 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Branched tetrasilane substituted phosphines – synthesis and characterisation of PhSi(SiiPr2)3P and {PhSi(SiMe2)3}2P14†
Michael
Feierabend
and
Carsten
von Hänisch
*
Fachbereich Chemie and Wissenschaftliches Zentrum für Materialwissenschaften (WZMW) of the Philipps-Universität Marburg, Hans-Meerwein-Straße, D-35032 Marburg, Germany. E-mail: haenisch@chemie.uni-marburg.de
First published on 10th March 2014
Abstract
The branched trichlorotetrasilane PhSi(SiMe2Cl)3 reacts with P7(SiMe3)3 leading to formation of a new oligophosphane {PhSi(SiMe2)3}2P14 (1), which consists of two PhSi(SiMe2)3 substituted P7 norbornane units. The phosphatetrasila[1.1.1]pentane derivative PhSi(SiiPr2)3P (3) was obtained from the reaction of PhSi(SiiPr2Cl)3 (2) with Li3P.
Silicon–phosphorus compounds are still of considerable interest due to their versatile molecular structures and bonding properties. Moreover, they are useful synthons for the formation of several other phosphorus as well as silicon compounds.1 Silyl groups are also able to stabilise highly reactive phosphorus species such as P73−, the silylderivatives of which (e.g. P7(SiMe3)3 or P7{Si(SiMe3)3}3) are much less reactive during oxidation or hydrolysis.2 As shown in recent studies, bridging silyl substituents such as bidentate silyl or siloxane groups have a major impact on the structures and properties of oligophosphanes.3 Thus, we decided to investigate the extent to which tripodal silyl groups are able to stabilise new Pn-compounds.4
Herein, we report on the application of the compound PhSi(SiMe2Cl)3 as a substituent for P73−. Moreover, we present the synthesis of the sterically demanding branched trichlorotetrasilane PhSi(SiiPr2Cl)3 as well as its reaction with Li3P.
After work-up of the reaction of P7(SiMe3)3 with PhSi(SiMe2Cl)3 in DME, {PhSi(SiMe2)3}2P14 (1) was obtained as yellow crystals in 53% yield. The latter was formed by dimerisation of the P7 cage through substitution of the silyl groups and represents a silyl derivative of the so far unknown Zintl anion P146−. The P14 framework of compound 1 consists of two P7 norbornane cages. In both P7 norbornane subunits, one P5 ring is substituted by the branched silyl frame in 1, 2 and 4 positions (Fig. 1). These two P5Si4 fragments show the same structure as the P9 cage in Hittorf's phosphorus, and they are connected through a central P4 ring. Until now, no comparable P14 substructure has been observed in molecular compounds. The known Zintl ion P144− consists of two P7 nortricyclan cages connected by one P–P bond.5 P14iPr4, consists – like compound 1 – of two P7 norbornane units, which are connected by three P–P bonds via P5 rings in 1, 2, and 4 positions (Scheme 1).6 The P14 unit in 1, however, represents a part within the phosphorus strands in [(CuI)8P12].7
 | ||
| Fig. 1 Molecular structure of 1; thermal ellipsoids represent a 50% probability level, hydrogen atoms are not shown, selected bond lengths (pm) and angles (°): Si1–Si2 233.5(4), Si1–Si3 232.9(5), Si1–Si4 234.24(4), Si2–P1 229.3(4), Si3–P2 229.9(4), Si4–P4 228.3(4), P1–P2 225.4(4), P1–P5 221.0(4), P2–P3 221.65(4), P3–P4 217.6(4), P4–P5 219.9(4), P5–P6 218.6(4), P6–P7 229.4(4), P6–P7′ 224.0(4); Si2–Si1–Si3 98.88(16), Si2–Si1–Si4 107.90(14), Si3–Si1–Si4 106.70(15), P1–Si2–Si1 102.70(15), P2–Si3–Si1 102.57(15), P4–Si4–Si1 110.70(16). | ||
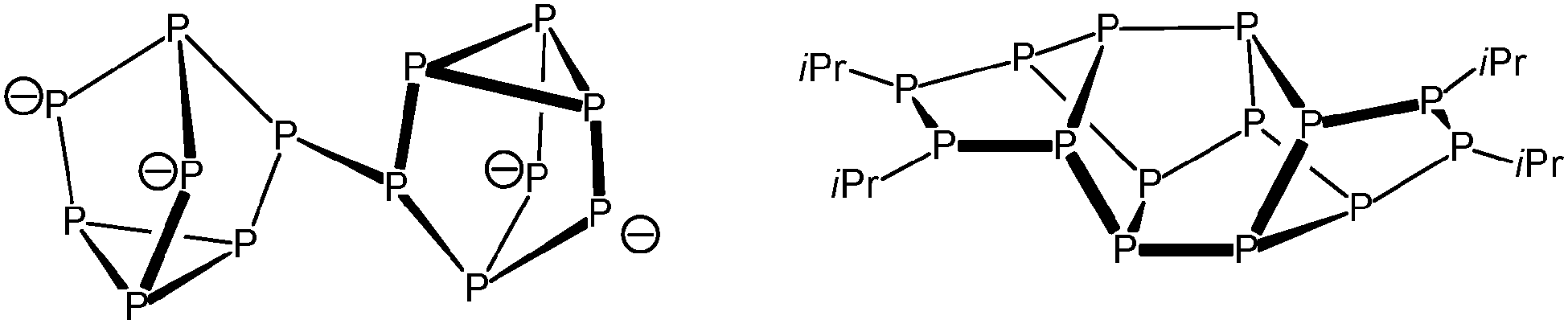 | ||
| Scheme 1 Structurally characterised ionic/molecular compounds with P14 units. | ||
The 31P{1H} NMR spectrum of 1 shows four signals at 50.9, 9.1, 2.4 and −42.6 ppm corresponding to the four different phosphorus positions within the molecule. Unfortunately, these signals are broad, independent of the temperature and solvent. So, the fine structure can be resolved only for the signal at 50.9 ppm, which shows a triplet structure and represents the symmetrically surrounded phosphorus atoms P4 and P4′.
Some years ago, several groups reported on their attempts to synthesise a 1-phospha-2,3,4,5-tetrasila[1.1.1]pentane.8 These experiments, however, always led to the formation of other bicyclic silylphosphines as shown in Scheme 2.
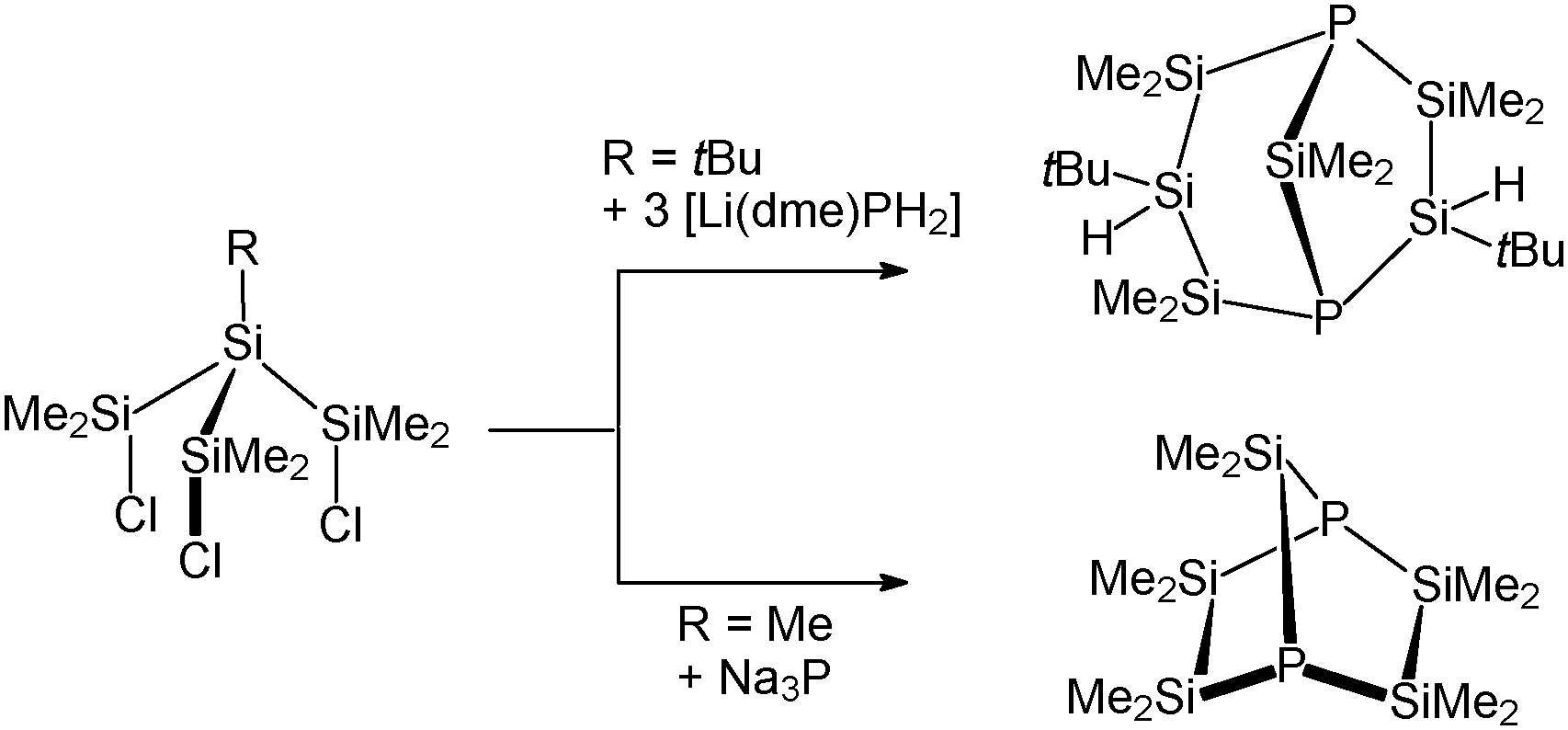 | ||
| Scheme 2 Reported attempts to synthesise a 1-phospha-2,3,4,5-tetrasila[1.1.1]pentane and the obtained products.8 | ||
In order to avoid decomposition of the Si4 substructure, we synthesised the sterically more demanding substituted branched trichlorotetrasilane PhSi(SiiPr2Cl)3 (2) as starting compound. 2 was obtained in a two-step synthesis by a reductive coupling of PhSiCl3 and iPr2SiHCl with elementary lithium and subsequent chlorination with trichloroisocyanuric acid (TCCA) (Scheme 3).9
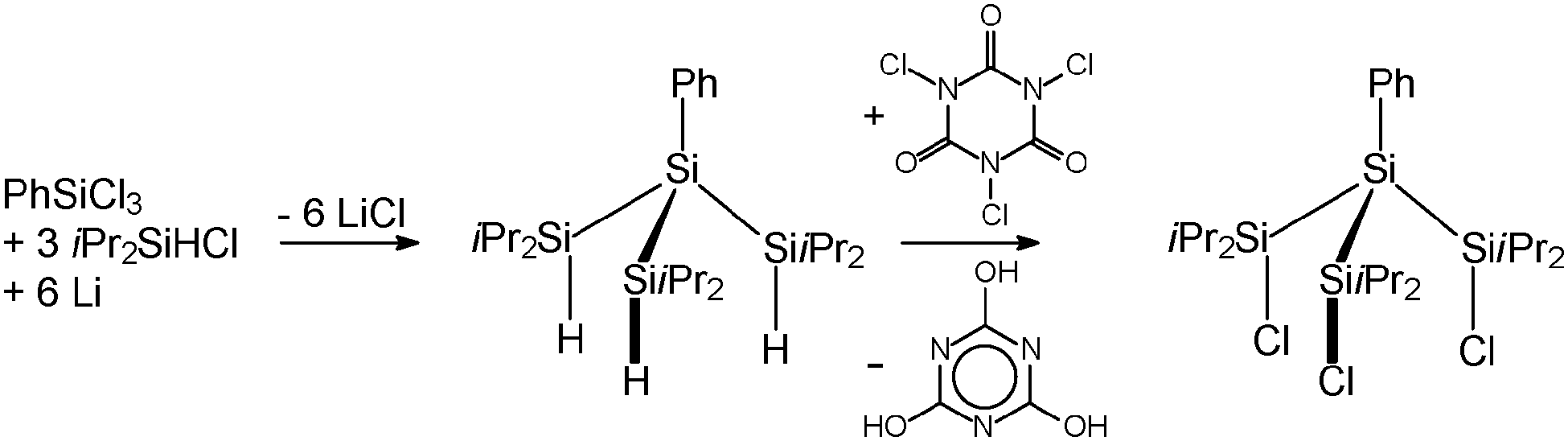 | ||
| Scheme 3 Synthesis pathway of compound 2. | ||
After work-up, the reaction of 2 with Li3P in THF provides the target molecule as a white crystalline solid in small but reproducible yields (Fig. 2). Other products could not be characterised to date. The crystal structure analysis shows the strained molecular structure with endocyclic bond angles all below 90°: Si–Si–Si: 81.6–82.5°, Si–Si–P: 80.2–80.6° and Si–P–Si: 83.3–83.6°. The bond lengths in compound 3, however, are only slightly longer than the usual single bond between these elements (Fig. 2). Worth mentioning are the short distances between the atoms Si2, Si3 and Si4 (307.7(9)–309.0(12) pm) and between Si1 and P (301.2(10) pm), which are significantly shorter than the sum of the van der Waals radii.
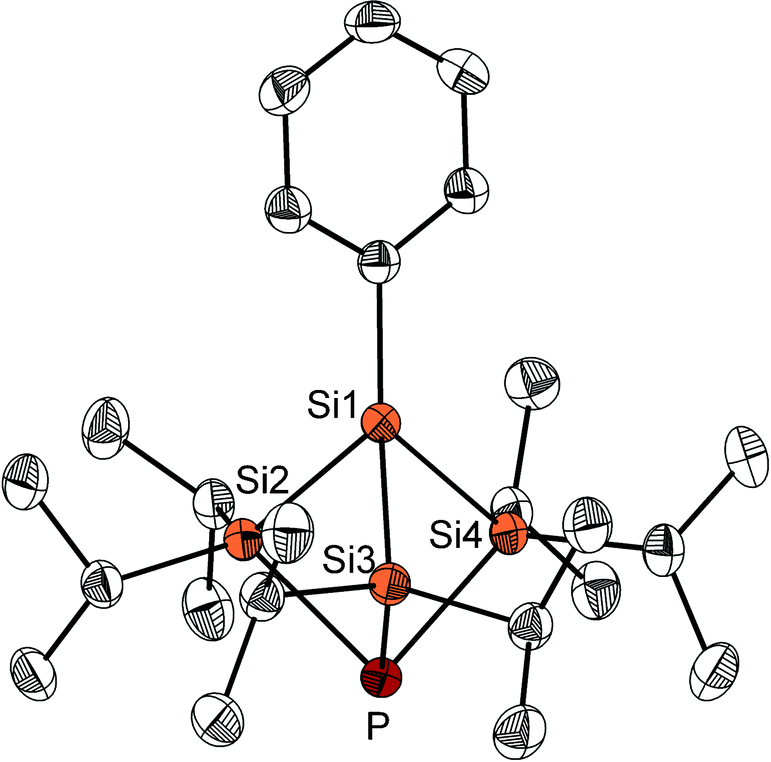 | ||
| Fig. 2 Molecular structure of 3; thermal ellipsoids represent a 50% probability level, hydrogen atoms are not shown, selected bond lengths (pm) and angles (°): Si1–Si2 235.46(8), Si1–Si3 235.01(14), Si1–Si4 233.61(12), Si2–P 230.05(14), Si3–P 232.56(9), Si4–P 231.60(11); Si–Si1–Si 81.61(4)–82.52(4), Si1–Si–P 80.19(4)–80.68(4), Si–P–Si 83.30(4)–83.60(4). | ||
The 31P NMR spectrum of compound 3 shows a typical upfield shift for silylphosphines at −241.7 ppm. In the 29Si{1H} NMR, two doublet signals can be observed at 15.4 and −58.2 ppm. The signal at 15.4 ppm corresponds to the SiiPr2 groups and shows a remarkably large 1JSi,P coupling constant of 53.2 Hz. This large coupling constant suggests a high s-orbital contribution to the Si–P σ-bonds and matches the results of DFT calculations (see ESI†), which show a high p-orbital character of the phosphorus lone pair and a symmetric bonding orbital with significant contributions of Si1–Si4 and P atomic s-orbitals. For comparison, the similar substituted but planar compound P(SiiPr3)3 shows a 1JSi,P coupling of only 9 Hz.10
All working procedures were conducted under rigorous exclusion of oxygen and moisture using a Schlenk line and an argon atmosphere. Solvents were dried and freshly distilled before use. NMR spectra were recorded using a BRUKER AVANCE 300 or a BRUKER DRX 400. The structural analysis was carried out using appropriate single crystals on an automatic diffractometer (STOE-IPDS-2T, STOE-IPDS-2 or BRUKER D8-Quest). The structures were solved and refined using SHELXS-9711 and SHELXL-2013.12 The presentation of crystal structures was effected by DIAMOND3.2. IR vibrational spectra were recorded using the BRUKER ALPHA ATR-FT-IR. The starting materials PhSi(SiMe2Cl)3,4 P7(SiMe3)3,2 and Li3P13 were prepared by reported methods.
Crystal data for 1: C24H46Si8P14·1.5 C7H8, 100 K, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 931.0(2), b = 1656.6(4), c = 1880.4(4) pm, α = 88.880(19)°, β = 87.593(19)°, γ = 77.702(18)°, V = 2830.9(11) Å3, Z = 2, density = 1.327 g cm3, μ = 0.619 mm−1, F(000) = 1174, GOF = 0.742, theta range: 1.26–24.00°, 12
, a = 931.0(2), b = 1656.6(4), c = 1880.4(4) pm, α = 88.880(19)°, β = 87.593(19)°, γ = 77.702(18)°, V = 2830.9(11) Å3, Z = 2, density = 1.327 g cm3, μ = 0.619 mm−1, F(000) = 1174, GOF = 0.742, theta range: 1.26–24.00°, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 809 reflections collected, 8177 unique (Rint = 0.1002). R1 (wR2) = 0.0716 (0.1586) for 530 parameters and 3183 reflections with I > 2σ(I).
809 reflections collected, 8177 unique (Rint = 0.1002). R1 (wR2) = 0.0716 (0.1586) for 530 parameters and 3183 reflections with I > 2σ(I).
Crystal data for 3: C24H47Si4P1, 100 K, triclinic, P, a = 916.4(3), b = 1020.7(3), c = 1644.1(9) pm, α = 92.88(4)°, β = 94.84(4)°, γ = 110.34(2)°, V = 1431.7(10) Å3, Z = 2, density = 1.111 g cm3, μ = 0.273 mm−1, F(000) = 524, GOF = 1.036, theta range: 1.25–25.00°, 9353 reflections collected, 4731 unique (Rint = 0.0205). R1 (wR2) = 0.0247 (0.0657) for 262 parameters and 4266 reflections with I > 2σ(I).
1: PhSi(SiMe2Cl)3 (0.42 g, 1.1 mmol) in dme (20 mL) was added dropwise to a solution of P7(SiMe3)3 (0.48 g, 1.1 mmol) in dme (20 mL) at −45 °C. The reaction mixture was allowed to warm up within 4 h to ambient temperature, the solvent was removed under reduced pressure and the residue was dissolved in 5 mL toluene. Subsequently, after 2 days yellow crystals of [{PhSi(SiMe2)3}2P14]·1.5 tol were obtained at 20 °C in a yield of 53% (0.29 g). Elemental analysis (%): calc. for C24H46Si8P14: C 29.03, H 4.67, found: C 29.09, H 5.07.
1H-NMR (thf-d8): δ/ppm = 0.35 (m, CH3, 12H), 0.78 (m, CH3, 12H), 0.92 (m, CH3, 12H), 7.28 (m, C6H5, 6H), 7.39 (m, C6H5, 4H). 13C{1H}-NMR (thf-d8): δ/ppm = 2.96 (m, CH3), 3.68 (m, CH3), 128.24 (s, C6H5), 128.66 (s, C6H5), 129.11 (s, C6H5), 136.56 (s, C6H5). 29Si{1H}-NMR (thf-d8): δ/ppm = −4.3 (m, Si(CH3)2), −5.8 (m, Si(CH3)2), −77.5 (s, SiC6H5). 31P-NMR (thf-d8): δ/ppm = 51.0 (t, 1JPP = 328.1 Hz), 9.1 (m), 2.4 (m), −42.6 (m). MS(ESI+) m/z (%) calc.: 992.8153 [M+ + H], found: 992.8265 (45).
PhSi(SiiPr2H)3: A solution of PhSiCl3 (12.63 g, 0.06 mol) and iPr2HSiCl (44.96 g, 0.29 mol) in thf (250 mL) was slowly dropped at ambient temperature to a vigorously stirred suspension of Li cuts (3.32 g, 0.48 mol) in thf (250 mL) over 3 h, and stirring was continued for 24 h. The suspension was poured into a mixture of ice and HCl (200 mL, 1 M) and n-pentane (100 mL) was added. The organic phase was separated, the aqueous layer was extracted twice with n-pentane (100 mL) and the combined organic phases were dried with MgSO4 and filtered. After evaporation of the solvents, the raw product was distilled fractionally under vacuum to afford PhSi(SiiPr2H)3 (10−2 mbar, 120 °C) in a yield of 62.8% (16.9 g).
1H-NMR (C6D6): δ/ppm = 1.18 (d, 3JHH = 7.4 Hz, CH(CH3)2, 18H), 1.20 (d, 3JHH = 7.4 Hz, CH(CH3)2, 18H), 1.42 (d, sep, 3JHH = 7.4 Hz, and 2.7 Hz, CH(CH3)2, 6H), 4.18 (t, 3JHH = 2.7 Hz, SiH, 3H), 7.11 (m, C6H5, 3H), 7.80 (m, C6H5, 2H). 13C{1H}-NMR (C6D6): δ/ppm = 13.96 (s, CH(CH3)2), 21.40 (s, CH(CH3)2), 22.98, (s, CH(CH3)2), 128.51 (s, C6H5), 129.29 (s, C6H5), 135.52 (s, C6H5), 138.42 (s, C6H5). 29Si-NMR (C6D6): δ/ppm = −6.4 (d, m, 1JSiH = 172.3 Hz, SiCH(CH3)2), −81,2 (s, SiC6H5). MS(APCI+) m/z (%) calc.: 449.2906 [M+ − H], found: 449.2903 (15); IR [cm−1]: 463(w), 584(w), 650(m), 698(s), 744(vs), 877(m), 917(m), 1003(m), 1067(m), 1233(vw), 1363(w), 1383(w), 1427(w), 1460(m), 2073(m, Si–H), 2861(m), 2940(m).
2: A solution of PhSi(SiiPr2H)3 (16.9 g, 0.038 mol) in thf (250 mL) was cooled down to −20 °C. Afterwards, TCCA (10 g, 0.043 mol) was slowly added and the solution was warmed up to ambient temperature. The solvent was removed under reduced pressure and the residue was dissolved in n-pentane (60 mL). The insoluble white precipitate was filtrated and washed two times with n-pentane (25 mL). The volume of the combined filtrates was reduced to 50 mL. After 4 days at −8 °C, colourless crystals of PhSi(SiiPr2Cl)3 were obtained, the yield being 71.0% (14.9 g). Elemental analysis (%): calc. for C24H47Si4Cl3: C 52.00, H 8.55, found C 52.01, H 8.97.
1H-NMR (C6D6): δ/ppm = 1.13 (d, 3JHH = 7.4 Hz, CH(CH3)2, 18H), 1.24 (d, 3JHH = 7.4 Hz, CH(CH3)2, 18H), 1.70 (sep, 3JHH = 7.4 Hz, CH(CH3)2, 6H), 7.10 (m, C6H5, 3H), 8.14 (m, C6H5, 2H). 13C{1H}-NMR (C6D6): δ/ppm = 19.08 (s, CH(CH3)2), 19.36 (s, CH(CH3)2), 20.14 (s, CH(CH3)2), 129.06 (s, C6H5), 129.32 (s, C6H5), 132.31 (s, C6H5), 138.85 (s, C6H5). 29Si{1H}-NMR (C6D6): δ/ppm = 33.4 (s, SiCH(CH3)2), −70.6 (s, SiC6H5). MS(APCI+) m/z (%) calc.: 517.2126 [M+ − Cl], found: 517.2126 [M+ − Cl] (100); IR [cm−1]: 424(w), 463(s), 553(vs), 599(s), 621(m), 655(m), 700(m), 735(m), 760(w), 875(s), 991(m), 1233(vw), 1365(w), 1427(w), 1461(m), 2867(m), 2948(m).
3: PhSi(SiiPr2Cl)3 (1.34 g, 2.42 mmol) in thf (10 mL) was added to a suspension of Li3P (0.13 g, 2.50 mmol) in thf (100 mL) at −30 °C. Subsequently, the reaction mixture was first warmed-up to room temperature and stirred for 16 hours and then heated to reflux for 4 days. Thereafter, the solvent was removed under reduced pressure, and the residue was dissolved in n-pentane (25 mL). After filtration, the volume of the solution was reduced to 2 mL and cooled down to −30 °C. Compound 3 was obtained as colourless crystals within 4 days in a yield of 8.8% (0.10 g). Elemental analysis (%): calc. for C24H48Si4P: C 60.19, H 9.89, found: C 60.24, H 10.03.
1H-NMR (C6D6): δ/ppm = 1.36 (d, 3JHH = 7.5 Hz, CH(CH3)2, 18H), 1.38 (d, 3JHH = 7.5 Hz, CH(CH3)2, 18H), 1.71 (sep, 3JHH = 7.5 Hz, CH(CH3)2, 6H), 7.09 (m, C6H5, 3H), 7.70 (m, C6H5, 2H). 13C{1H}-NMR (C6D6): δ/ppm = 18.91 (d, 2JCP = 5.2 Hz, CH(CH3)2), 20.92 (d, 2JCP = 1.1 Hz, CH(CH3)2), 21.36 (d, 3JCP = 1.5 Hz, CH(CH3)2), 128.67 (s, C6H5), 129.22 (s, C6H5), 133.40 (s, C6H5), 138.25 (s, C6H5). 29Si{1H}-NMR (C6D6): δ/ppm = 15.4 (d, 1JSiP = 53.2 Hz, Si3P), −58.2 (d, 2JSiP = 8.5 Hz, SiC6H5). 31P-NMR(C6D6): δ/ppm = −241.7 (s). MS(APCI−) m/z (%) calc.: 479.2565 [M + H], found: 479.2560 [M + H] (30).
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG). The authors gratefully acknowledge financial support from Evonik Industries AG, Creavis. The authors thank Mr Günther Thiele for his help with the cover picture and the DFT calculations.
Notes and references
- (a) T. Arnold, H. Braunschweig, J. O. C. Jimenez-Halla, K. Radacki and S. S. Sen, Chem.–Eur. J., 2013, 19, 9114–9117 CAS; (b) R. Rodriguez, T. Troadec, T. Kato, N. Saffon-Merceron, J.-M. Sotiropoulos and A. Baceiredo, Angew. Chem., Int. Ed., 2012, 51, 7158–7161 CrossRef CAS PubMed; (c) S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786–11789 CAS; (d) R. Rodriguez, D. Gau, Y. Contie, T. Kato, N. Saffon-Merceron and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 11492–11495 CAS; (e) G. Fritz and P. Scheer, Chem. Rev., 2000, 100, 3341–3401 CAS.
- (a) W. Hönle and G. H. von Schnering, Z. Anorg. Allg. Chem., 1978, 440, 171–182 Search PubMed; (b) G. Fritz and W. Hölderich, Naturwissenschaften, 1975, 62, 573–575 CAS; (c) H. Siegl, W. Krumlacher and K. Hassler, Silicon Chem., 1999, 139–145 CAS.
- (a) A. Kracke and C. von Hänisch, Eur. J. Inorg. Chem., 2011, 3374–3380 CAS; (b) P. Kopecky, C. von Hänisch, F. Weigend and A. Kracke, Eur. J. Inorg. Chem., 2010, 258–265 CAS; (c) S. Traut, C. von Hänisch and H.-J. Kathagen, Eur. J. Inorg. Chem., 2009, 777–783 CrossRef CAS; (d) C. von Hänisch, S. Traut and S. Stahl, Z. Anorg. Allg. Chem., 2007, 633, 2199–2204 CrossRef; (e) C. von Hänisch and E. Matern, Z. Anorg. Allg. Chem., 2005, 631, 1655–1659 CrossRef.
- C. von Hänisch and M. Feierabend, Z. Anorg. Allg. Chem., 2013, 639, 788–793 CrossRef.
- (a) V. Miluykov, A. Kataev, O. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Z. Anorg. Allg. Chem., 2006, 632, 1728–1732 CrossRef CAS; (b) N. Korber, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 124, 339–346 CrossRef.
- M. Baudler, H. Jachow, B. Lieser, K.-F. Tebbe and M. Fehér, Angew. Chem., 1989, 101, 1245–1247 CrossRef CAS.
- M. H. Möller and W. Jeitschko, J. Solid State Chem., 1986, 65, 178–189 CrossRef.
- (a) M. Driess, M. Reisgys and H. Pritzkow, Z. Anorg. Allg. Chem., 1998, 624, 1886–1890 CrossRef CAS; (b) G. M. Kollegger, U. Katzenbeisser, K. Hassler, C. Krüger, D. Brauer and R. Gielen, J. Organomet. Chem., 1997, 543, 103–110 CrossRef CAS.
- S. Varaprath and D. H. Stutts, J. Organomet. Chem., 2007, 692, 1892–1897 CrossRef CAS PubMed.
- (a) C. von Hänisch, Z. Anorg. Allg. Chem., 2001, 627, 1414–1416 CrossRef; (b) M. Driess and C. Monsé, Z. Anorg. Allg. Chem., 2000, 626, 2264–2268 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL, University of Göttingen, Germany, 2013 Search PubMed.
- G. Fritz and R. Blastoch, Z. Anorg. Allg. Chem., 1986, 535, 63–85 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: DFT calculations, crystallographic data of compound 2, proposed mechanism for the formation of compound 1 and 31PNMR spectrum of compound 1. CCDC 978105 (1), 978107 (2) and 978106 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc00165f |
| This journal is © The Royal Society of Chemistry 2014 |