 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereospecific conversion of alcohols into pinacol boronic esters using lithiation–borylation methodology with pinacolborane†
Stefan
Roesner
,
Christopher A.
Brown
,
Maziar
Mohiti
,
Alexander P.
Pulis
,
Ramesh
Rasappan
,
Daniel J.
Blair
,
Stéphanie
Essafi
,
Daniele
Leonori
and
Varinder K.
Aggarwal
*
School of Chemistry, Cantock’s Close, Bristol, BS8 1TS, UK. E-mail: v.aggarwal@bristol.ac.uk
First published on 26th February 2014
Abstract
The synthesis of primary and secondary pinacol boronic esters via lithiation–borylation of carbamates and benzoates with pinacolborane is described. This new protocol enables the highly selective synthesis of enantioenriched and geometrically defined boronic esters that cannot otherwise be accessed by alternative methodologies.
Organoboron compounds are versatile functional groups which, in addition to cross-coupling reactions, undergo a host of useful functional group transformations. The most common and indeed archetypal of these transformations is their stereospecific conversion into alcohols.1 The reverse process, the stereospecific conversion of an alcohol into a boronic ester, would itself be very useful since chiral alcohols are readily available, but such a transformation is unknown (Scheme 1A). Related, but non-selective processes have recently been reported. For example, alcohols have been converted into halides or tosylates and subsequently reacted with B2(pin)2 in the presence of Cu,2a,b Pd2c or Ni2d,e salts. These reactions are believed to involve the metal–boryl species and occur via radical intermediates, rendering them non-stereospecific (Scheme 1B).2
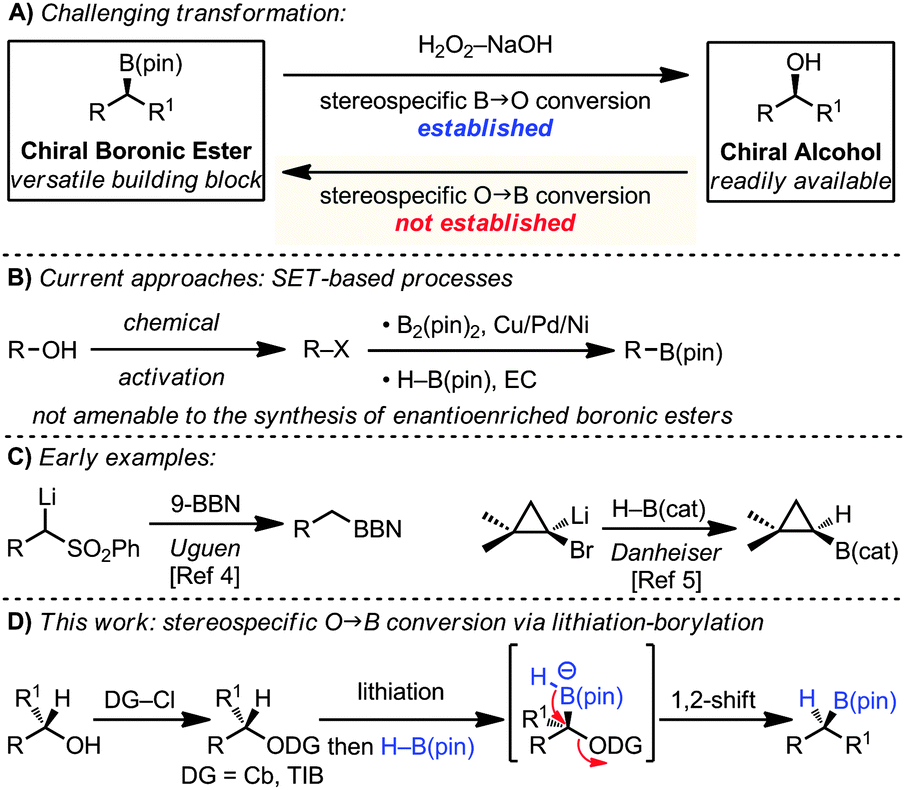 | ||
| Scheme 1 | ||
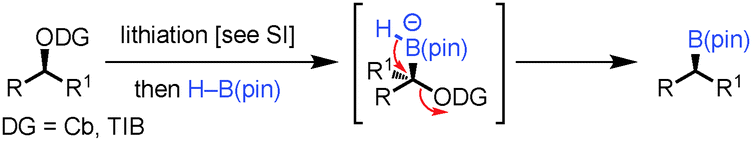
Our strategy for the conversion of alcohols into boronic esters is based on lithiation of carbamates or hindered benzoates3 and subsequent borylation with H–B(pin). Related reactions had been described with aryl and alkyl boronic esters in which aryl and alkyl groups migrated with expulsion of the carbamates or hindered benzoates.3 In the current strategy, a hydride would be required to migrate, a very different migrating group with very few reports in the literature.4,5 Uguen initially reported the conversion of phenylsulfones to primary alcohols by reaction of a lithiated sulfone with 9-BBN followed by oxidation.4 In this case the hydride migrates from a borane–ate complex, a much more facile process than from a boronic ester–ate complex. Danheiser showed that treatment of 1,1-dibromocyclopropanes with n-BuLi and H–B(cat) gave the cyclopropyl boronic esters via an intermediate boronate complex, followed by 1,2-hydride migration (Scheme 1C).5
In this communication we show that this strategy can also be applied to a range of carbamates and hindered benzoates enabling the near stereospecific conversion of readily accessible chiral, non-racemic alcohols into chiral boronic esters with high enantiomeric ratios (Scheme 1D).
Initial studies focused on the benzylic carbamate 1, which can be prepared in very high enantiopurity and on gram scale (ee 99%). Thus, after stereospecific lithiation6 two different boranes [H–B(pin) and H–B(cat)] were added leading to the corresponding ‘boron–ate’ complexes 2 and 3. We were delighted to find that upon warming boronic ester 4 [using H–B(pin)] was formed in high yield and enantiospecificity (92% yield, 98% ee, 98% es). Despite Danheiser's report (Scheme 1C),5 the use of H–B(cat) resulted in a very low yield of the boronic ester 4′ (11%)7 (Scheme 2). This method represents a valid alternative for the synthesis of 4 in very high enantiopurity as the metal-catalysed hydroboration of styrene proceeds with lower levels of enantioselectivity (98% ee vs. 92% ee).8a
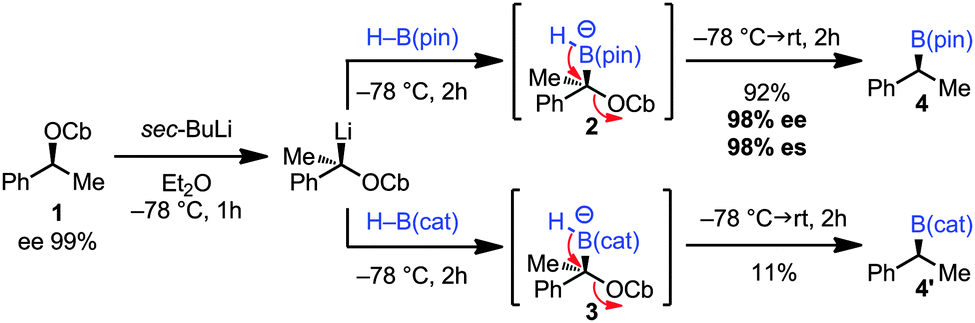 | ||
| Scheme 2 | ||
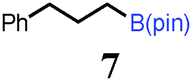
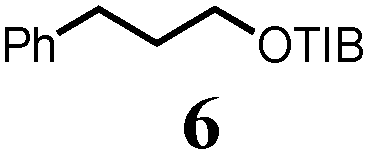
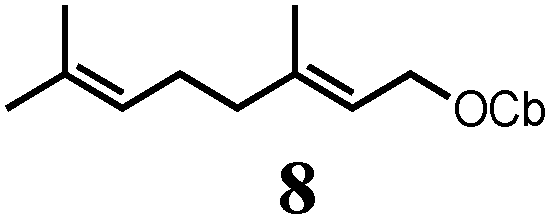
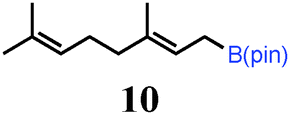
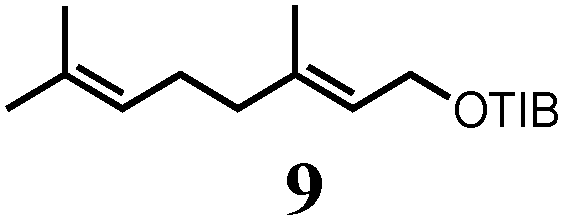
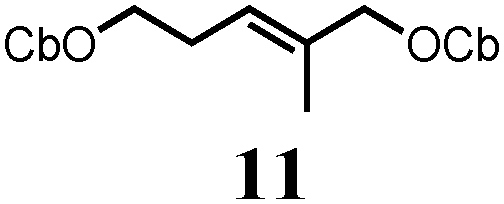
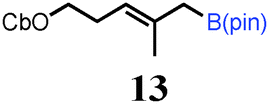
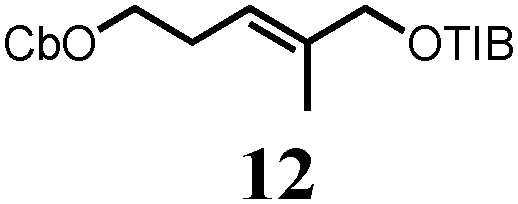
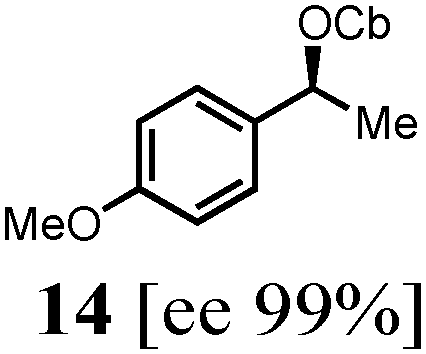
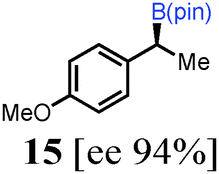
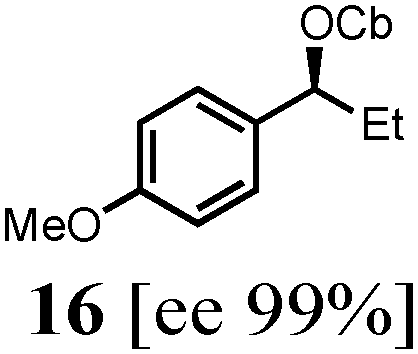
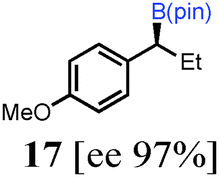
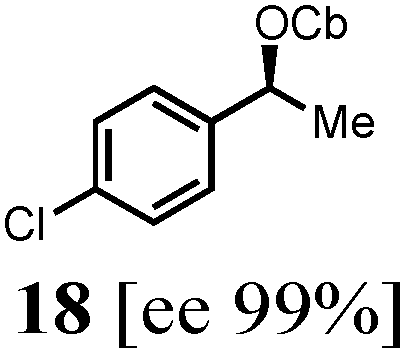
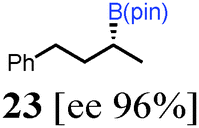
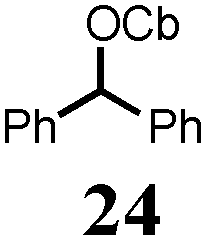
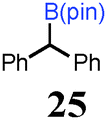
To probe the scope of this new 1,2-metallate rearrangement we then evaluated a selection of structurally different carbamates and benzoates with particular emphasis on accessing boronic esters that are difficult to prepare using other methods (Table 1). With primary aliphatic substrates 5 and 6, the benzoate derivative 6 was found to be better providing the desired product 7 in 83% yield (compare entries 1 and 2). Allylic boronic esters are very useful intermediates in organic synthesis that can undergo a broad range of reactions with aldehydes,9a,b ketones,9b imines9c and as Suzuki coupling partners.9d However, metal-catalysed conversion of allylic alcohols or related derivatives to allylic boronic esters is often accompanied by isomerisation of variable amounts of olefin.2 Thus, geraniol was converted into the corresponding carbamate 8 and benzoate 9 and subsequent lithiation–borylation gave the desired allylic boronic ester 10 in good yields and as a single isomer in both cases (entries 3 and 4).10 This method was extended to tri-substituted allylic boronic esters, a class of substrates that are especially problematic to be prepared using the metal-catalysed process.2c,d Thus, deprotonation of substrates 11 and 12 followed by addition of H–B(pin), gave the allylic boronic ester 13 in moderate yields but as a single isomer (entries 5 and 6).10 We then investigated the scope of secondary alcohol derivatives. This class of compounds is particularly relevant as they are readily available in high enantiomeric ratios. Enantioenriched chiral benzylic boronic esters can be accessed by the asymmetric metal-catalysed hydroboration of styrenes but certain derivatives give low selectivities.8 For example, the Cu(I)-TangPhos catalysed asymmetric hydroboration of p-chlorostyrene gives the boronic ester 19 in 87% ee.8a We started our investigations using the p-MeO substituted carbamate 14 that underwent the desired lithiation–borylation in a good yield and 95% es (15, entry 7). When the readily available ethyl-substituted and Cl-functionalised carbamates 16 and 18 were exposed to the reaction conditions, the desired boronic esters 17 and 19 were obtained with very high levels of enantiospecificity (98 and 99% respectively, entries 8 and 9). Reaction of the 2-naphthyl substituted carbamate 20 gave the desired boronic ester 21 in good yield and high ee (entry 10). Non-benzylic secondary alcohol derivatives can also be employed. Thus, lithiation of chiral benzoate 223e followed by addition of H–B(pin), gave dialkyl secondary boronic ester 23 with high ee (96%) and good yield. This methodology can also be applied to achiral secondary alcohols. For example, deprotonation of diaryl carbamate 24 and borylation gave the desired boronic ester 25 in high yield (entry 12).11
|
|
|||
|---|---|---|---|
| Entry | Carbamate/benzoate | Product | Yield (%) [es (%)] |
| 1 |
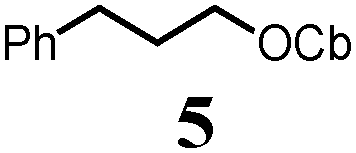
|
|
65 |
| 2 |
|
7 | 83 |
| 3 |
|
|
69 |
| 4 |
|
10 | 66 |
| 5 |
|
|
49 |
| 6 |
|
13 | 34 |
| 7 |
|
|
62 [95] |
| 8 |
|
|
64 [98] |
| 9 |
|
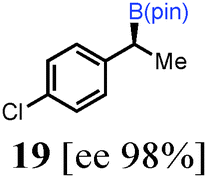
|
43 [99] |
| 10 |
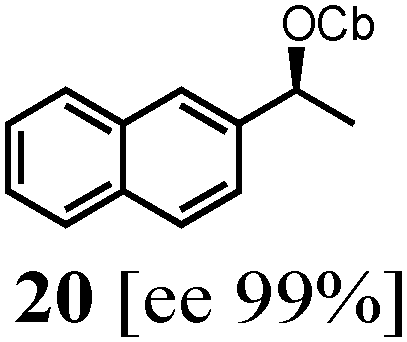
|
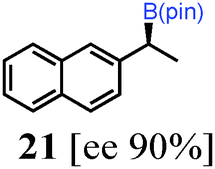
|
76 [91] |
| 11 |
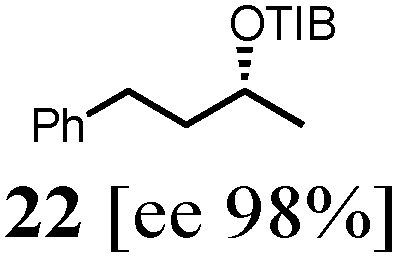
|
|
53 [98] |
| 12 |
|
|
79 |
The lithiation–borylation reactions of secondary benzylic carbamates are usually completely stereospecific.3c,d We were surprised by the slight erosion in enantiospecificity observed for the reaction of carbamates 14 and 20 (Table 1, entries 7 and 10). One possibility was that if 1,2-migration was slow then upon warming competing dissociation of the ate-complex back to the starting Li-carbamate might take place, which could be followed by racemisation of the Li-carbamate (Scheme 3A), a process we had observed with hindered boronic esters.3d
 | ||
| Scheme 3 | ||
In order to probe reversibility, we added Et–B(pin) after formation of the boron–ate complexes 26 but none of the Et-incorporated boronic ester 27 was observed (Scheme 3B). In a control experiment, reaction of Li-20 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of HB(pin) and EtB(pin) gave a 63:37 ratio of 21:27 showing that both boronic esters had similar reactivity towards Li-20 (see the ESI†). These experiments ruled out reversibility as a source of the erosion in ee.
:1 mixture of HB(pin) and EtB(pin) gave a 63:37 ratio of 21:27 showing that both boronic esters had similar reactivity towards Li-20 (see the ESI†). These experiments ruled out reversibility as a source of the erosion in ee.
Instead, we believe that the small erosion in ee arises from a strong preference of the boron–ate complex to adopt a conformation in which one of the oxygens of the pinacol ester is positioned anti-periplanar to the carbamate. Subsequent oxygen-assisted expulsion of the carbamate followed by hydride migration would then occur with overall retention of configuration.12 Initial computational studies support this hypothesis (Scheme 3C). The degree to which this minor pathway occurs is substrate dependent, but the dominant pathway remains the stereospecific 1,2-migration with inversion.
In conclusion we have reported the facile migration of hydride in lithiation–borylation reactions and we have applied it to the preparation of a variety of primary and secondary boronic esters. Using this protocol variously substituted boronic esters can now be obtained with very high levels of enantioselectivity and double bond geometry, where current methodology is limited.
We thank EPSRC and the European Research Council (FP7/2007-2013, ERC grant no. 246785) for support. R. R. thanks the Marie Curie Fellowship program (EC FP7, no. 329578).
Notes and references
- A. Pelter and K. Smith, “Oxidation of Carbon-Boron Bonds” Comprehensive Organic Synthesis, Elsevier, p. 593 Search PubMed.
- For Cu-based approaches, see: (a) H. Ito and K. Kubota, Org. Lett., 2012, 14, 890–893 CrossRef CAS PubMed; (b) C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef CAS PubMed . For Pd-based approaches, see: ; (c) G. Dutheuil, N. Selander, K. J. Szabò and V. K. Aggarwal, Synthesis, 2008, 2293–2297 CAS . For Ni-based approaches, see: ; (d) A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693–10697 CrossRef CAS PubMed; (e) J. Yi, J.-H. Liu, J. Liang, J.-J. Dai, C.-T. Yang, Y. Fu and L. Liu, Adv. Synth. Catal., 2012, 354, 1685–1691 CrossRef CAS . For a Mg-based approach, see: ; (f) C. Pintaric, S. Olivero, Y. Gimbert, P. Y. Chavant and E. Duñach, J. Am. Chem. Soc., 2010, 132, 11825–11827 CrossRef CAS PubMed.
- (a) J. L. Stymiest, G. Dutheuil, A. Mahmood and V. K. Aggarwal, Angew. Chem., Int. Ed., 2007, 46, 7491–7494 CrossRef CAS PubMed; (b) R. Larouche-Gauthier, C. J. Fletcher, I. Couto and V. K. Aggarwal, Chem. Commun., 2011, 47, 12592–12594 RSC; (c) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778–782 CrossRef CAS PubMed; (d) V. Bagutski, R. M. French and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 5142–5145 CrossRef CAS PubMed; (e) A. P. Pulis, D. J. Blair, E. Torres and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 16054–16057 CrossRef CAS PubMed.
- (a) D. Uguen, Bull. Soc. Chim. Fr. II, 1981, 99 CAS; (b) L. Liu, J. A. Henderson, A. Yamamoto, P. Brémond and Y. Kishi, Org. Lett., 2012, 14, 2262–2265 CrossRef CAS PubMed.
- R. L. Danheiser and A. C. Savoca, J. Org. Chem., 1985, 50, 2401–2403 CrossRef CAS.
- (a) A. Carstens and D. Hoppe, Tetrahedron, 1994, 50, 6097–6108 CrossRef CAS; (b) D. Hoppe, A. Carstens and T. Kramer, Angew. Chem., Int. Ed., 1990, 29, 1424–1425 CrossRef.
- Yield obtained after oxidation of 4′ to the alcohol.
- For selected examples, see: (a) D. Noh, H. Chea, J. Ju and J. Yun, Angew. Chem., Int. Ed., 2009, 48, 6062–6064 CrossRef CAS PubMed; (b) D. Noh, S. K. Yoon, J. Won, J. Y. Lee and J. Yun, Chem.–Asian J., 2011, 6, 1967–1969 CrossRef CAS PubMed; (c) C. M. Crudden, Y. B. Hleba and A. C. Chen, J. Am. Chem. Soc., 2004, 126, 9200–9201 CrossRef CAS PubMed; (d) S. A. Moteki, D. Wu, K. L. Chandra, D. S. Reddy and J. M. Takacs, Org. Lett., 2006, 8, 3097–3100 CrossRef CAS PubMed . Reviews: ; (e) I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS; (f) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS.
- For representative reviews, see: (a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207–2293 CrossRef CAS; (b) I. Marek and G. Sklute, Chem. Commun., 2007, 1683–1691 RSC; (c) T. R. Ramadhar and R. A. Batey, Synthesis, 2011, 1321–1346 CAS . For their use in Suzuki cross-couplings, see: ; (d) L. Chausset-Boissarie, K. Ghozati, E. LaBine, J. L. Y. Chen, V. K. Aggarwal and C. M. Crudden, Chem.–Eur. J., 2013, 19, 17698–17701 CrossRef CAS PubMed.
- (a) Pd-catalyzed borylation of geraniol and the mono-carbamate analog of 11 with B2(pin)2 was also possible but 10 and 13 were obtained as inseparable mixtures of isomers [see ESI†]; (b) For an electrochemical approach to 10, see: J. Godeau, C. Pintaric, S. Olivero and E. Duñach, Electrochim. Acta, 2009, 54, 5116–5119 CrossRef CAS PubMed.
- C. Pintaric, C. Laza, S. Olivero and E. Duñach, Tetrahedron Lett., 2004, 45, 8031–8033 CrossRef CAS PubMed.
- M. E. Jung, A. van den Heuvel, A. G. Leach and K. N. Houk, Org. Lett., 2003, 5, 3375–3378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, 1H and 13C NMR spectra of all new compounds, chiral HPLC and SFC traces. See DOI: 10.1039/c4cc00993b |
| This journal is © The Royal Society of Chemistry 2014 |