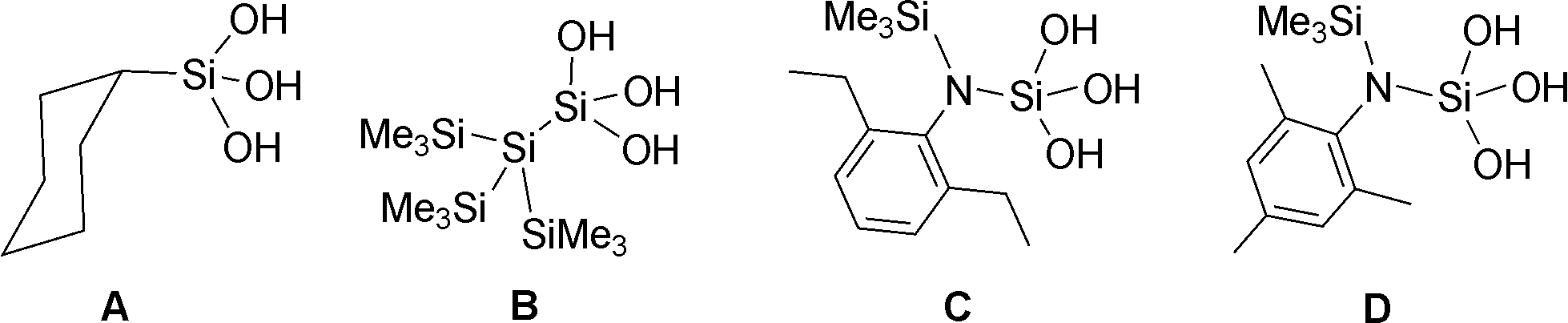 | ||
| Scheme 1 Aggregation of organosilanetriols. | ||
The first crystal structure of a silanetriol was determined in 1982 for cyclohexyl silanetriol, CySi(OH)3 (A) which was shown to form a double-sheet structure where the –Si(OH)3 groups aggregate in a head-to-head fashion.3 Subsequent structure determination of other silanetriols however revealed the existence of other types of aggregates. For example, the Si-bonded silanetriol (Me3Si)3Si–Si(OH)3 (B)4 forms a H-bonded close-shell polyhedral cluster containing six silanetriol molecules. It has been further demonstrated that the N-bonded silanetriol (C)2b also forms a cage structure, albeit with eight silanetriol molecules in an asymmetric fashion. Change of alkyl substituent on the aryl ring as in D results in a rare tubular-well structure where the Si(OH)3 moieties come together in a long tubular fashion (ESI,† Fig. S1).5 It has also been shown that these H-bonded architectures can be further modified by forming their amine complexes.2a
Organophosphonic acids also form very strong H-bonded structures as they can simultaneously act as strong H-bond donors and acceptors due to the presence of both P–OH and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. It has been shown that tert-butylphosphonic acid, one of the most well studied organophosphorous acids, forms two different types of aggregates depending on the solvent of the crystallisation (H-bonded polymer from THF and discrete hexameric cluster from CDCl3).6 The aggregation behaviour of mono, bis and tris phosphonic acids in the solid state (X-ray) and in solution has also been recently highlighted.7
O groups. It has been shown that tert-butylphosphonic acid, one of the most well studied organophosphorous acids, forms two different types of aggregates depending on the solvent of the crystallisation (H-bonded polymer from THF and discrete hexameric cluster from CDCl3).6 The aggregation behaviour of mono, bis and tris phosphonic acids in the solid state (X-ray) and in solution has also been recently highlighted.7
We have been employing a series of monoarylphosphates (Scheme 2) as synthons for the generation of metallophosphate clusters and frameworks.8 These studies have revealed that the reactions of phosphate monoesters with metal salts are dissimilar to phosphonic acids in many ways.9 Hence, it is of interest to examine the aggregation behaviour of monoarylphosphates with varied substituents on the para-position of the aryl ring. Some of these substituents offer additional H-bonding terminals which would in turn lead to interesting association both in solution and solid-state.
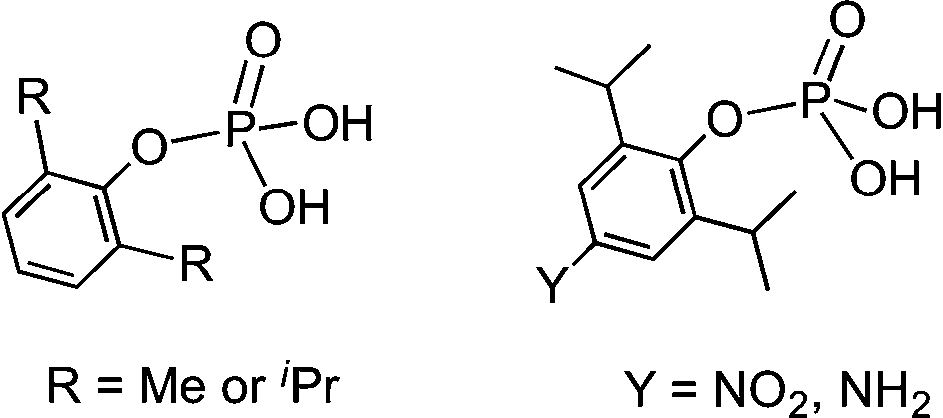 | ||
| Scheme 2 Monoaryl phosphates. | ||
Based on our earlier studies, where we have shown that the interactions of amines with the silanols can significantly alter the H-bonding network of parent silanols,2a,b the present investigation is aimed at the introduction of an amino group on the para-position of the aryl group and compare its aggregation behaviour vis-à-vis the parent compounds10 (Scheme 2). The synthesis of the target aminoaryl phosphate was achieved in two steps. The LiCl catalysed reaction of 2,6-diisopropyl-4-nitrophenol with POCl3 yields (4-NO2-2,6-Pri2C6H2O)P(O)Cl2 which on hydrolysis by aqueous acetone produces (4-NO2-2,6-Pri2C6H2O)P(O)(OH)2. In the second step the nitro compound was reduced by Pd/C in the presence of hydrazine hydrate in dry ethanol to yield the aminoaryl phosphate [(4-NH2-2,6-Pri2C6H2O)P(O)(OH)2] (adippH2; 1). Crude 1 was crystallized in methanol at room temperature to yield the first of the two pseudo-polymorphs of this compound, which has been formulated as [1·1/3H2O·MeOH] (1a) after determination of its structure by single crystal X-ray diffraction studies. Compound 1a undergoes a reversible single-crystal to single-crystal (SC–SC) transformation under vacuum to yield methanol desolvated form of 1a, [1·1/3H2O] (1b). On the other hand, recrystallisation of 1a in methanol results in the isolation of a second pseudo-polymorph [1·MeOH] (1c), which is devoid of water.§
Compound 1 has been fully characterized by analytical and spectroscopic methods including NMR and mass spectrometry and single crystal X-ray diffraction studies. Both 1a and 1c forms exhibit similar spectral behaviour (1b was not isolated in bulk, but has been observed as a SC–SC transformed single crystal). In the FT-IR spectrum characteristic absorptions for ν(N–H), νasy(C–H), νsy(C–H), νbent(N–H), and νP–O have been observed at 3285, 2964, 2866, 1608 and 979 cm−1, respectively (Fig. S2†). Two strong absorptions are observed in the ultraviolet region at 234 nm and 292 nm, while the fluorescence emission is observed at 350 nm (Fig. S3†). In the 1H NMR spectrum, the isopropyl CH3, isopropyl CH, and aromatic CH protons have been found to resonate at δ 1.05 (doublet), 3.43 (septet) and 6.25 ppm (singlet), respectively (Fig. S4†). The 31P NMR spectrum displays a single resonance at δ −4.24 ppm (Fig. S5†).
A methanol solution of 1 exhibits a peak in the positive ion ESI-HRMS at the exact molecular mass (Fig. S6†). The negative ion ESI LRMS spectrum is however more revealing in terms of the association of molecules of 1 in solution; the peaks observed at m/z 272.1 (100%), 545.2 (90%), and 831.3 (20%) correspond to [M − 1]−, [2M − 1]−, and [3M − 1]− ions, arising out of the presence of monomeric, dimeric, and trimeric forms of 1 in solution under electrospray mass spectral conditions (Fig. 1), as it has been observed for phosphonic acids tBuPO3H26 and MesCH2PO3H2.7
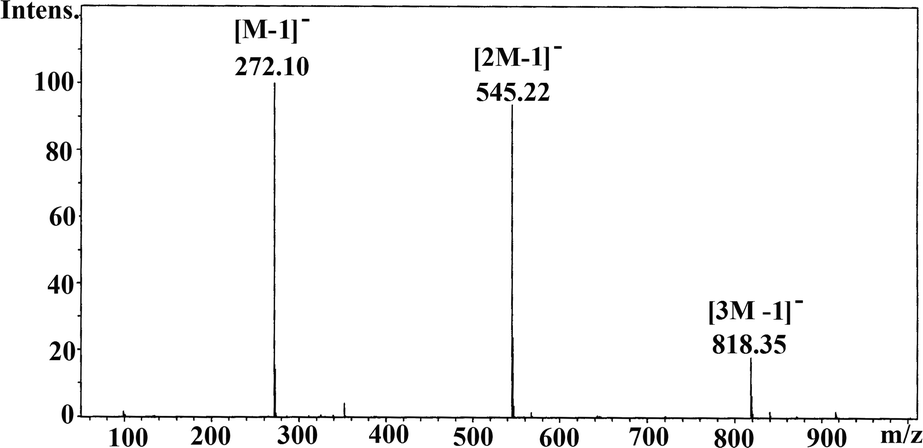 | ||
| Fig. 1 Negative ion mass spectrum (ESI) of 1a in methanol revealing the solution association behaviour. | ||
Single crystals of 1a have been obtained from reaction mixture by slow evaporation of solvent. 1a crystallizes in P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1c space group and its structure solution revealed that the asymmetric part contains one molecule of (4-NH2-2,6-Pri2C6H2O)P(O)(OH)2 (Fig. 2a), apart from a molecule of solvent methanol and one-third of a water molecule (Fig. 2). The bond lengths and angles observed for 1a are similar to those observed for other monoaryl phosphates described in the literature with tetrahedral geometry around central phosphorus atoms.10,11 What makes the structure of 1a different from other structurally characterized phosphate esters, phosphonic acids and silanetriols is the presence of a –NH2 group on the para-position of the aryl ring and the solvent water molecule sitting on the crystallographic three-fold axis.
1c space group and its structure solution revealed that the asymmetric part contains one molecule of (4-NH2-2,6-Pri2C6H2O)P(O)(OH)2 (Fig. 2a), apart from a molecule of solvent methanol and one-third of a water molecule (Fig. 2). The bond lengths and angles observed for 1a are similar to those observed for other monoaryl phosphates described in the literature with tetrahedral geometry around central phosphorus atoms.10,11 What makes the structure of 1a different from other structurally characterized phosphate esters, phosphonic acids and silanetriols is the presence of a –NH2 group on the para-position of the aryl ring and the solvent water molecule sitting on the crystallographic three-fold axis.
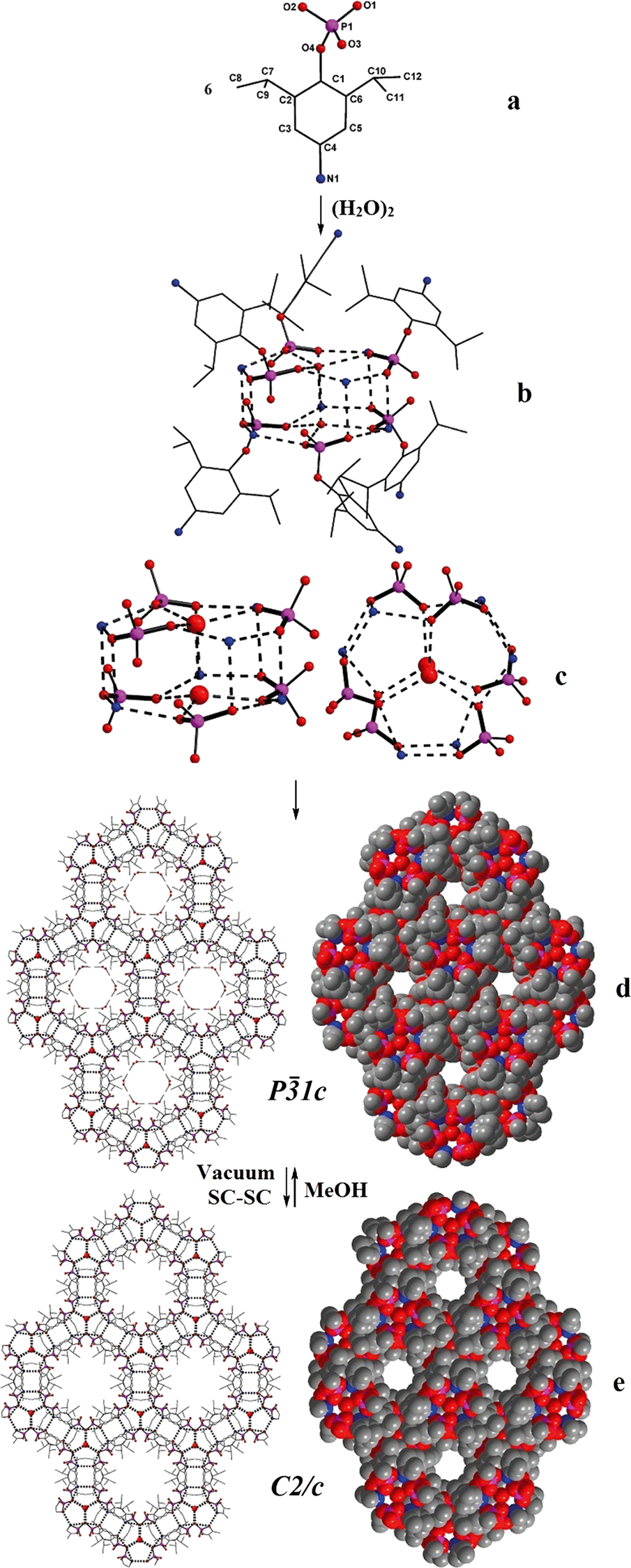 | ||
| Fig. 2 Structure evolution in 1a and its SC–SC transformation to 1b. (a) Ball and stick diagram of 1 in 1a; (b) dimeric water present on 3-fold axis connects six phosphate ligands each at the –PO3 and –NH3 ends to form a drum-like repeating unit; (c) core of the drum; (d) propagation of the drums to form a 3-D porous solid; (e) reversible removal of methanol and widening of pores by SC–SC conversion of 1a to 1b. | ||
Two adjacent water molecules along the 3-fold axis form a strong water-dimer with a O⋯O separation of only 2.979 Å (Scheme 3). This distance has an excellent match with the calculated value for an isolated water dimer in the ground state,12 as well as measured distances for water dimers found in inorganic and organic lattices.13 The water-dimers form the core of the hydrogen bonding network that produces a microporous framework for 1a. Starting from water-dimer, the formation of the microporous framework in 1a and its conversion to 1b is pictorially shown in Fig. 2. Each of the oxygen centres of this water dimer is surrounded by three –PO3 terminals leading to a hexameric phosphate assembly supported by (H2O)2 through H-bonding interactions (H2O⋯OP 2.698 Å) (Fig. 2b). Now six more molecules of phosphates approach this assembly through the –NH3 terminals to complete a H-bonded drum structure as depicted in Fig. 2c which is also schematized in Scheme 3.
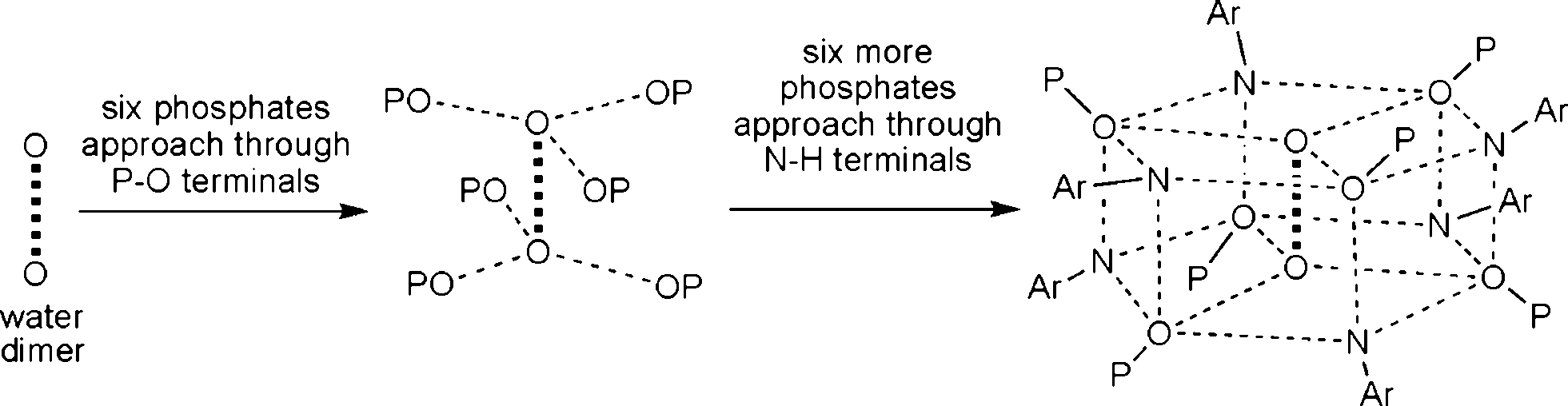 | ||
| Scheme 3 Schematic representation of the formation of a phosphate drum around the central water-dimer. | ||
Thus a water-dimer along with 12 monoaryl phosphates form the repeating unit in the structure of 1a. The free –PO3 and –NH3 ends of these repeating units in turn form another six such neighbouring drums (repeating units). This propagation is endless in all the three dimensions, since the phosphate molecule is functionalized at both ends (–PO3 and –NH3). The final result of this extensive H-bonded network is the formation of hexagonal channels/pores as shown in Fig. 2d. The unidimensional pores run parallel to the c-axis with a diameter of ~4 Å and hence 1a can be termed as an organophosphate microporous zeolitic framework. TGA analysis (Fig. S8†) of 1a shows that the weight loss starts just above the room temperature and about 10% of the material is lost by 200 °C due to removal of methanol and water molecules which leads to the collapse of the framework porosity. This has also been verified by the very poor surface area for 1a (4 m2 g−1) as determined by N2 gas adsorption studies after degassing the sample under high vacuum at 70 °C. Thus the porosity of 1a is retained only in the crystalline state and the framework is thermally too unstable.
Methanol from the channels of 1a can be fully removed through a SC–SC transformation under vacuum (Fig. 2 and Scheme 4). Evacuating the crystals of 1a for 3 hours (10−3 mmHg) did not result in any noticeable damage to the single crystal nature of the compound as revealed by the diffraction data obtained for an evacuated single crystal, which yielded a new crystal system and space group (monoclinic C2/c). Structure determination from these data clearly established the chemical constitution of the crystal to be [1·1/3H2O]3 (1b), a new structural form for adippH2 (Scheme 4). It is instructive to note that the prolonged evacuation under high vacuum results only in the loss of methanol and not water. As it can be seen from Fig. 2e, the overall architecture of the crystal packing remains intact during evacuation; the water dimers, the primary constituents of the crystal structure, remain persistent during the entire process of evacuation. Exposing crystals of 1b to methanol (soaking for a couple of minutes) converts 1b back to 1a with concomitant changes in the crystal system and space group (Scheme 4). Thus during both SC–SC (forward and backward) transformations, the water dimer remains intact and so the overall crystal structure and the porous nature of the framework.
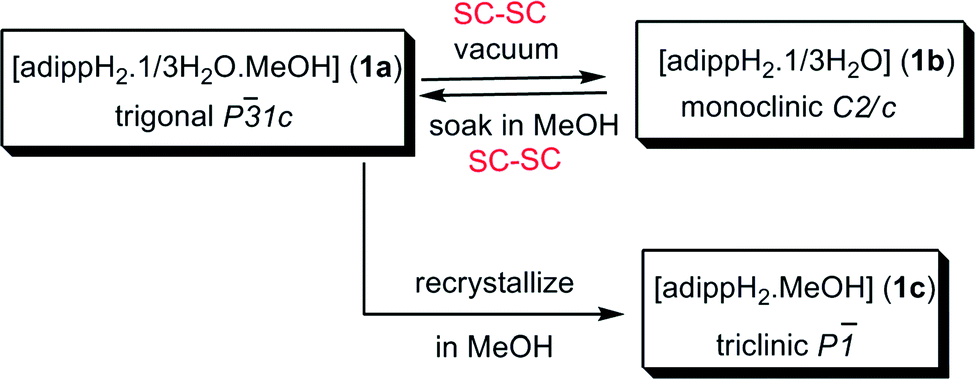 | ||
| Scheme 4 Various crystalline forms of adippH2. | ||
Since removal of water through SC–SC transformations could not be achieved, attempts have been made to re-crystallise 1a in methanol which resulted in crystals of [1·MeOH] (1c) (Scheme 4), whose morphology is different from the crystals of 1a and 1b. Determination of the crystal structure revealed that 1c crystallizes in triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Further, the asymmetric part of the unit cell contains a molecule of organophosphate and a molecule of methanol. Thus, during the re-crystallization process, the water present per asymmetric part in 1a has left the crystal lattice (Fig. 3). The absence of water-dimer in 1c leads to a supramolecular association which is essentially derived from coming together of four –PO3 and four –NH3 moieties through strong intermolecular O–H⋯O and O–H⋯N/O⋯H–N hydrogen bonding interactions (Fig. 3a). The spherical cages shown in Fig. 3a are connected to each other in the form of a 2-D sheet with the aryl group spacer in between. These 2D sheets are further connected through lattice methanol molecules, eventually giving rise to another 3-D H-bonded network (Fig. 3b).
space group. Further, the asymmetric part of the unit cell contains a molecule of organophosphate and a molecule of methanol. Thus, during the re-crystallization process, the water present per asymmetric part in 1a has left the crystal lattice (Fig. 3). The absence of water-dimer in 1c leads to a supramolecular association which is essentially derived from coming together of four –PO3 and four –NH3 moieties through strong intermolecular O–H⋯O and O–H⋯N/O⋯H–N hydrogen bonding interactions (Fig. 3a). The spherical cages shown in Fig. 3a are connected to each other in the form of a 2-D sheet with the aryl group spacer in between. These 2D sheets are further connected through lattice methanol molecules, eventually giving rise to another 3-D H-bonded network (Fig. 3b).
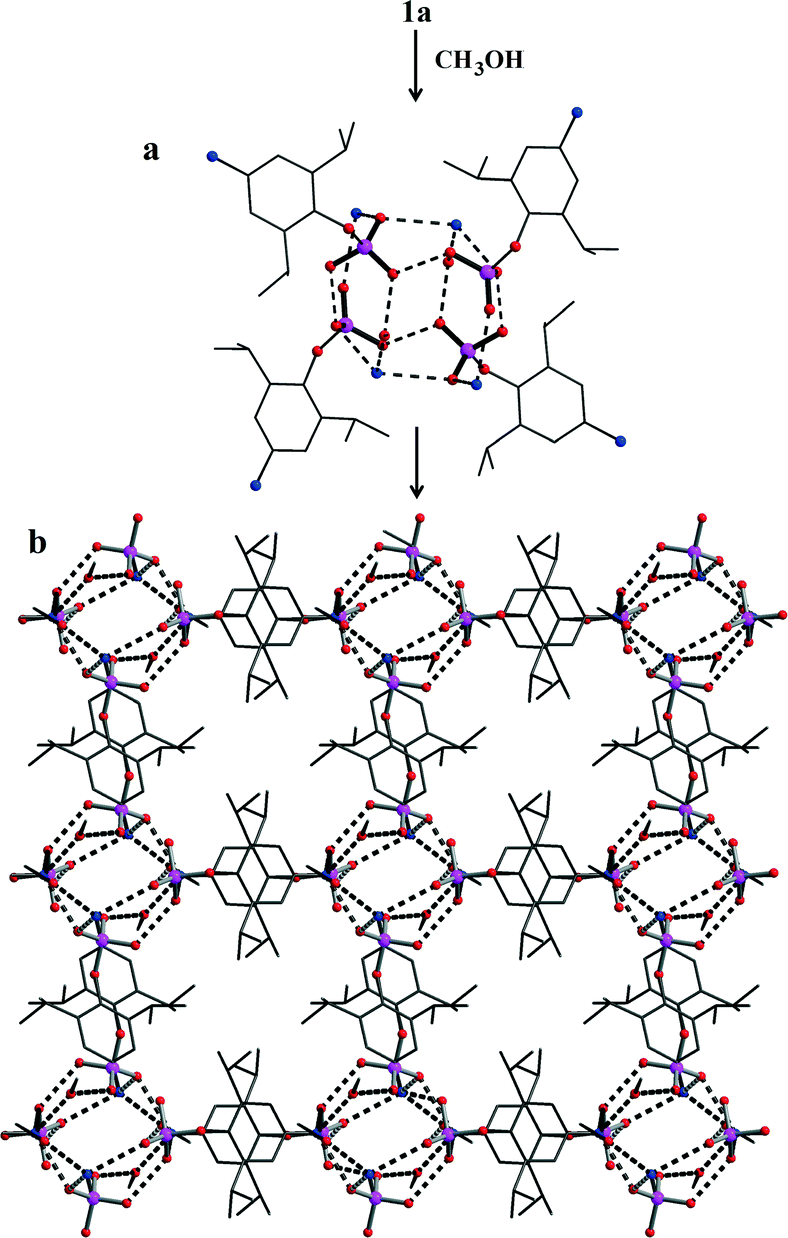 | ||
| Fig. 3 Structure of 1b; (a) formation of a cage-like repeating unit by H-bonding between four –PO3 and four –NH3 units; (b) propagation of the repeating units to form 2-D sheets which are connected by solvent methanol to form a 3-D non-porous solid. | ||
Conclusions
Introduction of an amino group within the structure of a monoaryl phosphate leads to H-bonded novel non-porous and porous 3-D framework structures. This work further demonstrates that pseudopolymorphism14 can be induced in a system by a simple recrystallization protocol in an anhydrous solvent. The rare water dimer in 1a acts as a template for the production of a porous structure, which undergoes reversible SC–SC transformations in vacuum and methanol to produce 1b. Further, it appears that the co-crystallization of 1 with a variety of potential H-bonding donor/acceptor molecules can lead to interesting supramolecular motifs with larger porous solids. Preliminary investigations reveal that addition of a dilute solution of DABCO to 1 in methanol produces peaks in ESI MS at m/z 386.2 (100%), 498.3 (65%), 771.4 (4%), which correspond to the formation of stable complexes [M+DABCO+1]+, [M+2DABCO+1]+, and [2M+2DABCO+1]+ (Fig. S8†). Similarly, addition of piperazine to 1 also leads to the formation of [M+pip+1]+ and [M+2pip+1]+ and [2M+2pip+1]+ ions (Fig. S9†). Isolation of these spectrally observed species and determination of their solid-state structures would reveal further interesting association patterns of 1.Acknowledgements
This work was supported by DST New Delhi and DAE (BRNS). RM thanks BRNS for the award of DAE-SRC Outstanding Investigator Awards grant which allowed the purchase of a single crystal CCD diffractometer. KS thanks CSIR for a research fellowship.Notes and references
- (a) R. Murugavel, V. Chandrasekhar and H. W. Roesky, Acc. Chem. Res., 1996, 29, 183 CrossRef CAS; (b) R. Murugavel, M. G. Walawalkar, M. Dan, H. W. Roesky and C. N. R. Rao, Acc. Chem. Res., 2004, 37, 763 CrossRef CAS PubMed; (c) R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Roesky, Chem. Rev., 1996, 96, 2205 CrossRef CAS PubMed; (d) R. Murugavel, A. Choudhury, M. G. Walawalkar, R. Pothiraja and C. N. R. Rao, Chem. Rev., 2008, 108, 3549 CrossRef CAS PubMed.
- (a) G. Prabusankar, R. Murugavel and R. J. Butcher, Organometallics, 2004, 23, 2305 CrossRef CAS; (b) G. Prabusankar, R. Murugavel and R. J. Butcher, Organometallics, 2005, 24, 2124 CrossRef CAS; (c) V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847 CrossRef CAS PubMed; (d) P. D. Lickiss, Polysilanol, in Chemistry of organic silicon compounds, ed. A. Rappoport, Chichester, UK, Wiley, 2001, pp. 695–744 Search PubMed.
- H. Ishida, J. L. Koenig and K. C. Gardner, J. Chem. Phys., 1982, 77, 5748 CrossRef CAS.
- S. S. Al-Juaid, N. H. Buttrus, R. I. Damja, Y. Derouiche, C. Eabom, P. B. Hitchcock and P. D. Lickiss, J. Organomet. Chem., 1989, 371, 287 CrossRef CAS.
- R. Murugavel, V. Chandrasekhar, A. Voigt, H. W. Roesky, H. G. Schmidt and M. Noltemeyer, Organometallics, 1995, 14, 5298 CrossRef CAS.
- M. Mehring, M. Schürmann and R. Ludwig, Chem.–Eur. J., 2003, 9, 837 CrossRef CAS PubMed.
- R. Murugavel and M. P. Singh, New J. Chem., 2010, 34, 1846 RSC.
- (a) R. Murugavel, S. Kuppuswamy, R. Boomishankar and A. Steiner, Angew. Chem., Int. Ed., 2006, 45, 5536 CrossRef CAS PubMed; (b) R. Murugavel, S. Kuppuswamy, N. Gogoi, R. Boomishankar and A. Steiner, Chem.–Eur. J., 2010, 16, 994 CrossRef CAS PubMed; (c) R. Murugavel, S. Kuppuswamy, N. Gogoi and A. Steiner, Inorg. Chem., 2010, 49, 2153 CrossRef CAS PubMed.
- R. Murugavel and C. N. R. Rao, Metal Phosphonate Chemistry: From Synthesis to Applications, ch. 11, Royal Society of Chemistry, 2012, p. 344 Search PubMed.
- A. Onoda, T. A. Okamura, H. Yamamoto and N. Ueyama, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o1022 CAS.
- A. N. Maity, Some aspects of Si-O-M and P-O-M chemistry, Master's Thesis, IIT-Bombay, 2003, (CCDC deposition number: 937943) Search PubMed.
- S. Scheiner, Ab initio studies of hydrogen bonds: the water dimer paradigm, Annu. Rev. Phys. Chem., 1994, 45, 23 CrossRef CAS PubMed.
- (a) P. J. Dutton, F. R. Fronczek, T. M. Fyles and R. D. Gandour, J. Am. Chem. Soc., 1990, 112, 8984 CrossRef CAS; (b) S. Manikumari, V. Shivaiah and S. K. Das, Inorg. Chem., 2002, 41, 6953 CrossRef CAS PubMed; (c) S. K. Ghosh and P. K. Bharadwaj, Inorg. Chem., 2003, 42, 8250 CrossRef CAS PubMed.
- (a) A. Nangia and G. R. Desiraju, Chem. Commun., 1999, 605 RSC; (b) C. Tedesco, L. Erra, I. Immediata, C. Gaeta, M. Brunelli, M. Merlini, C. Meneghini, P. Pattison and P. Neri, Cryst. Growth Des., 2010, 10, 1527 CrossRef CAS; (c) Y. Li, L. Li, Y. Zhu, X. Meng and A. Wu, Cryst. Growth Des., 2009, 9, 4255 CrossRef CAS; (d) T. L. Threlfall, Analyst, 1995, 120, 2435 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, crystallographic details, additional figures, and spectral characterization. CCDC deposition numbers 936275 (1a), 947939 (1b) and 936276 (1c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce40912k |
| ‡ These authors have made equal contributions to this work. |
§ Pd/C (100 mg of 10% w/w sample from Aldrich) was added to the solution of 2,6-diisopropyl-4-nitrophenylphosphate (1 g, 3.29 mmol) in dry ethanol (25 mL). The reaction mixture was heated under reflux in an inert atmosphere for 15 minutes and then hydrazine hydrate (15 mL) was added to the hot reaction mixture dropwise (Caution: exothermic reaction). The mixture was further heated under reflux for 24 h and cooled to room temperature under a stream of N2. The residue obtained after removal of solvent was crystallised from methanol to yield single crystals of [1·1/3H2O·MeOH] (1a). First and repeated recrystallizations of 1a from methanol results in 1c. Yield for 1a 0.57 g (46%). M.p. for 1a: 160 °C. IR (KBr, νmax/cm−1): 3285, 2964, 1608, 1095. HRMS calculated for C12H21NO4P (M + 1): m/z 274.1208; found m/z 274.1211. 1H NMR (DMSO-d6, 400 MHz, ppm) δ: 6.25 (s, Ar–CH, 1H), 3.43 (septet, iPr–CH, 2H), 1.05 (d, CH3, 12H). 31P NMR (DMSO-d6, 160 MHz, ppm) δ: −4.24 ppm. 13C NMR (DMSO-d6, 100.6 MHz, ppm) δ: 143.6 (C–NH2), 140.7 (C), 138.6 (C–O), 109.7 (CH), 26.0 (CH–iPr) and 23 (CH3–iPr) (Fig. S10†). Crystal data for 1a: C39H74N3O16P3, M = 933.92, trigonal, a = 19.098(4) Å, b= 19.098(4) Å, c = 16.333(3) Å, α = β = γ = 90°, V = 5159.1(2) Å3, T = 150(2) K. Space group P1c, Z = 4, 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 270 refl. measured, 3017 indep. refl. [R(int) = 0.0339]. The final R1 0.0638 [I > 2σ(I)]. Final wR2 0.1783 [I > 2σ(I)]; 0.1790 (all data). No attempts have been made to fix hydrogens on the water dimer since several positions have to be considered for each hydrogen around 3-fold axis. Crystal data for 1b: C36H62N3O13P3, M = 837.80, monoclinic, a = 19.394(2) Å, b= 35.680(3) Å, c = 15.180(1) Å, α = 90°, β = 90.311(15)°, γ = 90°, V = 10504(2) Å3, T = 150(2) K. Space group C2/c, Z = 8, 38532 refl. measured, 9210 indep. refl. [R(int) = 0.0748]. Final R1 0.1331 [I > 2σ(I)]. Final wR2 0.2882 [I > 2σ(I)]; 0.2947 (all data). Crystal data for 1c: C13H24NO5P, M = 305.30, triclinic, a = 11.946(5) Å, b= 11.952(5) Å, c = 12.210(5) Å, α = 116.916(7)°, β = 90.454(4)°, γ = 92.865(7)°, V = 1551.6(1) Å3, T = 150(2) K. Space group P, Z = 4, 12173 refl. measured, 5633 indep. refl. [R(int) = 0.0323]. Final R1 0.049 [I > 2σ(I)]. Final wR2 0.1051 [I > 2σ(I)]; 0.1131 (all data). 270 refl. measured, 3017 indep. refl. [R(int) = 0.0339]. The final R1 0.0638 [I > 2σ(I)]. Final wR2 0.1783 [I > 2σ(I)]; 0.1790 (all data). No attempts have been made to fix hydrogens on the water dimer since several positions have to be considered for each hydrogen around 3-fold axis. Crystal data for 1b: C36H62N3O13P3, M = 837.80, monoclinic, a = 19.394(2) Å, b= 35.680(3) Å, c = 15.180(1) Å, α = 90°, β = 90.311(15)°, γ = 90°, V = 10504(2) Å3, T = 150(2) K. Space group C2/c, Z = 8, 38532 refl. measured, 9210 indep. refl. [R(int) = 0.0748]. Final R1 0.1331 [I > 2σ(I)]. Final wR2 0.2882 [I > 2σ(I)]; 0.2947 (all data). Crystal data for 1c: C13H24NO5P, M = 305.30, triclinic, a = 11.946(5) Å, b= 11.952(5) Å, c = 12.210(5) Å, α = 116.916(7)°, β = 90.454(4)°, γ = 92.865(7)°, V = 1551.6(1) Å3, T = 150(2) K. Space group P, Z = 4, 12173 refl. measured, 5633 indep. refl. [R(int) = 0.0323]. Final R1 0.049 [I > 2σ(I)]. Final wR2 0.1051 [I > 2σ(I)]; 0.1131 (all data). |
| This journal is © The Royal Society of Chemistry 2014 |