 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-molecule magnet engineering: building-block approaches†
Kasper S.
Pedersen
*abc,
Jesper
Bendix
*c and
Rodolphe
Clérac
*ab
aCNRS, CRPP, UPR 8641, F-33600 Pessac, France. E-mail: clerac@crpp-bordeaux.cnrs.fr; Fax: +33 5 56 84 56 00; Tel: +33 5 56 84 56 50
bUniv. Bordeaux, CRPP, UPR 8641, F-33600 Pessac, France
cDepartment of Chemistry, University of Copenhagen, Denmark. E-mail: ksp@kiku.dk; bendix@kiku.dk
First published on 20th February 2014
Abstract
Tailoring the specific magnetic properties of any material relies on the topological control of the constituent metal ion building blocks. Although this general approach does not seem to be easily applied to traditional inorganic bulk magnets, coordination chemistry offers a unique tool to delicately tune, for instance, the properties of molecules that behave as “magnets”, the so-called single-molecule magnets (SMMs). Although many interesting SMMs have been prepared by a more or less serendipitous approach, the assembly of predesigned, isolatable molecular entities into higher nuclearity complexes constitutes an elegant and fascinating strategy. This Feature article focuses on the use of building blocks or modules (both terms being used indiscriminately) to direct the structure, and therefore also the magnetic properties, of metal ion complexes exhibiting SMM behaviour.
 Kasper S. Pedersen | Kasper S. Pedersen (b. in 1986, Allerød, Denmark) completed his studies in Inorganic Chemistry under the supervision of Prof. J. Bendix at the University of Copenhagen. From early in his studies he worked with Dr P. L. W. Tregenna-Piggott (✠) in Switzerland on spectroscopic studies of SMMs by inelastic neutron scattering. Currently, he is pursuing a PhD in the groups of Prof. J. Bendix and Dr R. Clérac. His current research interests include synthesis and investigations of molecule-based magnetic materials based on heavier transition elements and f-elements as well as spectroscopic investigations. |
 Jesper Bendix | Jesper Bendix (b. in 1965, Copenhagen, Denmark) received his PhD at the University of Copenhagen under the supervision of Prof. C. E. Schäffer in 1998. Following post-doctoral stays in Mülheim, Germany with Prof. K. Wieghardt, at Caltech, USA, with Prof. H. B. Gray, at the University of Utah with Prof. J. S. Miller, and in Berne, Switzerland, with Dr P. L. W. Tregenna-Piggott (✠), he returned to Copenhagen where he is currently heading the section for inorganic chemistry. His current research focuses on synthesis as well as electronic and structural studies of molecule-based magnetic materials and on the electronic structure of high-valent metal centres. |
 Rodolphe Clérac | Rodolphe Clérac (b. in 1971, Versailles, France) received his PhD at the University of Bordeaux 1 under the supervision of Prof. C. Coulon in 1997. After a short post-doctoral stay in the group of Prof. O. Kahn (ICMCB, Bordeaux), he joined Prof. K. R. Dunbar's group at Michigan State University (East Lansing, Michigan, USA) in 1998 and in 1999, he moved with Dunbar's group to Texas A&M University (College Station, Texas, USA) where he collaborated with Prof. F. A. Cotton. In 2000, he established his research group (Molecular Materials & Magnetism) at the Centre de Recherche Paul Pascal (CNRS) interested in the synthesis and physical properties of molecular materials. |
1. Introduction
Single-molecule magnets (SMMs), i.e. paramagnetic molecules exhibiting a magnet behaviour,‡ of intrinsic molecular origin in the absence of a magnetic order, have received considerable attention in chemistry, physics and materials science since their discovery in the early 1990s.1–5 The main interest arose from the possible applications of SMMs in data storage, quantum computing6 and molecule-based spintronics devices.7–9 Even if SMMs have not yet been employed for practical applications, their underlying physics and chemistry have a much broader perspective. For instance, these “nanomagnets” and related molecular magnetic complexes serve as simple models for understanding more complex magnetic materials (extended 1D, 2D or 3D networks, large aggregates,…). Moreover, significant advancements in nanostructuring and deposition of single molecules allowed the studies on individual SMMs and to probe their intrinsic magnetic properties outside the crystal lattice.10,11 These detailed studies of the SMM properties include the seminal observation of slow relaxation12 and quantum tunnelling13 of magnetization of magnetically isolated molecules covalently grafted to surfaces. Furthermore, studies of SMMs in solution,14,15 as well as nanostructured on surfaces,10,11,16,17 in junctions,18 films,19 porous materials20 or in multi-dimensional coordination networks have been undertaken.21–25Key to the possible applications of molecular magnetic systems is a thorough understanding of the design pathways towards specific structural motifs and the understanding of the related magnetic properties of the constituent molecular entities. SMMs can be roughly divided into two classes: mononuclear and polynuclear complexes. Mononuclear SMMs have only been reported in recent years with the first example being the [Ln(pc)2]− (Ln = DyIII, TbIII, H2pc = phthalocyanine) “double decker” complexes.26 After this ground-breaking discovery, a multitude of mononuclear lanthanide,27–38 and more recently, several 3d metal ion complexes behaving as SMMs have been reported.39–45,46–48 Additionally, SMM behaviour in photo-excited spin-crossover complexes has very recently also been reported.49,50 Common to the majority of these systems, the slow-relaxation of magnetization arises due to a strong uniaxial magnetic anisotropy of the paramagnetic metal ion. The second class of SMMs encompasses polynuclear, exchange coupled complexes in which the constituent metal ions may be transition metal (nd, n = 3 to 5) ions, lanthanides/actinides (nf, n = 4, 5), or both. The SMM signature was reported for the first time in a dodecanuclear {Mn12} complex that is the archetypal example of an exchange-coupled polynuclear SMM.1,5,51,52 This family of SMMs can be further sub-divided into two classes based on the employed synthetic approach. The synthesis of the first sub-class proceeds via a concerted association of metal ions through bridging ligands and with capping ligands to prevent polymerisation. The bridging ligands, most commonly oxide, hydroxide, alkoxides or phenolates obtained by deprotonation in the reaction medium, give pathways for magnetic exchange interactions between the constituent metal ions in the final polynuclear complex. The vast majority of SMMs have been obtained by this more or less serendipitous method53 and pivotal studies, which have paved the way for the current understanding of SMM physics (e.g. quantum tunneling of magnetization, QTM,54 and quantum coherence55), were discovered in such systems. Alternatively, the synthetic approach towards the second sub-class of polynuclear SMMs makes use of predesigned molecular building-blocks, which are able to associate directly in solution. In that respect, two kinds of precursors exist, namely M–L ligand donors and M′ ligand acceptors, which react and form M–L–M′ motifs.
The remaining and non-trivial question is now: how to define a building-block and to understand how the structure of the building-blocks influences the final polynuclear complex topology and eventually the magnetic properties? If these questions can be satisfactorily answered, SMMs can be tailored to specific applications by chemical design. In this Feature article, we review the recent efforts to design SMMs using building-block approaches. Instead of a comprehensive review of the vast literature, we have been selective and discuss several explanatory examples of different uses of building-blocks with various bridging ligands.
In most of the cases, the observation of an SMM behaviour is attributed to the presence of a large spin ground state (ST) and a strong easy-axis magnetic anisotropy.56 The large spin ground state is secured by the magnetic superexchange mechanism, which couples constituent spin centres (Si), more or less strongly as described by the phenomenological Heisenberg–Dirac–van Vleck (HDvV) spin-Hamiltonian:
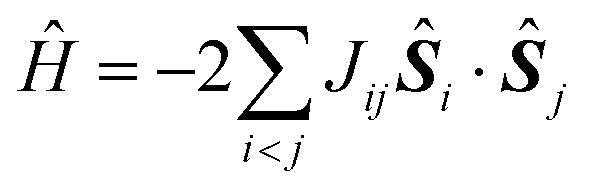 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 62 are the characteristic parameters commonly reported for an SMM. Most of the time, an experimental “effective” barrier (Δeff) smaller than the expected one (Δ, on the basis of ST and D) is obtained due to quantum tunnelling of magnetization (QTM) through the potential barrier via excited MS states.2 Indeed this is the case of the above example (Fig. 1) for which the observed energy barrier (Δeff) extracted from ac susceptibility measurements is only 16 K, which is much lower than the calculated value (25.5 K). However, application of a small dc field (800 Oe) puts the ±MS levels out of resonance and thereby increases Δeff to 23 K. The QTM is governed by non-diagonal terms entering into the Hamiltonian, giving rise to a mixing of MS states. In the vast majority of the characterized SMMs, the symmetry is lower than axial and the anisotropy part of the spin Hamiltonian to second order reads Ĥ = D(ŜZ2 − ⅓ST(ST + 1)) + E(ŜX2 − ŜY2) where |E| ≤ ⅓|D|. The E term has the effect of mixing the MS states differing by ΔMS = ±2. In some cases, this anisotropy description might not be sufficient to explain the relaxation and thus higher order terms, despite their small parameter values, have to be taken into account.2
62 are the characteristic parameters commonly reported for an SMM. Most of the time, an experimental “effective” barrier (Δeff) smaller than the expected one (Δ, on the basis of ST and D) is obtained due to quantum tunnelling of magnetization (QTM) through the potential barrier via excited MS states.2 Indeed this is the case of the above example (Fig. 1) for which the observed energy barrier (Δeff) extracted from ac susceptibility measurements is only 16 K, which is much lower than the calculated value (25.5 K). However, application of a small dc field (800 Oe) puts the ±MS levels out of resonance and thereby increases Δeff to 23 K. The QTM is governed by non-diagonal terms entering into the Hamiltonian, giving rise to a mixing of MS states. In the vast majority of the characterized SMMs, the symmetry is lower than axial and the anisotropy part of the spin Hamiltonian to second order reads Ĥ = D(ŜZ2 − ⅓ST(ST + 1)) + E(ŜX2 − ŜY2) where |E| ≤ ⅓|D|. The E term has the effect of mixing the MS states differing by ΔMS = ±2. In some cases, this anisotropy description might not be sufficient to explain the relaxation and thus higher order terms, despite their small parameter values, have to be taken into account.2
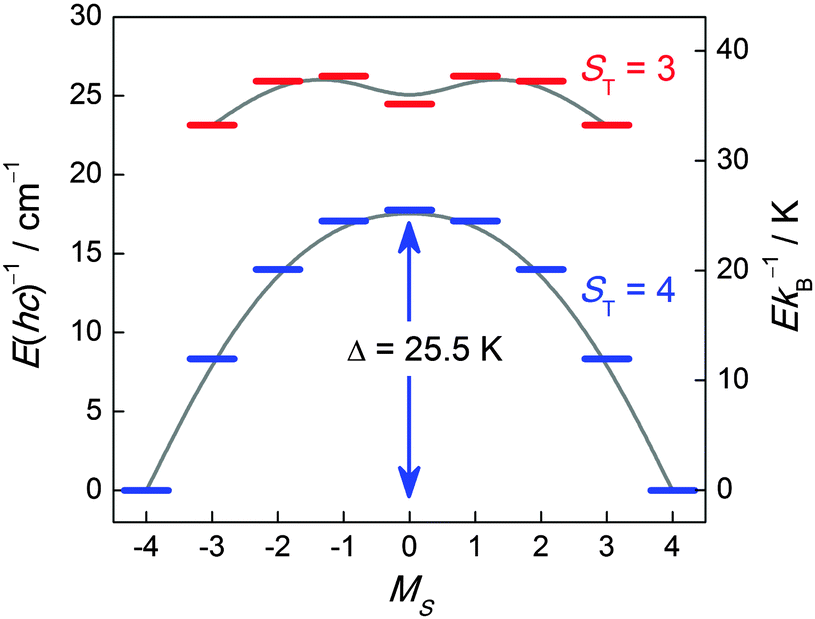 | ||
| Fig. 1 Energy level diagram of the two lowest spin-multiplets of an ST = 4 SMM ([MnIII2(saltmen)2(ReO4)2]) from ref. 61 (saltmen = N,N′-(1,1,2,2-tetramethylethylene)bis(salicylideneiminate)). The energy level diagram was calculated with JMn–Mn/kB = +2.7 K and D/kB = −4.0 K. Only the two energetically lowest spin manifolds are shown and solid lines are a guide for the eye. | ||
For the reasons stated above, the maximization of both D and ST seems crucial for the successful preparation of SMMs. Synthetically, it has been proven very difficult to obtain a large ST ground state by ferromagnetic interactions whilst simultaneously perfectly aligning anisotropy axes of each magnetic site. The largest SMM reported to date is an aesthetic {MnIII84} nanoscopic wheel which, despite its high nuclearity, exhibits only a relatively small spin ground state of ∼6 and a modest energy barrier of 18 K.63 The largest ST is found in a ferromagnetically coupled mixed-valence {MnII7MnIII12} complex exhibiting a record ST = 83/2 ground state but no SMM properties are observed due to an almost perfect compensation of the MnIII local anisotropy tensors.64,65 One of the successes in the realm of polynuclear SMMs has been a family of {Mn6} complexes, some of which exhibit an energy barrier higher (Δeff/kB = 86.4 K for [MnIII6O2(Et-sao)6(O2CPh(Me)2)2(EtOH)6], Et-saoH2 = 2-hydroxyphenylpropanone oxime) than the celebrated {Mn12} SMM (Δeff/kB = 61 K).1,66,67 Indeed targeting very large spin ground states in pursuit of effective SMMs is not necessarily the most fruitful approach since the overall anisotropy decreases as ST−2, leading to a SMM energy barrier almost independent of ST for sufficiently large ST values.68–70 Recently, M vs. H hysteresis loops at unprecedented temperatures (up to 14 K, 0.9 mT s−1) have been reported for dinuclear lanthanide complexes bridged by the exotic paramagnetic N23− radical,71,72 and a record anisotropy barrier for polynuclear SMMs of more than 600 K was observed for a {Dy4K2} hexanuclear complex.||73 Particularly in the latter case, these promising results rely rather on the strong single-ion magnetic anisotropy of the lanthanide ions than on the spin ground state of the molecule. However, the use of spin architectures employing multiple spin centres remains a viable route to prepare individual molecules with interesting magnetic properties, while simultaneously exploiting and optimizing the existing knowledge of preparative coordination chemistry. Importantly, this also constitutes the most realistic approach towards a good understanding of the interaction of magnetic molecules (irrespective of their nuclearity) with extended structures, e.g. surfaces.
2. Topological control
The rational synthesis of polynuclear metal complexes using a bottom-up approach based on building blocks or modules is by no means a new idea nor restricted to magnetic systems.74 However, due to the intimate relationship between structure and magnetic properties, this approach is particularly relevant for polynuclear magnetic systems. In order for the building blocks to be able to direct or template the desired structure of a polynuclear system some prerequisites need to be fulfilled to avoid the synthesis of non-expected products that might be thermodynamically favored. One of the most important aspects is to consider modules with a sufficient degree of robustness to maintain their structure-directing abilities under the assembly conditions. This somewhat vague property reflects the relative nature of the robustness concept in connection with synthesis, balancing ligand exchange kinetics between the different precursors and with the harshness of the conditions required for the assembly of the targeted polynuclear system.A second prerequisite for the building blocks to function as structure directing entities is a built-in preference for a specific coordination geometry at metal centers as well as at the bridging ligands. Octahedral coordination is predominant for the transition metal ions, and this is especially true for the kinetically robust systems. For a bridging ligand, the simplest conceivable geometry is to linearly connect two metal ions. This is true for the ubiquitous cyanide bridges, but also to quite some extent for fluoride, but not for oxide, when acting as bridging ligands. This tendency is supported by the histograms of Fig. 2 showing the crystallographically determined M–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C angle (where M is a transition metal ion) and, for comparison, the M–F/O–M′ angles in unsupported fluoride/oxide-bridged molecules and networks. The relative numbers are striking and reflect the extensive scientific work in cyanide chemistry. Consequently, M–NC–M′ motifs with robust octahedral metal ions, which are reminiscent of the Prussian blue compounds,76 are frequently used to design polynuclear complexes. Many molecular species obtained from building blocks of different denticities can be conceptually considered as fragments of a three-dimensional Prussian blue structure.77
C angle (where M is a transition metal ion) and, for comparison, the M–F/O–M′ angles in unsupported fluoride/oxide-bridged molecules and networks. The relative numbers are striking and reflect the extensive scientific work in cyanide chemistry. Consequently, M–NC–M′ motifs with robust octahedral metal ions, which are reminiscent of the Prussian blue compounds,76 are frequently used to design polynuclear complexes. Many molecular species obtained from building blocks of different denticities can be conceptually considered as fragments of a three-dimensional Prussian blue structure.77
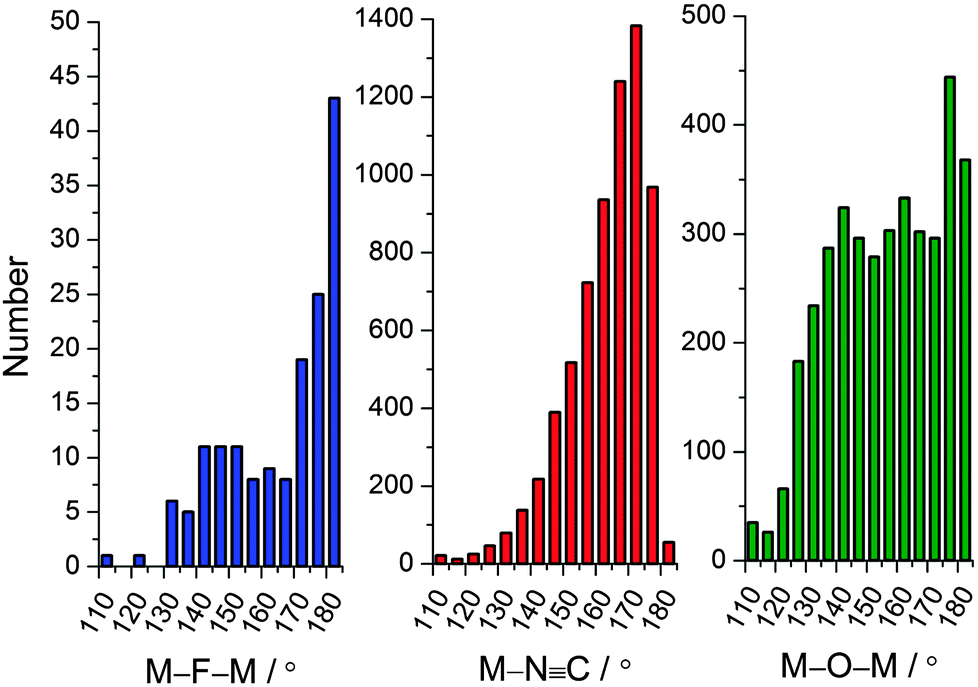 | ||
| Fig. 2 Histograms showing the numbers of the structurally characterized (Cambridge Structural Database)75 unsupported M–F–M′ (left), M–NC (middle) and unsupported M–O–M′ (right) bridging angles. | ||
The robustness of a given building block can derive from either the metal center (e.g. the most robust ones being d3 and diamagnetic low-spin d6 metal ions) or from the use of polydentate, and possibly, rigid ligands. Due to the limited choice of d3 or d6 systems, the use of polydentate ligands is the most efficient approach to enforce robustness and additionally to allow further geometrical preferences based on ligand design.
It should be mentioned that in this Feature article, the definition of the building blocks will be restricted to a molecular entity encompassing at least one metal ion. However, in a broader perspective, it is also useful to note that a less intuitive definition of the building block concept considers a metal-free bridging ligand as a building block directing the geometry of the whole system by its robust structure. This definition significantly widens the modular description but allows for a unified view encompassing common polynuclear topologies directed by the ligand structure. This last aspect is well illustrated by the large number of ring structures obtained using bridging carboxylate ligands.78 Indeed, the vast majority of the reported SMMs also falls in this extended definition and will not be included in this article; instead the reader is directed to excellent reviews by Aromi,3 Winpenny,79 Christou,80 Tang,81 and Powell82 for a detailed discussion of SMM topologies and their molecular control by ligand design.
In the context of molecule-based magnetism, the bridging ligand, in addition to guiding the structure, also needs to be compact enough to mediate efficient magnetic interactions. From the synthetic point of view, it is also preferable to choose bridging ligands with moderate basicity. Bridging ligands that are too reactive would limit the range of possible partners and conditions (e.g. solvents) since their structural integrity may be compromised, emphasizing further the relative nature of the robustness concept.
An essential aspect in engineering building blocks is to provide intrinsic magnetic characteristics essential to contribute to the final magnetic properties. To obtain SMMs, the building blocks usually contribute with Ising-like magnetic anisotropy or a large spin but other interesting additional properties like photomagnetism or luminescence can also be implemented. Building blocks may come as either homoleptic complexes, for which the cyanide (see Section 3) and oxalate (see Section 4) complexes are the most common, or as heteroleptic systems for which a large variety of ligand combination have been employed with a strong predominance of cyanide-based complexes. For the heteroleptic systems, the coordination sphere of the octahedral complexes, which can be cis-/trans- or fac-/mer-stereoisomers, directs towards different polynuclear structures.
The polymerization of building block units into multi-dimensional structures is a common synthetic problem for chemists who want to engineer molecular objects. In most of the cases, the successful synthesis of polynuclear complexes involves precursor units with strongly directional coordination abilities, which must be assisted by an appropriate choice of capping ligands. This choice is by no means trivial. First, the capping ligands often provide the solubility requirements for the subsequent assembly. Furthermore, the nature of the capping ligand may have dramatic structure-directing properties imposed by second coordination sphere interactions and packing effects. On the other hand, coordination polymers of SMMs are certainly another interesting research area that has led to magnetically interesting systems such as single-chain magnets (SCMs).21,83–86 Indeed, some SCM systems can be considered as one-dimensional polymers of SMM repeating units, allowing the modelling of the complex dynamics of Ising-type SCMs,22,84,87–89 on the basis of the known properties of the isolated SMMs.
3. Cyanide-based precursors
By far, the cyanide-based homo- or heteroleptic precursors are the most ubiquitous building blocks that have been used to design SMMs, high-spin or simple magnetic molecules (“0D”), extended magnetic networks such as chains (1D), sheets (2D) and three–dimensional (3D) lattices. Shatruk, Avendano and Dunbar reviewed comprehensively the chemistry of polynuclear cyanidometallates in 2009 and herein we will focus mainly on results obtained since.76 To quote these authors: “The shape adopted by the cyanide-bridged core in these clusters is dictated by the topology of the available coordination sites”. This sentence describes precisely what will be the theme of the following paragraphs.76The interest in cyanide-based SMMs is in direct line with the famous Prussian blue and its analogues, which have been intensively studied, in particular by the groups of Girolami,90 Verdaguer,91 and Miller.92 In these systems, an experimental and detailed theoretical understanding91,93 of the magnetic interaction through bridging cyanide ligands has been achieved in relation to the involved metal ions and structural/geometrical parameters. This knowledge of the exchange mechanisms was fundamental in order to engineer cyanide-encompassing molecular analogues with tailor-made magnetic properties. The popularity of the cyanide ion was also boosted by the availability and robustness of many cyanide complexes and the strong tendency of cyanide to bridge between transition metal centres. As discussed in the previous paragraph, cyanide often imposes an almost linear bridging mode that facilitates the design and prediction of specific topologies of the resulting polynuclear complexes. Moreover, the use of cyanide makes the heavier transition metals (4d, 5d) accessible to SMM materials. These elements exhibit some advantages over 3d metal ions as the presence of more diffused 4d/5d orbitals may give rise to stronger exchange interactions and significant magnetic anisotropy due to the strong spin–orbit coupling, as will be discussed in the next paragraphs.94–96 Remarkably, some of the Prussian blue analogues have shown interesting properties such as pronounced magnetic interactions leading to high ordering temperatures,90 charge-transfer and photomagnetic effects,97 all of which could possibly be, or have been, realized in molecule-based systems.
Homoleptic cyanidometallates
Homoleptic cyanide-complexes are known to possess coordination numbers ranging from 2 to 8, which allow them to bridge several metal ions.76,98,99 In SMM syntheses, only those having 6, 7 or 8 cyanide ligands have been employed with a majority of systems based on hexacyanide complexes.74,100 Paramagnetic hexacyanidometallates(III), [M(CN)6],3− are well-known for Ti to Fe,101–103 Mo,104 Ru,105 Os,106 and Ni (in solution).107 This series represents a unique opportunity to systematically investigate homologous SMMs incorporating transition metal ions with different d-orbital occupations; ideally with predictable structures, magnetic anisotropies and nature of the magnetic interaction.93,100,108 For example, if [Cr(CN)6]3− is coordinated to a NiII ion through a strictly linear cyanide bridge, the magnetic interaction is of ferromagnetic nature due to the orthogonality of the spin-bearing orbitals of CrIII [t2g3 (Oh)] and those of NiII [t2g6eg2 (Oh)]. However, such predictions do not necessary guarantee the successful synthesis of ferromagnetically coupled CrIII–CN–NiII complexes as small deviations from idealized geometries may give rise to, at first sight, counterintuitive results.One of the first examples of an SMM incorporating a homoleptic cyanidometallate was indeed a {CrIIINiII6} complex: [CrIII(CN)6][NiII(tetren)]6(ClO4)9 (1)109 (tetren = tetraethylenepent-tetraethylene-pentamine) having a close-to-octahedral {CrIII(μ-CN)6NiII6} central core. Ferromagnetic NiII–CrIII coupling interactions (JNi–Cr/kB = +12.1 K) give rise to an ST = 15/2 ground state. Even though NiII often possesses strong magnetic anisotropy, the proximity of the complex to octahedral symmetry is expected to significantly decrease the overall magnetic anisotropy and hence only a very small anisotropy barrier was found (Δeff/kB ≈ 6 K, τ0 = 1.1 × 10−11 s).100 The first established SMM incorporating a homoleptic cyanidometallate building block was a trigonal bipyramidal (TBP) complex, {[MnII(tmphen)2]3[MnIII(CN)6]2} (2, tmphen = 3,4,7,8-tetramethyl-1,10-phenanthroline) reported by Dunbar and co-workers.110,111 Herein, each of three facially-oriented cyanide ligands of the [Mn(CN)6]3− moiety links to a {Mn(tmphen)2}2+ unit as depicted in Fig. 3. Since the MnII magnetic anisotropy is negligibly small, the presence of a spin-relaxation barrier arises due to anisotropic MnIII–MnII exchange interactions through the bridging cyanides induced by the unquenched orbital angular momentum of the low-spin MnIII (t2g4) in octahedral symmetry (vide infra).112,113 In 3d metal ions, the orbital angular momentum is generally quenched by the presence of a low-symmetry ligand field. However, in systems incorporating hexacyanidometallates the main perturbation of the d-orbitals arises from the strong octahedral ligand field and thereby leaves the orbital angular momentum unquenched to a large extent even in polynuclear complexes with a low overall symmetry.114 Dunbar and co-workers reported several other TBP complexes but none showing SMM properties, while on the other hand, some of them showed remarkable spin-crossover and photomagnetic behaviour.111,115
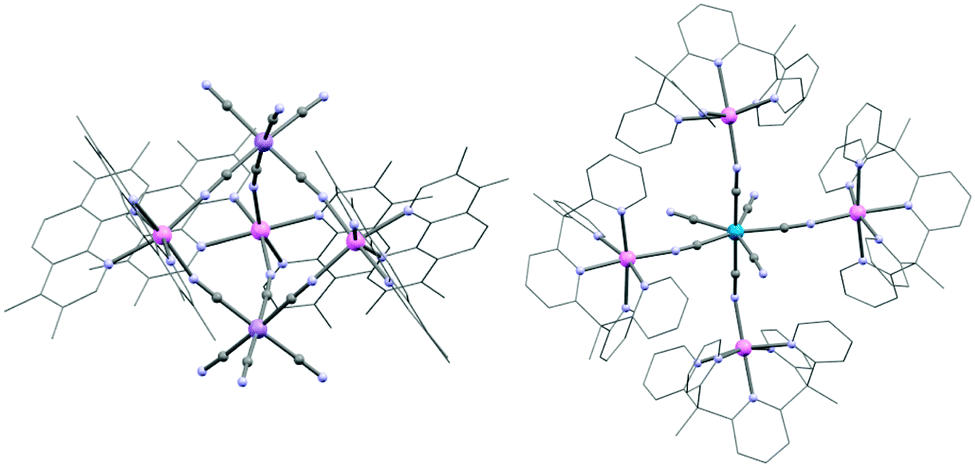 | ||
| Fig. 3 Molecular structures of 2 (left) and 14 (right). Colour code: Re, marine; MnIII, purple; MnII, pink; N, blue; C, grey. The C skeleton is shown as wireframe. Hydrogens, counterions and co-crystallized solvent molecules have been omitted for clarity. The latter two sentences apply to all the figures of this Feature article. | ||
The groups of Long, Miyasaka and Clérac reported similar trinuclear {MnIIIFeIIIMnIII} SMMs in which two MnIII Schiff-base (SB) complexes “sandwich” a trans-bridging [Fe(CN)6]3− moiety.87,116,117 In these compounds, the co-axial orientation of the MnIII ZFS tensors and the ferromagnetic Mn–Fe interaction (JMn–Fe/kB = +6.5 K) both contribute to the SMM behaviour.116 (NEt4)[Mn2(rac-salmen)2(MeOH)2FeIII(CN)6] (3) (rac-salmen2− = rac-(methylethylene)bis-salicylideneiminate, Fig. 4) has the higher spin-relaxation barrier (Δeff/kB) of 14 K (τ0 = 2.5 × 10−7 s).88,117,118
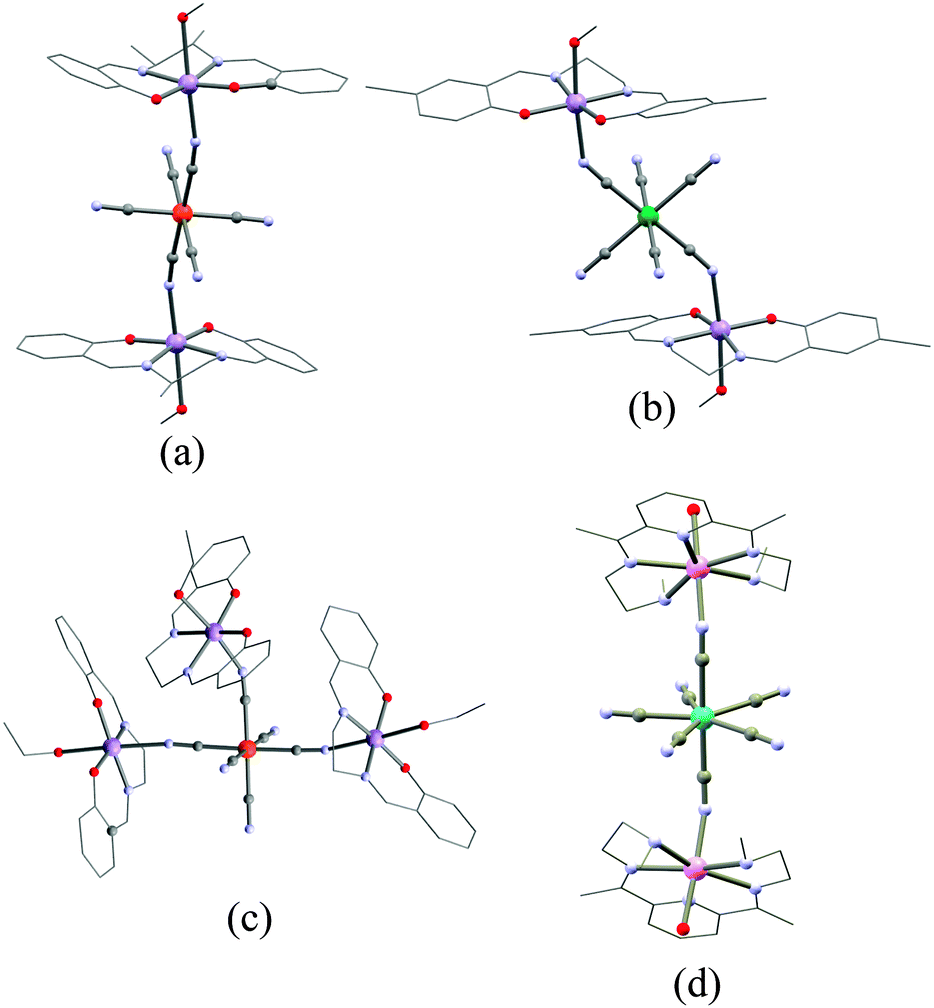 | ||
| Fig. 4 Molecular structures of 3 (a), 7 (b), 9 (c) and 19 (d). The main structural difference between 3 and 7 lies in the Mn–N–C angle being 165° and 145° degrees, respectively. Colour code: Os, green; Mo, turquoise; MnIII, purple; MnII, pink; Fe, orange; O, red; N, blue; C, grey. | ||
The {Mn(SB)}+ complexes are widely used modules to design SMMs due to the relatively strong magnetic anisotropy originating from the ZFS of the MnIII (d4) metal ion in tetragonal ligand fields imposed by the Jahn–Teller (JT) elongation.119 A comprehensive discussion of the {Mn(SB)}+ chemistry can be found in ref. 120 and some SMM highlights are presented in the next sections. Using the synthetic approach developed for 3 with different central hexacyanidometallate moieties, an isostructural series of SMMs has been described based on [Cr(CN)6]3− (4),121 [Fe(CN)6]3− (5),122 [Ru(CN)6]3− (6),123 and [Os(CN)6]3− (7)122 building blocks, “sandwiched” between two [MnIII(5-Brsalen)(MeOH)]+ (5-Brsalen2− = ethylene-bis(5-bromosalicylidene)iminate) units. The molecular structure of the [Mn2(5-Brsalen)2(MeOH)2M(CN)6]− unit is very close to that found in K[Mn2(5-Brsalen)2(H2O)2M(CN)6]·2H2O116,118 but the presence of NEt4+ counterions and methanol capping ligands on MnIII ligands leads to more magnetically isolated complexes and unquestionable SMM properties. From the viewpoint of the detailed understanding of the magnetic properties, 4 is the simplest system to analyse due to the orbitally non-degenerate ground state of the [Cr(CN)6]3− building block. The MnIII–CrIII interaction is antiferromagnetic, thereby giving rise to an ST = 5/2 ground state with a spin-relaxation barrier due to the intrinsic magnetic anisotropy provided by the MnIII sites. A detailed study of this SMM combining magnetic measurements, frequency-domain Fourier-transform THz-EPR spectroscopy and inelastic neutron scattering (INS) was reported to gain insight into the low-lying energy states of 4.121 Specifically, the analysis of both spectroscopic and magnetic data led to the following set of parameters: JMn–Cr/kB = +6.90 K and D/kB = −5.25 K. A similar analysis of the isostructural complex 8, incorporating diamagnetic [Ir(CN)6]3−, yielded DMn/kB = −5.35 K and EMn/kB = +0.30 K demonstrating that the intrinsic properties of the {Mn(SB)}+ unit are unaltered.124 Complex 4 displays clear frequency-dependent maxima in the out-of-phase component of the dynamic (ac) susceptibility with an SMM energy barrier of 18 K (τ0 = 2 × 10−8 s), which is slightly lower than the spectroscopically determined value of 26 K. This observation might be the result of QTM via the first excited state located at 18 K (MS = ±3/2, ST = 5/2). In 5–7, the theoretical treatment is more complicated as the exchange interactions become largely anisotropic as a result of the first-order orbital angular momentum present within the ground 2T2g(nd5) term (Oh).114,125–127 The transformation properties of the orbital angular momentum operator, ![[L with combining circumflex]](https://www.rsc.org/images/entities/b_char_004c_0302.gif) , leads to non-zero matrix elements, 〈Γ||Γ〉, only for Γ = 2S+1T1g or 2S+1T2g.128 Importantly, the orbital contributions to the superexchange mechanism render the HDvV Hamiltonian inapplicable.114,125 For 2T2g(nd5), the strong coupling of the fictitious l = 1 orbital momentum associated with a T term and the S = 1/2 spin momentum, lifts the 6-fold degeneracy giving a lower-lying E′1g(1/2) Kramers doublet (j = 1/2) of the octahedral double group (Oh*).128 Taking 7 as an example, the simultaneous modelling of the dc susceptibility, magnetization, INS and frequency-domain Fourier-transform EPR spectra by means of a nearest neighbour spin-Hamiltonian yielded the following principal component parameters Jxx/kB = 13(1) K, Jyy/kB = −25(1) K and Jzz/kB = 24(1) K.129 The averaged parameters show an increase in the values extracted for the isostructural complex 6, incorporating [Ru(CN)6]3−, corroborating the common theorem that descending in a transition metal group gives rise to an increase in the magnetic interaction due to increasingly diffuse magnetic orbitals.94,95,130 The energy separation between the ground j = 1/2 doublet and j = 3/2 state is given by
, leads to non-zero matrix elements, 〈Γ||Γ〉, only for Γ = 2S+1T1g or 2S+1T2g.128 Importantly, the orbital contributions to the superexchange mechanism render the HDvV Hamiltonian inapplicable.114,125 For 2T2g(nd5), the strong coupling of the fictitious l = 1 orbital momentum associated with a T term and the S = 1/2 spin momentum, lifts the 6-fold degeneracy giving a lower-lying E′1g(1/2) Kramers doublet (j = 1/2) of the octahedral double group (Oh*).128 Taking 7 as an example, the simultaneous modelling of the dc susceptibility, magnetization, INS and frequency-domain Fourier-transform EPR spectra by means of a nearest neighbour spin-Hamiltonian yielded the following principal component parameters Jxx/kB = 13(1) K, Jyy/kB = −25(1) K and Jzz/kB = 24(1) K.129 The averaged parameters show an increase in the values extracted for the isostructural complex 6, incorporating [Ru(CN)6]3−, corroborating the common theorem that descending in a transition metal group gives rise to an increase in the magnetic interaction due to increasingly diffuse magnetic orbitals.94,95,130 The energy separation between the ground j = 1/2 doublet and j = 3/2 state is given by ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) ζnl, where ζnl is the one-electron spin–orbit coupling parameter. ζnl scales dramatically with the atomic number and is approximately 700 and 4000 K for Fe and Os, respectively.131 When [M(CN)6]3− building blocks are parts of a polynuclear complex, the symmetry is no longer octahedral and low-symmetry ligand field effects become often of importance. For the [Os(CN)6]3− unit that exhibits a strong ligand field of ΔO/kB ≈ 56000 K (∼39000 cm−1),106 small ligand field effects are unlikely to alter the j = 1/2 ground state or induce significant quantum mixing as the separation from the j = 3/2 state is quite large: ζOs(III) ≈ 6000 K. For lighter atoms such as FeIII in a [Fe(CN)6]3− environment, small ligand field effects are able to mix j = 3/2 into the ground state. Using the angular overlap model, Tregenna-Piggott et al. estimated the 2T2g energy splitting of a {Fe(CN)6}3−trans-bridging unit to yield three Kramers doublets at 0, 850 and 1450 K.116 The intrinsic complicated magnetic properties of the [Fe(CN)6]3− and [Mn(CN)6]3− building blocks and their unexplored [V(CN)6]3− and [Ti(CN)6]3− analogues, make them less predictable – but fascinating – magnetic modules to design SMMs.132 Interestingly, Δeff for complexes 5 to 7 was found to increase upon descending in the group 8 of the periodic table, emphasizing the promising and largely unexplored properties of 4d and 5d metals in the quest for new SMMs.133
ζnl, where ζnl is the one-electron spin–orbit coupling parameter. ζnl scales dramatically with the atomic number and is approximately 700 and 4000 K for Fe and Os, respectively.131 When [M(CN)6]3− building blocks are parts of a polynuclear complex, the symmetry is no longer octahedral and low-symmetry ligand field effects become often of importance. For the [Os(CN)6]3− unit that exhibits a strong ligand field of ΔO/kB ≈ 56000 K (∼39000 cm−1),106 small ligand field effects are unlikely to alter the j = 1/2 ground state or induce significant quantum mixing as the separation from the j = 3/2 state is quite large: ζOs(III) ≈ 6000 K. For lighter atoms such as FeIII in a [Fe(CN)6]3− environment, small ligand field effects are able to mix j = 3/2 into the ground state. Using the angular overlap model, Tregenna-Piggott et al. estimated the 2T2g energy splitting of a {Fe(CN)6}3−trans-bridging unit to yield three Kramers doublets at 0, 850 and 1450 K.116 The intrinsic complicated magnetic properties of the [Fe(CN)6]3− and [Mn(CN)6]3− building blocks and their unexplored [V(CN)6]3− and [Ti(CN)6]3− analogues, make them less predictable – but fascinating – magnetic modules to design SMMs.132 Interestingly, Δeff for complexes 5 to 7 was found to increase upon descending in the group 8 of the periodic table, emphasizing the promising and largely unexplored properties of 4d and 5d metals in the quest for new SMMs.133
Related to the above systems based on {Mn(SB)}+ units, a “T-shaped” SMM, [MnIII(salen)(EtOH)]3[FeIII(CN)6] (9, Fig. 4c) was also reported (salen2− = N,N′-ethylene-bis(salicylidene-iminate)).134 The nearly perpendicular orientation of the MnIII JT axes reduces the overall magnetic anisotropy and the complex has a smaller energy barrier than the related system 3. When the assembly of {Mn(SB)}+ and [Cr(CN)6]3− is pursued to its logical end, a heptanuclear complex is formed, [Cr(μ-CN)6MnIII6(salen)6(EtOH)6] (10).119 For this complex, the nearly complete cancellation of D by the almost perpendicular JT axes results in the absence of SMM behaviour. The [Cr(CN)6]3− module was also combined with an S = 2 FeII ion placed in a macrocyclic pentadentate ligand yielding a linear ferromagnetically (JFe–Cr/kB = 5.41 K) coupled {FeII2CrIII} complex (11, {[{Fe(LN3O2)(H2O)}2Cr(CN)6][ClO4]}·3H2O; LN3O2 = 3,12,18-triaza-6,9-dioxabicyclo[12.3.1]octadeca-1(18),14,16-triene) with a large Δeff/kB = 44.3 K (τ0 = 1.4 × 10−9 s).135 The magnetic anisotropy in this SMM originates from the hepta-coordinated FeII (S = 2) unit (see Scheme 1m) for which DFe/kB amounts to −6.7 K.
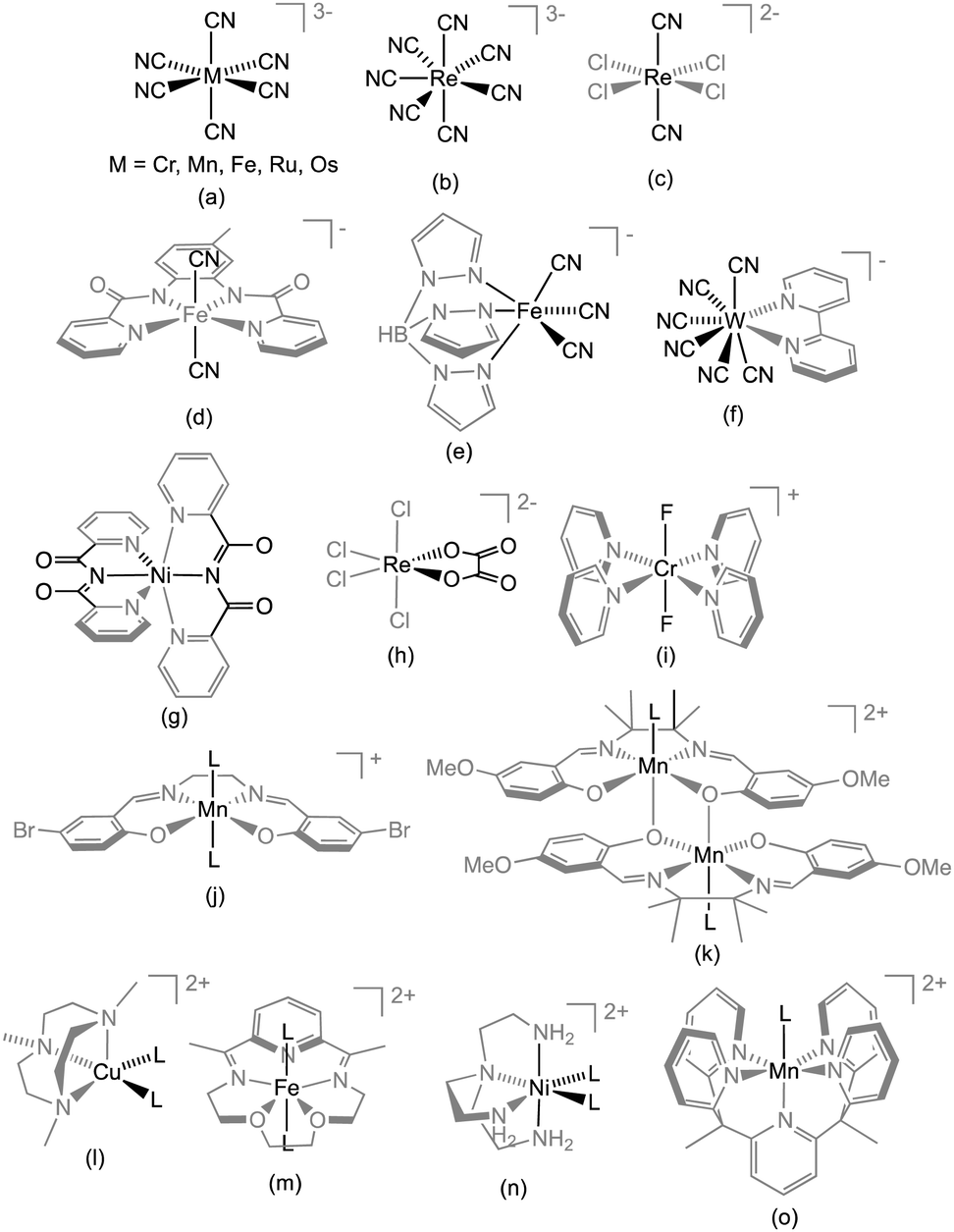 | ||
| Scheme 1 Representative examples of donor-type (a–i) and acceptor-type modules (j–o), which have all been employed to design SMMs. For the latter type, “L” designates the accessible coordination site(s). | ||
Glaser et al. extended the hexacyanidometallate approach by exploiting phloroglucinol-derived (= 1,3,5-trihydroxybenzene) salen ligands to synthesize a heptanuclear {MnIII6CrIII} SMM (12, Fig. 5, [{(talentBu2)Mn3}2{Cr(CN)6}(MeOH)3(CH3CN)2](BPh4)3·4CH3CN·2Et2O; H6talentBu2 = 2,4,6-tris{1-[2-(3,5-di-tert-butylsalicylaldimino)-2-methylpropylimino]-ethyl}-1,3,5-trihydroxybenzene) exhibiting an effective barrier of 25.4 K.136,137 The main difference of this system from 10 lies in a trigonal distortion of the octahedral geometry resulting in a non-cancellation of the magnetic anisotropy and thereby in the observation of the SMM behaviour.137 Fitting of the χT vs. T data at high temperatures allowed an estimation of JMn–Cr at about −7.2 K (with ST = 21/2). This value is close to the one found for 4 despite the more linear Mn–N–C angle of 160–162° in 12. The synthesis of C3 symmetrical SMMs is particularly appealing as the QTM is commonly governed by the rhombic E term that vanishes in the trigonal symmetry. Nevertheless, higher order terms of the anisotropy allowed in the C3 symmetry might still govern the QTM despite their small values. Exchanging [Cr(CN)6]3− by [Fe(CN)6]3− affords the analogous {MnIII6FeIII} complex showing weak characteristics of SMM behaviour.138 However, substituting for [Os(CN)6]3− yields {MnIII6OsIII} with stronger anisotropy and slower relaxation of the magnetization due to anisotropic MnIII–OsIII exchange interactions.139
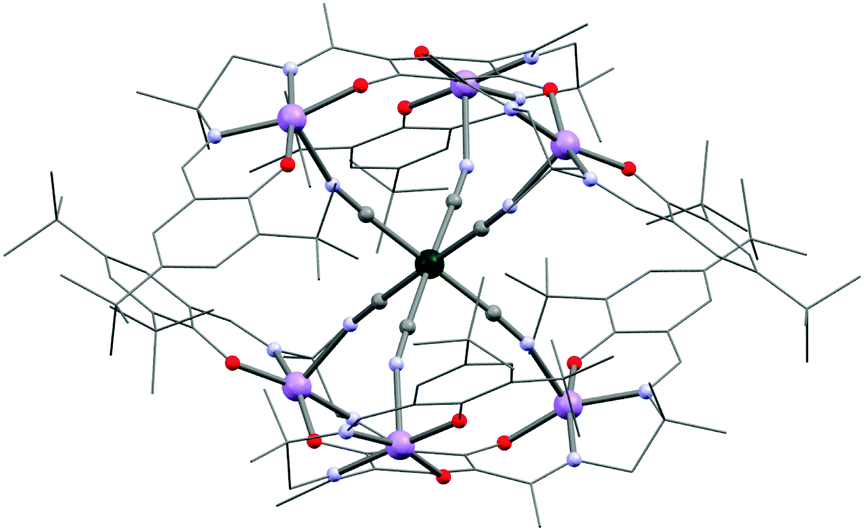 | ||
| Fig. 5 Molecular structure of 12. Solvent molecules located on axial 6th position of the MnIII sites and counterions have been omitted for clarity. Colour code: Mn, purple; Cr, dark green; O, red; N, pale blue; C, grey. | ||
Recently, the same group reported an analogous {MnIII6MnIII} complex encompassing a central low-spin [Mn(CN)6]3− module ([{(talentBu2)(MnIII(MeOH))3}2{MnIII(CN)6}](lac)3·10.5MeOH, 13, lac− = lactate).140 Thanks to the lactate counterions that favour the occurrence of hexagonal and cubic packings, the {MnIII6MnIII} complex adopts an S6 crystallographic symmetry. Notably, a trigonal distortion of [Mn(CN)6]3− does not quench the effective l = 1 orbital angular momentum but the spin–orbit coupling leads to a nonmagnetic ground state for the j = 0 [Mn(CN)6]3− central unit.140,141 Despite the “blocked” exchange pathway through the essentially diamagnetic [Mn(CN)6]3− unit (at low temperature), the {MnIII6MnIII} complex exhibits an unusual double M vs. H hysteretic behaviour. The weak intra-{talentBu2MnIII3} MnIII–MnIII antiferromagnetic interactions stabilize an S = 2 intermediate spin state for both {talentBu2MnIII3} units, which interact weakly ferromagnetically leading to an ST = 4 ground state. Slow dynamics of this ground state is observed around zero-dc field but above 3.4 T, zero-field excited spin states become the lowest in energy giving rise to the second hysteresis loop.
Among the cyanide-based SMMs, the {ReIVMnII4} complex (14; [(PY5Me2)4Mn4Re(CN)7](PF6)5·6H2O; PY5Me2 = 2,6-bis(1,1-bis(2-pyridyl)ethyl)-pyridine) based on the pentagonal bipyramidal [ReIV(CN)7]3− module stands apart with a large barrier of 47 K (τ0 = 2.4 × 10−8 s).142,143 This system, depicted in Fig. 3, incorporates bulky [(PY5Me2)MnII]2+ units (Scheme 1o), which limit the nuclearity of the complex. At first look, the presence of an SMM behaviour in 14 seems surprising as the magnetic anisotropy of MnII is well-known to be very weak and ZFS is obviously meaningless for S = 1/2 systems like [ReIV(CN)7]3−. Indeed, the (NBu4)3[Re(CN)7] precursor exhibits a strongly anisotropic g tensor with g∥ = 3.66 and g⊥ = 1.59 (from X-band EPR) reflecting the unquenched orbital angular momentum of the 2E1′′(d3) ground state in the idealized D5h symmetry.144 Therefore the strong magnetic anisotropy of 14 is likely due to anisotropic ReIV–MnII exchange interactions, which have been described for isoelectronic [Mo(CN)7]4−–MnII systems in the frame of the superexchange theory.145 Similar complexes with {NiII4ReIV} (15; [(PY5Me2)4Ni4Re(CN)7](PF6)5) and {CuII4ReIV} (16; [(PY5Me2)4Cu4Re(CN)7](PF6)5) cores also exhibit slow relaxation of the magnetization although with significantly reduced Δeff's.143 While Δeff/kB = 24 K and τ0 = 1.4 × 10−7 s for 15, only a small frequency dependence of a non-zero χ′′(T) with νac ≤ 1.5 kHz was observed for 16 suggesting a much smaller Δeff.143 The potentially interesting magnetic properties of the [Mo(CN)7]4− module incorporated into molecular systems have been studied by Dunbar, Wang and co-workers. The first complex incorporating this moiety was [Mn(LN5)(H2O)2]2[{Mo(CN)7}8{Mn(LN5)}10{Mn(LN5)(H2O)}4]·xH2O (LN5 = 2,13-dimethyl-3,6,9,12,18-pentaazabicyclo-[12.3.1]octadeca-1(18),2,12,14,16-pentaene). This {MnII14MoIII8} (18) complex exhibits a large spin ground state (ST = 31), but instead of possessing SMM properties, it shows a 3D ferrimagnetic ordering at low temperature.146 Very recently, the same group reported on three trinuclear MnIIL–[MoIII(CN)7]–MnIIL complexes where L is a pentadentate ligand.147 In particular, the quasi-linear MnII–NC–MoIII–CN–MnII complex, [Mn(LN5Me)(H2O)]2[Mo(CN)7]·6H2O (19, LN5Me = 2,6-bis(3,6-diazahept-2-ene-2-yl)pyridine, Fig. 4d), exhibits clear SMM properties with Δeff/kB = 58.5(4) K and τ0 = 2.0(3) × 10−8 s. These characteristics make this complex the current record holder in terms of Δeff for cyanide-based SMMs. Additionally, this system exhibits a large M vs. H hysteresis loop at low temperatures with a coercive field of 2.0 T (with a 0.05 T s−1 sweeping rate) at 1.8 K. Octacyanidometallates are known for WIV/V, MoIV/V, NbIII/IV and ReV metal ions. The incorporation of these units into coordination networks and their resulting magnetic properties have been the topics of reviews by Sieklucka and co-workers.148–151 Using these octacyanidometallate building blocks, Dunbar and co-workers isolated TBP complexes similar to the ones described earlier (2, Fig. 3, left), [NiII(tmphen)2]3[WV(CN)]2 (20), but no M vs. H hysteresis loop was observed down to 40 mK.152 Only a few reports on SMMs based on [MV(CN)8]3− building blocks (MV = MoV, WV, ReV (S = 0)) have been reported. These include large polynuclear complexes with stoichiometries such as {NiII9MoV6} (21, ST = 12; [Ni{Ni(bpy)(H2O)}8{Mo(CN)8}6]·12H2O),153 {NiII9WV6} (22, ST = 12; [Ni{Ni(bpy)(H2O)}8{W(CN)8}6]·23H2O,152,154) and site-substituted ReV analogues ([Co9(CH3OH)24{W(CN)8}5{Re(CN)8}]·xCH3OH·yH2O, 23),155 but only thin evidence of slow magnetic relaxation has been observed. Another family of heterometallic systems encompass mixed 3d–5d–4f species incorporating paramagnetic octacyanometallates,156–159 some of which exhibit SMM behaviour.159–161 Herein, the 3d–4f back-bone is based on bicompartmental Schiff-base ligands derived from o-vanillin and diamine ligands, which accommodate a CuII ion in a salen-type environment.162,163 With the phenolates and the methoxy groups, this unit constitutes a chelating metallo-ligand for lanthanide ions, which, subsequently, may coordinate the octacyanometallate by either the CuII or the LnIII ion.
Heteroleptic cyanidometallates
Detailed reviews of the use of di- and tri-cyanidometallate precursors in the design of polynuclear systems have recently been published by Wang et al.164,165 Using these modules, the first indications of SMM behaviour in a cyanide-bridged system was found in a {MoIII6MnII} complex (24; K[(Me3tacn)6MnMo6(CN)18](ClO4)3; Me3tacn = N,N′,N′′-trimethyl-1,4,7-triazacyclononane) complex incorporating fac-[MoIII(Me3tacn)(CN)3] units.166 Each of these modules coordinates through only one cyanide ligand to the central MnII ion leading to an approximately prismatic structure (Fig. 6). The intra-complex MoIII–MnII antiferromagnetic interactions (JMo–Mn/kB = −9.6 K) yield an ST = 13/2 spin ground state. Fitting of the M vs. H/T data revealed an Ising-type magnetic anisotropy of D/kB = −0.47 K.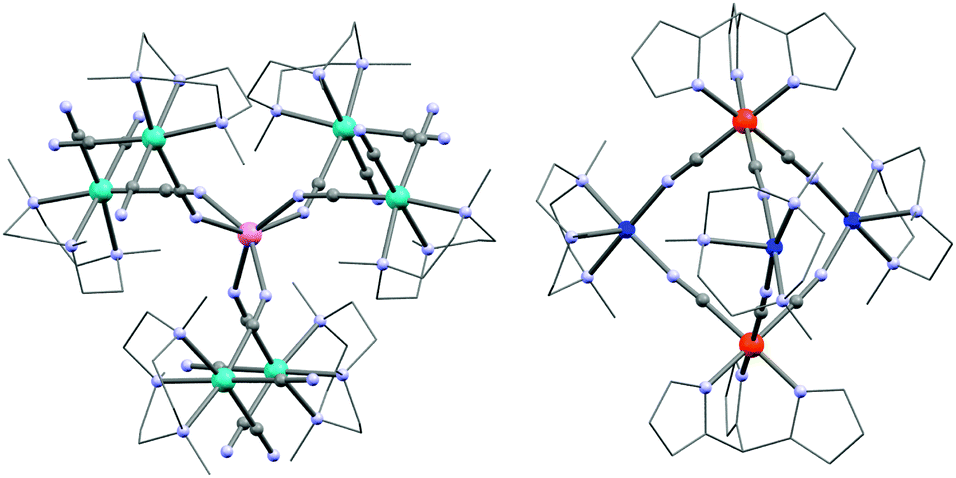 | ||
| Fig. 6 Molecular structure of 24 (left) and 29 (right). Colour code: Mo, turquoise; Cu, marine; Fe, orange; Mn, pink; N, pale blue; C, grey. | ||
Notably, the isostructural {CrIII6MnII} (25; K[(Me3tacn)6MnCr6(CN)18](ClO4)3) complex did not exhibit SMM properties,167 likely due to a stronger magnetic anisotropy exhibited by the MoIII unit over the CrIII building block. For d3 ions (t2g3), like MoIII, in an axially perturbed ligand field, the magnetic anisotropy is primarily induced by the mixing of the 4A2g(Oh) ground state with the excited 4T2g(Oh) state via spin–orbit coupling.168 If only this mixing is taken into account, the D parameter scales as ζnl2. For 3d3 metal ion such as V2+, Cr3+ and Mn4+ for which the spin–orbit coupling is relatively weak, this second-order contribution to the magnetic anisotropy is often negligible, whereas this effect becomes important for 4d and 5d transition metal ions.
The trans-[ReCl4(CN)2]2− building block reported by Long and co-workers is another interesting example of a heteroleptic cyanide-based 5d metal ion module.169 This building block is unique in the sense of being the sole example of a paramagnetic mixed halide–cyanide complex. In addition, it offers both strong magnetic anisotropy as well as effective mediation of super-exchange.170,171 The latter property is well illustrated in the (NBu4)[TpCuReCl4(CN)2]·1.33CH3CN chain system (26, Tp− = hydrotris(pyrazol-1-yl)borate) exhibiting the strongest ferromagnetic interaction mediated by a cyanide bridge (JCu–Re/kB = 41 K) reported until now.96 Remarkably, the CuII JT axis in 26 is not placed along the Cu–NC axes and the short Cu–N bond lengths facilitate the pronounced interaction. The reaction of trans-[ReCl4(CN)2]2− with [(TPA2C(O)NHtBu)FeII(CF3SO3)]+ (TPA2C(O)NHtBu = 6,6′-(pyridin-2-ylmethylazanediyl)bis(methylene)bis(N-tert-butylpicolinamide)) affords a dinuclear cyanido-bridged SMM (27; (TPA2C(O)NHtBu)FeReCl4(CN)2).172 The pentagonal bipyramidal FeII precursor (S = 2) has (as the triflato complex) a strong magnetic anisotropy reflected by its ZFS parameters: D/kB = 11 K and |E|/kB = 3.2 K. ac susceptibility measurements reveal only an increase of χ′′(νac) without a maximum (with νac ≤ 1.5 kHz) indicating a small Δeff. Despite the promising FeII–ReIV ferromagnetic interactions and the strong magnetic anisotropy of the building blocks, the non-collinearity of anisotropy tensors might be responsible for the small overall anisotropy of the final complex emphasizing the necessity to control the geometry of the designed polynuclear SMMs. Several other magnetic systems based on the trans-[ReCl4(CN)2]2− module have been reported but most of them are chains (that are commonly observed for trans-dicyanidometallates) and SCM compounds.169,171 Only a few other similar building blocks based on 4d/5d metal ions are known including trans-[RuIII(acac)2(CN)2]− (acac = acetylacetonate),173 [MIII(salen)(CN)2]− (M = Ru,174 Os175), and trans-[RuIII(8-quin)2(CN)2]− (quin = 8-quinolinolate),176 but none of them have been used to design SMMs yet. Returning to the first row transition metal ions, a particularly exotic complex is obtained with the trans-dicyanidometallate [FeIII(bpmb)(CN)2]− module: [MnIII(salen)]6[FeIII(bpmb)(CN)2]6·7H2O (28, H2bpmb = 1,2-bis(pyridine-2-carboxamido)-4-methylbenzene). Instead of forming a chain system, a twelve membered wheel is crystallized as shown in Fig. 7.177,178 As the magnetic anisotropy is dictated by the JT distorted MnIII ions, the overall magnetic anisotropy of 28 is accordingly small leading to SMM properties with an effective energy barrier of only 7.5 K.
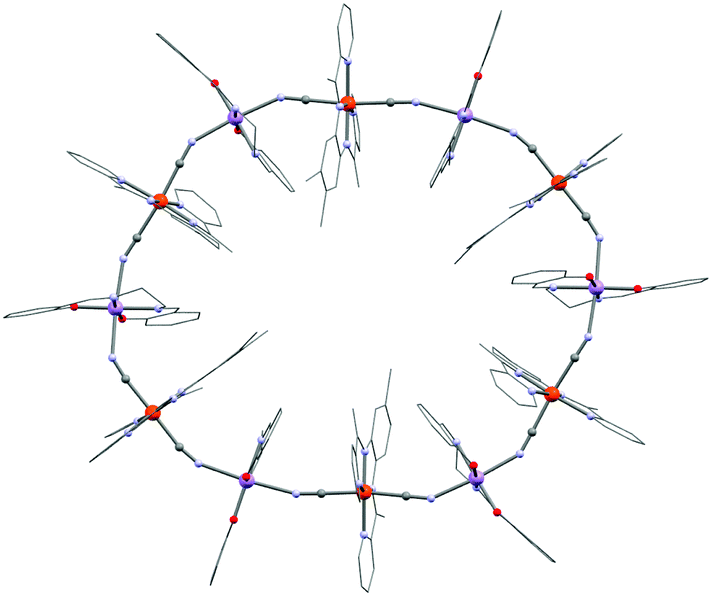 | ||
| Fig. 7 Molecular structure of 28. Colour code: Fe, orange; Mn, purple; O, red; N, pale blue; C, grey. | ||
Several facial tricyanidoiron(III) complexes with the auxiliary ligand sphere occupied by various pyrazolylborate ligands, fac-[LFeIII(CN)3]−, have been utilized as modules to synthesize SMMs.179–187 Most of these SMM systems encompass NiII ions and commonly give rise to square-based structures. However, the reaction of the simple [TpFeIII(CN)3]− module with [(Me3tacn)CuII(H2O)2](ClO4)2 affords a remarkable TBP complex (29, [Tp2(Me3tacn)3Cu3Fe2(CN)6](ClO4)4·2H2O Fig. 6).179 The apparent preference of the CuII ion to penta-coordinated geometry prevents the formation of a molecular square or cube structure as the Me3tacn ligand blocks three facial coordination sites. The CuII (S = 1/2) and low-spin FeIII (S = 1/2) magnetic centres couple ferromagnetically (JCu–Fe/kB = 12 K) stabilizing an ST = 5/2 ground state that combined with a relatively strong magnetic anisotropy (D/kB = −8.2 K obtained from fitting of reduced magnetization data) induces SMM properties with Δeff/kB = 23 K (τ0 = 4.8 × 10−8 s). As the local spins are all S = 1/2, the magnetic anisotropy is likely the result of the orbital angular momentum of the low-spin FeIII modules. A structural analogue is obtained when [TpFeIII(CN)3]− is reacted with [NiII(cyclen)](BF4)2 (cyclen = 1,4,7,10-tetraazacyclododecane) giving a {FeIII2NiII3} SMM (30; [(cyclen)Ni]3[TpFe(CN)3]2(BF4)4·4H2O). The TBP geometry is induced by the cis configuration of the accessible coordination sites of the NiII building block imposed by the small cavity of the cyclen ligand.188 This complex displays intra-molecular ferromagnetic interactions (JNi–Fe/kB = +7.8 K, ST = 4) and shows the onset of χ′′(T) peaks above 1.8 K suggesting SMM properties.
Other fac-tricyanido building blocks such as [ReII(triphos)(CN)3]− (triphos = 1,1,1-tris(diphenylphosphinomethyl)ethane) have been studied by Dunbar and co-workers who have reported a {MnII4ReII4} (31; [MnCl]4[Re(triphos)(CN)3]4) SMM189,190 and polymeric systems191 based on this module. The structure of the parent complex, 31, is a molecular cube as depicted in Fig. 8. Other divalent metal ions such as FeII, CoII, NiII and ZnII have been incorporated into analogous systems, but only the MnII complex was shown to be an SMM (Δeff/kB = 13 K, τ0 = 3.25 × 10−7 s).192 The static magnetic properties are dominated by the antiferromagnetic interactions between ReII (S = 1/2) and MnII (S = 5/2) spins but a detailed analysis of the experimental magnetic data becomes highly complicated due to orbital contributions to the magnetic exchange mechanism and a large temperature independent paramagnetism (TIP) of the ReII ion.193
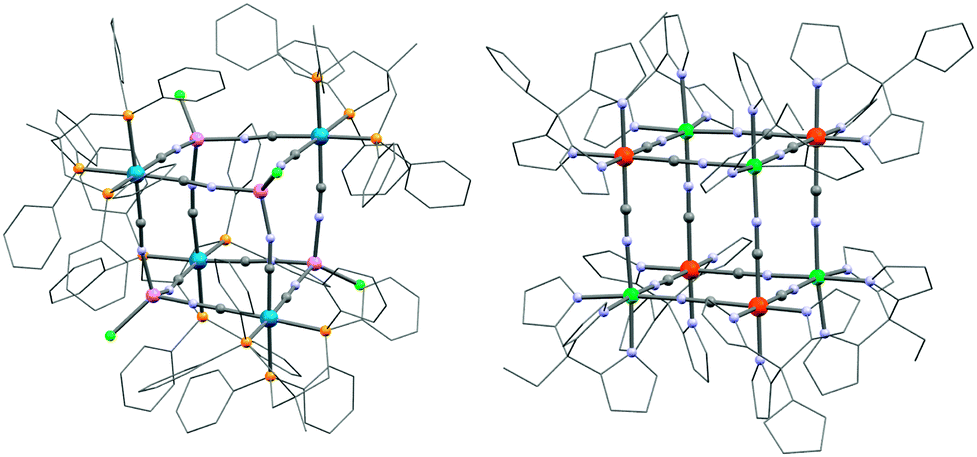 | ||
| Fig. 8 Molecular structures of 31 (left) and 33 (right) cubes. Colour codes: Re, marine; Ni, turquoise; Fe, orange; Mn, pink; Cl, green; P, yellow; N, pale blue; C, grey. | ||
Glaser and co-workers extended their strategy of molecular recognition with triple-salen ligands by exchanging [Cr(CN)6]3− with fac-[CrIII(Me3tacn)(CN)3]. The three (fac) positions occupied by the Me3tacn ligand around the CrIII ion force the fac-[CrIII(Me3tacn)(CN)3] module to coordinate only one {MnIII3} triple-salen moiety (32; [(talentBu2)(Mn(MeOH))3][(Me3tacn)Cr(CN)3](ClO4)3).194 For this system, the χ′′(T) data only show weakly frequency-dependent onsets of peaks between 1.8 and 2.5 K suggesting a lower spin-relaxation barrier than in the {MnIII6CrIII} complex (12, Δeff/kB = 25.4 K) as expected when lowering the spin ground state from ST = 21/2 (12) to 7/2 (32).
The groups of Holmes, Oshio and Zuo reported {FeIII4NiII4} molecular cubes exhibiting SMM properties.195–199 All the reported examples are based on cyanido-based FeIII modules with tris(pyrazol-1-yl)borate capping ligand derivatives, whereas a plethora of ligands, primarily amines, have been employed for the more labile NiII moiety. The prototypical example, {[(pzTp)Fe(CN)3]4[Ni(tpe)]4}[OTf]4·10DMF·Et2O, (33, Fig. 8) reported by Holmes and co-workers involves the [(pzTp)FeIII(CN)3]− building block (pzTp− = tetra(pyrazol-1-yl)borate; OTf− = trifluoromethanesulfonate) and a NiII site with a 2,2,2-tris(pyrazolyl)ethanol (tpe) capping ligand.196 The FeIII–NiII ferromagnetic coupling (J/kB = +9.5(5) K) yields an ST = 6 ground state as found for the other analogues.195–199 Fitting of the M vs. H/T data allowed an estimation of D/kB at about −0.33 K (and thus Δ/kB = |D|ST2 ≈ 12 K) that corroborates the experimental finding of only a small Δeff. It is worth mentioning that a similar {FeIII4NiII4} complex (34; [(tach)4(H2O)12Ni4Fe4(CN)12]Br8·18H2O) based on the tach (1,3,5-triaminocyclohexane) capping ligand was synthesized by Long and co-workers but no slow relaxation of magnetization was reported.200 Recently, Oshio and co-workers reported a mixed-valence cube complex Na[(Tp)4FeIII2FeII2(CN)12NiII4(L)4](BF4)3 (35) incorporating a redox-active ligand: L = α-(4′-methyl-4,5-dimethylthio-tetrathiafulvalene-5′-thio)-α′-[tris-2,2,2-(1-pyrazolyl)ethoxy]-p-xylene. Unfortunately this complex, which was not structurally characterized, only exhibits a small frequency-dependent increase of the χ′′(T) data between 1.8 and 3 K.199 An analogous {Fe4Co4} cube complex, {[(pzTp)Fe(CN)3]4[Co(tpe)]4}(ClO4)4·13DMF·4H2O (36), was also reported but instead of exhibiting SMM properties, it displays temperature- and light-induced magnetic bistability201 controlled by an intra-molecular electron transfer and two different redox configurations: i.e. the paramagnetic {FeIII4CoII4} and diamagnetic {FeII4CoIII4} states.
From a strategic point of view, the nearly cubic structure of the previously described SMMs does not appear to be the ideal geometry to obtain SMM properties (due to a near compensation of magnetic anisotropy tensors). Therefore, researchers have been trying to reduce the {FeIII4NiII4} cubes into less-symmetrical smaller fragments like defect cubanes, squares and trinuclear complexes. Interestingly, the use of facial tricyanido modules also stabilizes molecular square SMMs with alternating FeIII and NiII,181,182,184,202–205 or CuII metal ions.206 In these FeIII–NiII systems illustrated by [Tp*Fe(CN)3]2[Ni(DMF)4]2(OTf)2·2DMF (37) in Fig. 9 (Tp* = hydridotris(3,5-dimethylpyrazol-1-yl)borate),181 the FeIII–NiII interaction is ferromagnetic with JNi–Fe coupling constants up to +10.1 K,184 giving rise to an ST = 3 ground state. It was argued that the distortion of the NiII coordination sphere does not significantly alter the SMM properties suggesting that orbital contributions from the S = 1/2 FeIII module is the main origin of the SMM properties in these molecular FeIII–NiII squares.207 Their effective SMM energy gaps are relatively moderate with the largest reported value being Δeff/kB = 29.0(4) K.202
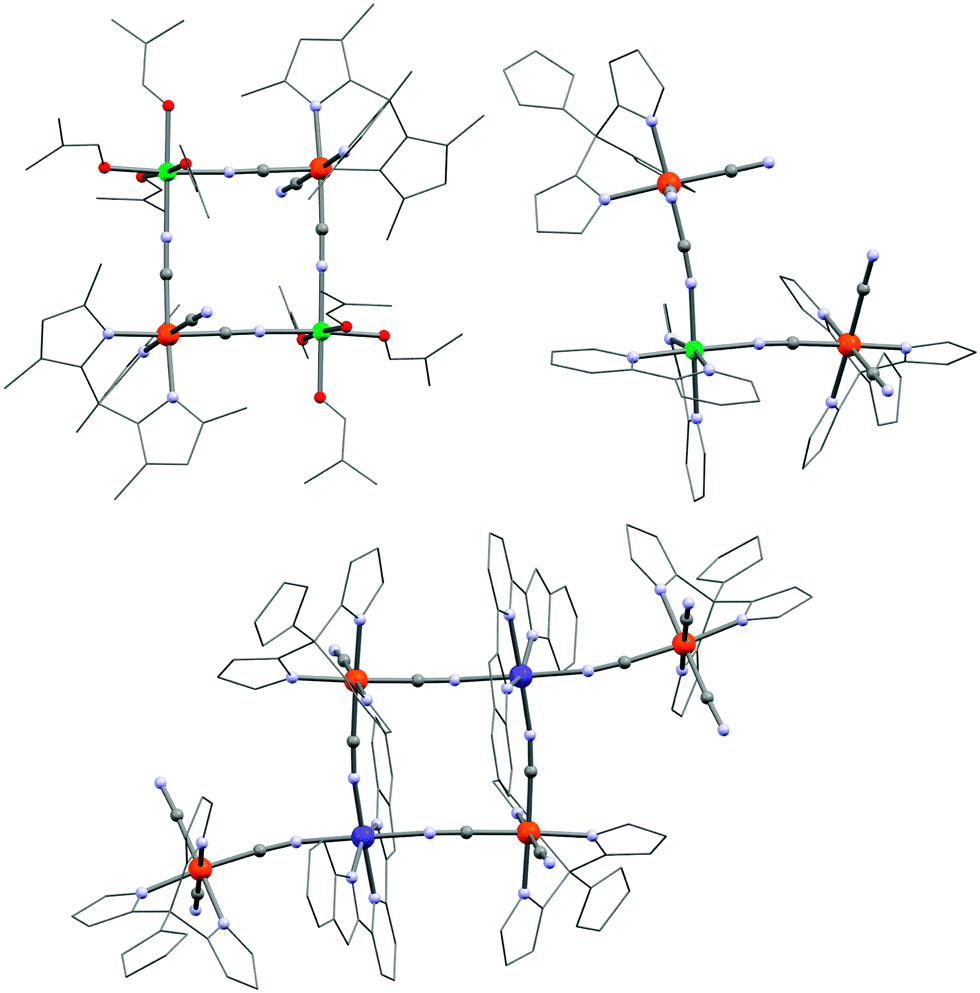 | ||
| Fig. 9 Molecular structures of 37 (top, left), 38 (top, right) and 42 (bottom, in the {FeII2CoIII2FeIII2} state obtained at T = 100 K). Colour codes: Ni, turquoise; Co, purple; Fe, orange; O, red; N, pale blue; C, grey. | ||
Defect square {FeIII2NiII} SMMs were also obtained, for example by reaction of (NEt4)[(pzTp)FeIII(CN)3] with NiII and bpy (38, {[(pzTp)Fe(CN)3]2[Ni(bpy)2]}·2H2O; bpy = 2,2′-bipyridine).183 Interestingly, this complex shown in Fig. 9 exhibits a clear SMM signature by ac susceptibility measurements despite the small ST = 2 ground state induced by the intra-complex ferromagnetic interactions (JFe–Ni/kB = 7.0(2) K). In the absence of a static dc field, the energy barrier is about 12.0 K (τ0 = 4 × 10−7 s) that increases up to 20.6 K (τ0 = 2 × 10−8 s) in a small dc field (2000 Oe). Such trinuclear SMMs can themselves be viewed as building-blocks for larger molecular {FeIII2NiII}n structures where n = 2 or 3. Although not synthetically assembled from pre-isolated trinuclear precursors, an exo-cyclic {FeIII4NiII2} SMM (39; {[(Tp*Me)Fe(CN)3]4[Ni(DMF)3]2}·4DMF·H2O; Tp*Me = tris(3,4,5-trimethylpyrazole)borate) and a fused system of exo-cyclic squares {FeIII6NiII3} (40; {[(Tp*Me)Fe(CN)3]6[Ni(MeOH)3]2[Ni(MeOH)2]}·3H2O·8MeOH) have been reported.208 In these two complexes, the NiII sites connect three [(Tp*Me)Fe(CN)3]− modules. Their remaining positions are occupied by rather labile solvent molecules, which do not apply any particular geometrical constraints on the coordination sphere. The energy barriers for these {FeIII2NiII}n SMMs are slightly higher than for the trinuclear parent complex, 38, with Δeff/kB = 15.6 K and 17.7 K in zero-dc field and 26 K and 24.5 K in 1500 and 600 Oe for 39 and 40, respectively.
The use of the same FeIII module, [(Tp*Me)FeIII(CN)3]−, and geometrically constraining the NiII coordination sphere by the tetradentate tren ligand (tren = tris(2-aminoethyl)amine), leaving only two cis-positions accessible, leads to an octanuclear {FeIII4NiII4} complex (41; {[(Tp*Me)Fe(CN)3]4[Ni(tren)]4[ClO4]4}·7H2O·4MeCN).185 Its complicated molecular structure can be viewed as an “unwrapped” version of the {FeIII4NiII4} cube. This low-symmetric complex exhibits one of the largest effective energy barriers (33 K) for any cyanide-based 1st row transition metal SMM.
As previously mentioned for a {Fe4Co4} cube complex,201 a few examples of molecular {FeIII/II2CoII/III2} squares have been reported209–215 to exhibit photo- and thermally-assisted intra-molecular charge transfer similar to the effect observed in three-dimensional Fe/Co Prussian Blue analogues.97 The principle of this phenomenon is based on the reversible interconversion of diamagnetic {FeIILS(μ-CN)CoIIILS} pairs into paramagnetic {FeIIILS(μ-CN)CoIIHS} pairs by light irradiation and thermal energy. Very recently, SMM properties in a photo-induced state were observed for the first time in the hexanuclear complex, [(pzTp)4Fe4(CN)6(μ-CN)6Co2(bimpy)2]·2nPrOH·4H2O216 (42; bimpy = 2,6-bis(benzimidazol-2-yl)pyridine) shown in Fig. 9. The use of the tridentate bimpy ligand permits only three vacant sites on the Co metal ions in a mer-position allowing the central {FeIII/II2CoII/III2} square to be decorated by two [(pzTp)FeIII(μ-CN)(CN)2]− units. These exo-cyclic moieties are permanently in a LS (t2g5) state, whereas the {Fe2Co2} square consists of (i) HS CoII (S = 3/2) and LS FeIII (S = 1/2) metal ions above 250 K and (ii) LS CoIII (S = 0) and LS FeII (S = 0) sites below 200 K, as evidenced from the χT vs. T data and single-crystal X-ray crystallography at different temperatures. Upon an 808 nm irradiation at 5 K, the χT product increases to 20.3 cm3 K mol−1 as a result of an intra-square electron transfer from a diamagnetic central {CoIII2FeII2} core to an exchange-coupled {CoII2FeIII2} unit. Before irradiation, ac susceptibility measurements did not detect any sign of slow relaxation of magnetization in 42, but in its photo-excited state, clear frequency-dependent χ′′(T) peaks were observed, suggesting that this complex was the first photo-switchable SMM. The associated spin-reversal barrier was estimated at about 26 K in a static field of 500 Oe.
In contrast to the fac-tricyanido systems, mer-tricyanido modules have received much less attention.217–222mer-Tricyanide complexes are well-known especially for iron(III) as illustrated by the [FeIII(bpca)(CN)3]− (bpcaH = bis(2-pyridylcarbonyl)amine),223 and [FeIII(pcq)(CN)3]− (pcqH = 8-(pyridine-2-carboxamido)quinoline) building blocks.217,224 As for the trans-dicyanido modules (vide supra), these units are favoring one-dimensional assembly unless steric constraints from the other building blocks impose otherwise. To the best of our knowledge, no SMMs incorporating these mer-tricyanido building blocks have been reported so far.
4. Non-cyanide based precursors
Undoubtedly, cyanide remains the coordination chemist's favourite bridging ligand in the quest for new SMMs and functional materials. The cyanide-based complexes often possess three essential properties: (i) integrity in solution, (ii) relatively linear bridging modes between metal ions (Fig. 2) and (iii) a redox-activity that does not alter the complex structure. None of these properties are indeed exclusive to the cyanide-based building blocks, and magnetic modules based on other type of bridging groups should be considered in the design of SMM systems.Halide and pseudo-halide based modules
Although a few examples of SMMs with chloride bridges have been reported,225 no examples assembled from modules can be said to exist. Building-block examples based on pseudohalides (other than cyanides) including azide and (iso)thiocyanate are also extremely rare226,227 and no examples of SMMs synthesized by a modular approach have been reported. Gao, Lau and co-workers reported the mer-[RuIII(sap)(N3)3]− module (H2sap = N-salicylidene-o-aminophenol) but its reaction with NiII or CoII afforded polynuclear complexes incorporating diamagnetic RuVI ions.228 Isoelectronic three-atom ligands such as cyanate, thiocyanate or azide have been used to prepare complexes of paramagnetic transition metal ions but no SMM systems incorporating these modules have been prepared so far. Remarkably, relatively strong exchange interactions have been observed in NiII–SCN–CrIII and NiII–SCN–MoIII linkages but SMMs incorporating such units have not been reported.226At this stage of this Feature article, it is natural to wonder if the modular approach can use a single atom as a magnetic bridge to design SMMs. An obvious choice would be oxide or hydroxide based building blocks but they are not easy to employ due to the strong basicity of most paramagnetic complexes. In principle, a reasonable approach could involve complexes with metal ions in a high oxidation state, which should have less basic oxide, or, eventually, nitride complexes. Only two examples of SMMs incorporating oxide-based modules have been reported, but both involve diamagnetic ReV metal ions.229,230 Building blocks based on fluoride as the potential bridging ligand to synthesize magnetic materials are also largely unexplored.231–236 Being isoelectronic to oxide, the incorporation of fluoride is not obvious. In addition to its basicity, the oxide group has the tendency to form non-linear bridges (Fig. 2) between two or more metal ions making it less appealing as the bridging ligand. These two effects appear less pronounced for fluoride. Several mononuclear 3d metal ion fluoride complexes are known and commonly fluoride leads to linear or almost linear bridges.237 The main synthetic problem of these fluoride complexes arises from the inherent lability of many fluoride complexes. However, this issue can be overcome by using, for instance, kinetically robust CrIII fluoride complexes,232 or by enforcing robustness with selected auxiliary ligands as discussed earlier. Being a “hard” ligand, fluoride has a strong preference for “hard” metal ions such as lanthanides. Further discussions about the controlled design of 3d–4f SMM systems with fluoride-based modules will be presented in the last paragraph of this section dedicated to lanthanide and actinide based building-blocks. We recently reported (PPh4)2[ReF6]·2H2O (43) incorporating a close-to-octahedral [ReF6]2− anion to exhibit slow relaxation of the magnetization.60 This interesting module in 43 has a large zero-field splitting of D/kB = +34.0 K and |E|/kB = 3.7 K as determined from inelastic neutron scattering and high-field EPR spectroscopy.60 The strong magnetic anisotropy combined with the ability to bridge several metal centres make homoleptic fluoride-complexes, such as [ReF6]2−, interesting, but completely unexplored modules for SMMs.
Oxalate-based modules
Trisoxalatometallate(III)s, [M(ox)3]3−, have been widely employed for assembling magnetic materials.238 This interest is motivated by the strong preference of the oxalate group to bridge two metal ions in a double-chelate fashion. However, the propensity of the [M(ox)3]3− unit to form extended systems makes these precursors less suitable to design SMMs, unless sterical constraints imposed by capping ligands are introduced on the acceptor metal ions. This problem can also be overcome by turning to heteroleptic oxalate systems with an appropriate choice of capping ligands. For instance, an interesting ReIV module, [ReIVCl4(ox)]2−, has been reported.239 The d3-configuration of the ReIV ion gives a kinetically robust and hydrolytically stable building block. In combination with NiII metal ions, a propeller-shaped {NiIIReIV3} complex ((NBu4)4[Ni{ReCl4(ox)}3], 44; Fig. 10) is formed.240,241 Modelling of the χT vs. T data gave a ferromagnetic NiII–ox–ReIV interaction (JRe–Ni/kB) of +12 K. It is interesting to note that the field dependence of the magnetization for the (PPh4)2[ReCl4(ox)] precursor reveals a strong magnetic anisotropy that has been estimated at D/kB ≈ 86 K.239 Notably, Martínez-Lillo et al. recently reported NBu4+ salts of [ReCl4(ox)]2− and [ReBr4(ox)]2−, to display SMM properties in small dc fields.242 For 44, an out-of-phase χ′′ signal was clearly visible in zero dc field, but under 2000 Oe, the spin-relaxation slows down as expected in presence of significant QTM. In a recent communication, the same authors reported a {GdIIIReIV4} four-bladed propeller ((NBu4)5[Gd{ReBr4(μ-ox)}4(H2O)]·H2O), but no slow relaxation of the magnetization was observed.243 So far, the only example of SMM behaviour in a 3d transition metal oxalate complex is {[CrIII(bpy)(ox)2]2CoII(Me2phen)}·4H2O (45; Me2phen = 2,9-dimethyl-1,10-phenanthroline) that is obtained by the reaction of the [CrIII(bpy)(ox)2]− module with CoII in the presence of the Me2phen ligand. Only an onset of χ′′(T) peaks is observed above 1.8 K, suggesting SMM properties and a small energy barrier.244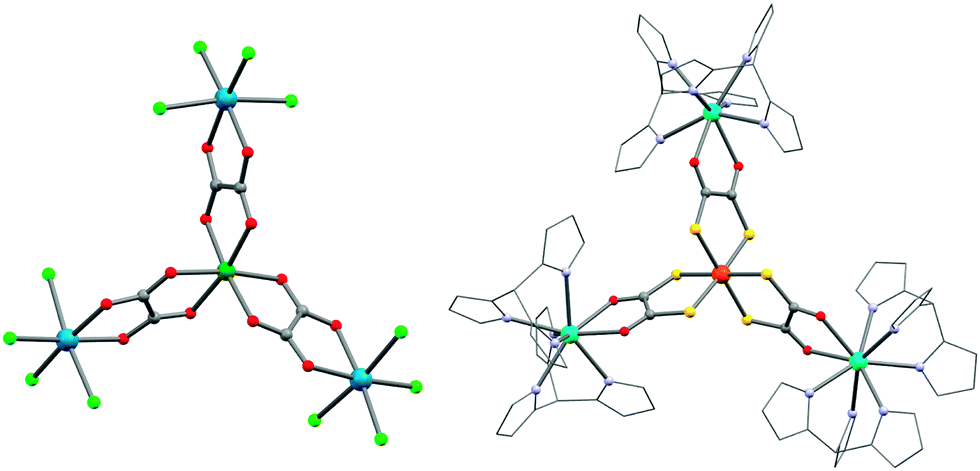 | ||
| Fig. 10 Molecular structures of 44 (left) and 56 (right). Colour codes: Dy, turquoise; Re, marine; Ni, dark green; Fe, orange; Cl, light green; S, yellow; O, red; N, light blue; C, grey. | ||
Acceptor modules
Some of the most commonly used modules to design SMMs are acceptor type units made of a MnIII ion and a Schiff base (SB) ligand (see e.g.Scheme 1j). Polynuclear complexes based on these Schiff base complexes (abbreviated {Mn(SB)}+) have been reviewed by Miyasaka et al.120 and along this Feature article we have already shown several examples of SMMs incorporating these units (Fig. 4, 5 and 7). The Schiff base ligands are often tetradentate to the MnIII site occupying equatorial positions and allowing the two remaining apical positions to be accessible for further coordination for example with a cyanide group.88,116–119,121–123 The {Mn(SB)}+ units constitute a class of very useful modules for SMM synthesis due to their relatively strong magnetic anisotropy reflected in a large negative value of D estimated up to −6 K.124,245 In solution, the mononuclear {Mn(SB)}+ unit is in equilibrium with an “out-of-plane” dinuclear form (see Scheme 1k) in which the phenolate oxygens bridge two {Mn(SB)}+ moieties along the JT axes.120 Depending on the structural parameters, the MnIII–MnIII interaction through the bis-phenolate oxygens in this dinuclear module is often ferromagnetic in nature due to an accidental orthogonality of the dz2 orbital occupied by one electron and the empty dx2–y2 orbital of the other MnIII centre. Due to this ferromagnetic interaction, that leads to an ST = 4 spin ground state, and the co-axial ZFS MnIII tensors, the [MnIII2(saltmen)2(ReO4)2] complex (46; saltmen2− = N,N′-(1,1,2,2-tetramethylene)bis(salicylideneiminate)) was found to be an SMM (Fig. 1).61 Since then, several other {Mn2(SB)2}2+ SMMs have been reported.246,247 These out-of-plane {Mn2(SB)2}2+ building blocks have also been employed extensively to obtain photomagnetic chains,248 SCMs84,87–89 and two-dimensional networks by assembly with appropriate modules.22,249–252The terminal labile positions of the {MnIII2(SB)2}2+ module can be decorated by other building blocks, for instance the S = 1/2 [WV(CN)6(bpy)]− unit yielding a {MnIII2WV2} linear complex ([W(bpy)(CN)6]2[Mn(L)]2·3H2O with L = N,N′-bis(2-hydroxyacetophenylidene)-1,2-diaminopropane, 47; Fig. 11).253 Overall, 47 is ferromagnetically coupled, JW–Mn/kB = +1.2 K and JMn–Mn/kB = +1.4 K, leading to an ST = 5 ground state. Although the energy barrier of 32 K is relatively large, the τ0 preexponential factor is small, 5.1 × 10−12 s, and thus the relaxation of the magnetization is observed using the ac technique only below 3 K.
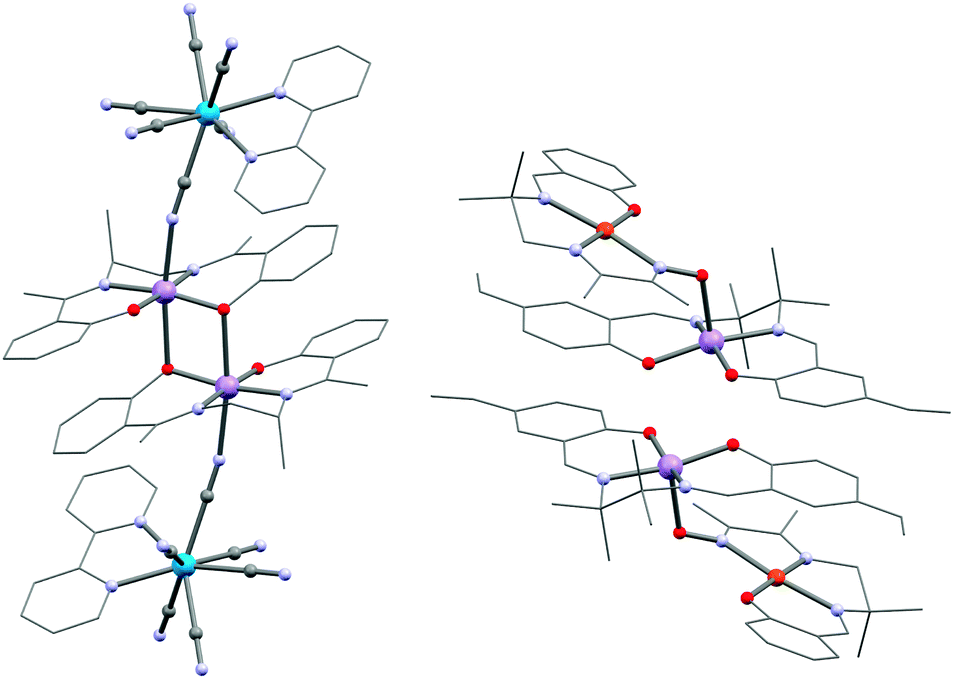 | ||
| Fig. 11 Structure of 47 (left) and 51 (right). For 51, the apical Mn⋯O separations are very long (3.081(2) Å) and concomitantly is the MnIII–MnIII interaction only weak (JMn–Mn/kB = +1.7(1) K). | ||
Other building blocks such as aldoximates have been also associated with {MnIII2(SB)2}2+ moieties as exemplified by the series of tetranuclear [MnIII(5-Rsaltmen)NiII(pao)(bpy)2]2(ClO4)4 complexes (48; pao = pyridine-2-aldoximate) where R can be H, Cl, Br or OMe.254 The relatively strong antiferromagnetic MnIII–NiII interactions (−26 K < JMn–Ni/kB < −24 K depending on the system) leads to a relatively small ST = 2 ground state for the complexes, which do not show any slow relaxation of the magnetization in zero-dc field above 1.8 K. However, two related trinuclear NiII-aldoximate SMMs, [Mn2(5-Rsaltmen)2Ni(pao)2(phen)](ClO4)2 (R = Cl (49), Br (50); phen = 1,10-phenanthroline), have been reported.255 The intra-complex antiferromagnetic interactions are also relatively large, JMn–Ni/kB ≈ −24 K, but ac susceptibility measurements reveal SMM properties with Δeff/kB ≈ 18 K and τ0 ≈ 10−7 s for both systems. It is worth mentioning that these SMMs are indeed the elementary units of the archetypal single-chain magnets: [Mn2(saltmen)2Ni(pao)2(py)2](ClO4)2, and the analogous systems.84 In [MnIII2(5-MeOsaltmen)2CuII2L2](CF3SO3)2·2H2O (51; L = 3-{2-[(2-hydroxybenzylidene)amino]2-methyl-propylimino}-butan-2-one-oximate) incorporating a central {Mn2(SB)2}2+ core decorated by two CuII-aldoximate units (Fig. 11), clear SMM properties are detected.256 Similarly, the reaction of a manganese(II) complex [MnII(5-MeOsaltmen)]·nH2O with N,N′-dicyano-1,4naphthoquinonediimine (DCNNQI) affords MnIII-radical complexes with a [MnIII2(5-MeOsaltmen)2(DCNNQI˙−)2] core (52).257 The MnIII–radical interaction is antiferromagnetic (JMn–rad/kB < −23 K) and much stronger than the ferromagnetic MnIII–MnIII interaction (JMn–Mn/kB < +2.0 K) leading to an ST = 3 ground state. The ac susceptibility measurements reveal the SMM properties of these complexes with frequency-dependent in-phase and out-of-phase components (νac ≤ 1.5 kHz, T ≥ 1.8 K) as well as a sweep rate dependence of the M vs. H hysteresis at 0.4 K.
Beside the {Mn(SB)}+ modules, similar acceptor building blocks to design SMMs are relatively rare and only a few other examples, which have already been described in the previous paragraphs, are shown in Scheme 1(l–o). Recently, mononuclear transition metal complexes with “unconventional” coordination numbers and geometries have been reported to display SMM properties due to a very strong magnetic anisotropy.40,46,258 This new category of mononuclear SMMs is currently the topic of a very competitive subject with a rapidly growing number of published systems.39–48,259 It sounds reasonable to think that in the close future some of these complexes could be employed as acceptors or, in some cases, donor modules to design new polynuclear SMMs with remarkable characteristics.
Lanthanide and actinide based building-blocks
Obtaining a topological control of coordination architectures with f-block elements is notoriously difficult due to their high coordination numbers and the lack of ligand field stabilization. Indeed the coordination geometries are mainly governed by the sterical hindrances of the (metallo-)ligands and crystal packing effects.260 In addition, even very weak ligand field perturbations may have a significantly strong influence on the SMM properties.38,261–263 Despite the obvious downsides from the viewpoint of the chemical design, lanthanide-based complexes have received an immense attention in recent years as they have served as key ingredients in several high barrier 3d–4f or pure 4f SMMs.71,72,264–266 Although the observation of SMM properties in most lanthanide complexes is inherently related to the ligand field of the isolated lanthanide ion,262,263,267,268 effects of even small ligand field perturbations and exchange interactions have shown to be of crucial importance in the observation of magnetization slow dynamics.38,269 For these reasons, the molecular design of lanthanide-based SMMs requires the ligand field of the lanthanide ions to be as preserved as possible. This is clearly not an easy task even if the use of multi-dentate chelating ligands or other particularly rigid ligands, with or without functional groups susceptible to bridge adjacent magnetic centers, might be an approach to explore. Recently, Murugesu and Long reported COT-based Er and Dy SMMs (COT = cycloocta-1,3,5,7-tetraenediide), which exhibit M vs. H hysteresis loops at temperatures of up to 10 K (for [Er(COT)2]− with a field sweep rate of 0.78 mT s−1). Indeed such rigid complexes appear to be promising modules for higher-nuclearity systems.36 Although serendipitous approaches have been use extensively to synthesize 4f metal ion based SMMs,27 lanthanide building blocks do exist and the modular strategy has also been applied successfully to a limited number of systems.270,271 A very common building block is the [Dy(hfac)3(H2O)2] complex (hfac = 1,1,1,5,5,5-hexafluoroacetylacetonate). The two coordinated water molecules are easily replaced either by azeotropic distillation or directly in the synthesis of polynuclear systems. Lanthanide-based SMM complexes are reported with coordinating nitronyl nitroxide radicals,272,273 organic linkers274,275 and transition metal complexes.276–279 For instance, [NiII(bpca)2] (Scheme 1g; Hbpca = bis(2-pyridylcarbonyl)amine) is able to coordinate bidentately to one or two lanthanide ions in an “acac-like” fashion (Fig. 12).280,281 Both [Dy(hfac)3Ni(bpca)2Dy(hfac)3]·CHCl3 (53) and the iron(II) analogue 54 incorporating low-spin diamagnetic [FeII(bpca)2], show SMM properties but the latter has a higher energy barrier (9.7 K vs. 4.9 K) despite the ferromagnetic Dy–Ni interactions in 53. This result illustrates well that a simple design of lanthanide-based SMMs is currently not easily accessible.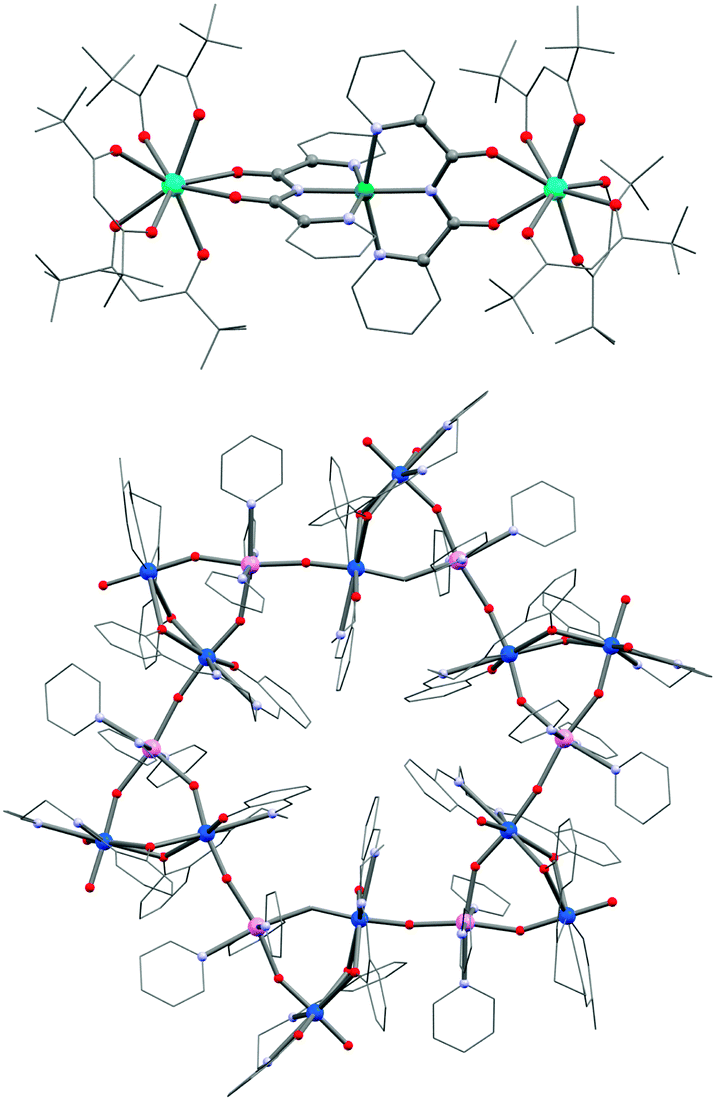 | ||
| Fig. 12 Molecular structure of 53 (top) and 59 (bottom). U, marine; Dy, pink; Ni, green; Mn, pink; Cr, green; O, red; N, light blue; C, grey. | ||
Indeed, only a very few polynuclear lanthanide complexes, which exhibit SMM properties, were obtained using a molecular building block approach. Recent examples are the {Dy3CoIII} (55) and {Dy3FeIII} (56) propellers ([MDy3Tp6(dto)3]·4CH3CN·2CH2Cl2; M = CoIII or FeIII; dto = dithiooxalate dianion) reported by Tang and co-workers.282 In these complexes, three dithiooxalate dianions bridge by the sulfur end to the “softer” CoIII or FeIII metal ions while the “hard” DyIII sites prefer to coordinate to the donor oxygens (Fig. 10).282 It is worth noting that lanthanide oxalates are extremely insoluble and only one lanthanide-based SMM featuring oxalate bridging has been reported so far ([(Tp)4Dy2(μ-ox)]·2CH3CN·CH2Cl2).283 Interestingly, the SMM barrier of 55 (52 K) is higher than for the exchange coupled complex 56 that corroborates the argumentation given by Sessoli and co-workers for 53 and 54.281 Winpenny and co-workers have elegantly used CrIII “horseshoe” modules to obtain mixed chromium(III)–lanthanide(III) complexes but none of them were reported to be SMMs.284 Bendix's group has recently demonstrated the possibility of controlling to some extent the topology of lanthanide-based complexes using fluoride bridges.232,233,236 The strong preference of fluoride to stabilize linear bridges seems to dictate the polynuclear complex arrangement.231,234 For instance, cis-difluoride, trans-difluoride and fac-trifluoride complexes can form linear rod-like, square-like and pyramidal molecular systems, respectively, which show SMM properties for some of them.230,232 As an illustration, the trinuclear fluoride-bridged SMM (57), [Dy(hfac)3(H2O)CrF2(py)4Dy(hfac)3(NO3)], is obtained from the assembly of trans-[CrF2(py)4]+ and [Dy(hfac)3(H2O)2] modules. The magnetization dynamics was too fast to be able to observe a χ′′ maximum (with νac ≤ 1.5 kHz), but muon-spin rotation spectroscopy reveals a small energy barrier of about 4.2 K (τ0 = 5.3(4) × 10−8 s). In addition, the study of 57 by X-ray magnetic circular dichroism allowed a direct estimation of the nature and magnitude of the exchange interaction.233 This information can hardly be obtained from bulk magnetic measurements and is therefore, generally, not known for lanthanide SMMs.280
Actinide based complexes are gaining an increasing attention in the SMM community due to their strong magnetic anisotropy, like lanthanide ions, but also because they offer the possibility of stronger exchange interaction due to the less localized 5f orbitals over the 4f orbitals of the lanthanides.285 Until now, actinide SMMs are known only for U and Np systems. But most of the few published studies have been dedicated to mononuclear UIII systems286–290 for which it was recently argued that the SMM properties are intrinsic to the trivalent uranium centre.291 A single example of a mononuclear NpIV system has been reported: neptunocene, [Np(COT)2] (58).292 Only two examples of exchange coupled polynuclear uranium SMMs are reported so far.293,294 The first one reported by Liddle and co-workers is an inverted-sandwich arene-bridged diuranium(III) species that shows a frequency dependent ac susceptibility signal below 5 K.293 The second example is an UV-based {MnII6UV12} wheel complex (59; [{[UO2(salen)]12Mn(py)3}6]; Fig. 12) prepared by one-electron reduction of [UVIO2(salen)] with [CoIICp*2] (Cp* = pentamethylcyclopentadienide) and subsequent assembly with MnII metal ions.294 The six-coordinate [UVO2(salen)]− unit dimerizes to form an approximate pentagonal bipyramidal local symmetry around the UV sites. This complex exhibits an M vs. H hysteresis loop below 4 K (with an 4 mT s−1 average field sweep rate) and a relaxation time that is thermally activated with a large barrier of 142(7) K and a very small τ0 = 3(2) × 10−12 s. In 2010, Magnani and co-workers reported the first neptunium SMM, {NpVIO2Cl2}{NpVO2Cl(THF)3}2 (60), exhibiting an exotic trinuclear neptunium(V,VI) core. The slow dynamics of the magnetization of this complex was studied using the ac technique leading to an 140-K energy barrier.50,295
5. Conclusions and perspectives
Combining relevant and interesting physical properties in a controlled manner in the same material is an important topic for modern chemists. In particular, the engineering of new multifunctional materials associating magnetism with e.g. photo-activity, electro-activity, porosity, conductivity or other properties controlled by an external stimuli, are being currently targeted by many research groups. Potentially, coordination chemistry provides the necessary tools for designing such new materials in rational and methodical approaches. However, the development of the preparative coordination chemistry is still behind the organic chemistry and consequently, it remains difficult for coordination chemists to design and synthesize, at will, polynuclear metal ion complexes or coordination polymers. Inspired from the protective groups in organic chemistry that direct the reactivity in particular positions, coordination chemists increase their structural control on the final material by using precursors with reduced degrees of freedom. Along this line, the used building-blocks are often carrying capping or strongly coordinating ligands to be able to direct the coordination properties and the final assemblies. Nevertheless, it is important to keep in mind that serendipitous self-assembly reactions have provided many systems of crucial importance to the development of the field of molecular magnetism and that the limitations of the modular approach with respect to structural design of polynuclear complexes are still important.By creating complicated structures in a hierarchical fashion, the preparative coordination chemist can rely on an existing understanding of the first and second coordination sphere complexations and the well-understood relative robustness of coordination complexes. Furthermore, this modular strategy offers an efficient transfer of the metal ion properties, imposed by the first coordination sphere, to extended structures. Thus, this approach allows us to control not only the spatial arrangements, but also the electronic structure of complicated systems. In this context, many combinations of metal ion modules and bridging ligands are unexplored to design new molecule-based magnetic materials including SMMs and related materials.
In parallel with the use of known building-blocks, the quest for new magnetic modules should not be forgotten as they constitute the basis of this synthetic strategy. In particular, it would be very interesting to exploit the recent examples of mononuclear SMMs and photo-switchable SMMs as modules for higher nuclearity SMM-based architectures. As evidenced in this Feature article by the current limited number of modules used to elaborate SMMs, chemists should continue to develop this step-by-step approach in concert with more serendipitous syntheses, which have led, and will also lead, to many magnetically interesting systems without clearly identified building-block precursors. However, we believe that the described modular synthetic strategy, that efficiently used the existing knowledge of coordination chemistry, offers the best chances to premeditate and control the physical properties of the resulting coordination structures.
Acknowledgements
We are grateful to all our co-workers, past students and friends who have contributed to our scientific adventures. Special thanks to Dr D. Woodruff for valuable discussions. In addition, the authors thank the Danish Ministry of Science, Innovation and Higher Education for the EliteForsk travel stipend to K.S.P., the Conseil Régional d'Aquitaine, the Université of Bordeaux, the Danish research Councils, the French Embassy in Denmark (IFD, French-Danish Research Collaboration Program), the CNRS and the ANR.Notes and references
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS PubMed.
- G. Aromi and E. K. Brechin, in Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, 2006, pp. 1–67 Search PubMed.
- G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66–71 CrossRef CAS.
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Y. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- J. Lehmann, A. Gaita-Arino, E. Coronado and D. Loss, J. Mater. Chem., 2009, 19, 1672–1677 RSC.
- J. Camarero and E. Coronado, J. Mater. Chem., 2009, 19, 1678–1684 RSC.
- D. Gatteschi, A. Cornia, M. Mannini and R. Sessoli, Inorg. Chem., 2009, 48, 3408–3419 CrossRef CAS PubMed.
- A. Cornia, M. Mannini, P. Sainctavit and R. Sessoli, Chem. Soc. Rev., 2011, 40, 3076–3091 RSC.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M. A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed.
- M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M. A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS PubMed.
- C. Schlegel, E. Burzurí, F. Luis, F. Moro, M. Manoli, E. K. Brechin, M. Murrie and J. van Slageren, Chem.–Eur. J., 2010, 16, 10178–10185 CrossRef CAS PubMed.
- R. Inglis, J. Bendix, T. Brock-Nannestad, H. Weihe, E. K. Brechin and S. Piligkos, Chem. Sci., 2010, 1, 631–636 RSC.
- A. Cornia, A. F. Costantino, L. Zobbi, A. Caneschi, D. Gatteschi, M. Mannini and R. Sessoli, in Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, Springer-Verlag Berlin, Berlin, 2006, pp. 133–161 Search PubMed.
- N. Domingo, E. Bellido and D. Ruiz-Molina, Chem. Soc. Rev., 2012, 41, 258–302 RSC.
- R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357–360 CrossRef CAS PubMed.
- M. Clemente-León, H. Soyer, E. Coronado, C. Mingotaud, C. J. Gómez-García and P. Delhaès, Angew. Chem., Int. Ed., 1998, 37, 2842–2845 CrossRef.
- T. Coradin, J. Larionova, A. A. Smith, G. Rogez, R. Clérac, C. Guérin, G. Blondin, R. E. P. Winpenny, C. Sanchez and T. Mallah, Adv. Mater., 2002, 14, 896–898 CrossRef CAS.
- C. Coulon, H. Miyasaka and R. Clérac, in Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, 2006, pp. 163–206 Search PubMed.
- I.-R. Jeon and R. Clérac, Dalton Trans., 2012, 41, 9569–9586 RSC.
- L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750–4758 RSC.
- H.-L. Sun, Z.-M. Wang and S. Gao, Coord. Chem. Rev., 2010, 254, 1081–1100 CrossRef CAS PubMed.
- W.-X. Zhang, R. Ishikawa, B. K. Breedlove and M. Yamashita, RSC Adv., 2012, 3, 3772–3798 RSC.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874–8875 CrossRef CAS PubMed.
- P.-E. Car, M. Perfetti, M. Mannini, A. Favre, A. Caneschi and R. Sessoli, Chem. Commun., 2011, 47, 3751–3753 RSC.
- S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef CAS PubMed.
- S.-D. Jiang, B.-W. Wang, G. Su, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448–7451 CrossRef CAS PubMed.
- M.-E. Boulon, G. Cucinotta, J. Luzon, C. Degl'Innocenti, M. Perfetti, K. Bernot, G. Calvez, A. Caneschi and R. Sessoli, Angew. Chem., Int. Ed., 2013, 52, 350–354 CrossRef CAS PubMed.
- J.-L. Liu, K. Yuan, J.-D. Leng, L. Ungur, W. Wernsdorfer, F.-S. Guo, L. F. Chibotaru and M.-L. Tong, Inorg. Chem., 2012, 51, 8538–8544 CrossRef CAS PubMed.
- K. R. Meihaus, J. D. Rinehart and J. R. Long, Inorg. Chem., 2011, 50, 8484–8489 CrossRef CAS PubMed.
- M. A. AlDamen, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, C. Martí-Gastaldo, F. Luis and O. Montero, Inorg. Chem., 2009, 48, 3467–3479 CrossRef CAS PubMed.
- J. J. Le Roy, M. Jeletic, S. I. Gorelsky, I. Korobkov, L. Ungur, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 3502–3510 CrossRef CAS PubMed; K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952–17957 CrossRef PubMed; J. J. Le Roy, I. Korobkov and M. Murugesu, Chem. Commun., 2014, 50, 1602–1604 RSC.
- E. Lucaccini, L. Sorace, M. Perfetti, J.-P. Costes and R. Sessoli, Chem. Commun., 2014, 50, 1648–1651 RSC.
- K. S. Pedersen, L. Ungur, M. Sigrist, A. Sundt, M. Schau-Magnussen, V. Vieru, H. Mutka, S. Rols, H. Weihe, O. Waldmann, L. Chibotaru, J. Bendix and J. Dreiser, Chem. Sci., 2014, 5, 1650–1660 RSC.
- W. H. Harman, T. D. Harris, D. E. Freedman, H. Fong, A. Chang, J. D. Rinehart, A. Ozarowski, M. T. Sougrati, F. Grandjean, G. J. Long, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 18115–18126 CrossRef CAS PubMed.
- J. M. Zadrozny, M. Atanasov, A. M. Bryan, C.-Y. Lin, B. D. Rekken, P. P. Power, F. Neese and J. R. Long, Chem. Sci., 2013, 4, 125–138 RSC.
- D. E. Freedman, W. H. Harman, T. D. Harris, G. J. Long, C. J. Chang and J. R. Long, J. Am. Chem. Soc., 2010, 132, 1224–1225 CrossRef CAS PubMed.
- P.-H. Lin, N. C. Smythe, S. I. Gorelsky, S. Maguire, N. J. Henson, I. Korobkov, B. L. Scott, J. C. Gordon, R. T. Baker and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 15806–15809 CrossRef CAS PubMed.
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS PubMed.
- S. Mossin, B. L. Tran, D. Adhikari, M. Pink, F. W. Heinemann, J. Sutter, R. K. Szilagyi, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2012, 134, 13651–13661 CrossRef CAS PubMed.
- Y.-Y. Zhu, C. Cui, Y.-Q. Zhang, J.-H. Jia, X. Guo, C. Gao, K. Qian, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Chem. Sci., 2013, 4, 1802–1806 RSC.
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577–581 CrossRef CAS PubMed.
- F. Habib, O. R. Luca, V. Vieru, M. Shiddiq, I. Korobkov, S. I. Gorelsky, M. K. Takase, L. F. Chibotaru, S. Hill, R. H. Crabtree and M. Murugesu, Angew. Chem., Int. Ed., 2013, 52, 11290–11293 CrossRef CAS PubMed.
- J. M. Zadrozny, J. Telser and J. R. Long, Polyhedron, 2013, 64, 209–217 CrossRef CAS PubMed.
- X. Feng, C. Mathonière, I.-R. Jeon, M. Rouzières, A. Ozarowski, M. L. Aubrey, M. I. Gonzalez, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2013, 135, 15880–15884 CrossRef CAS PubMed.
- C. Mathonière, H.-J. Lin, D. Siretanu, R. Clérac and J. M. Smith, J. Am. Chem. Soc., 2013, 135, 19083–19086 CrossRef PubMed.
- T. Lis, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2042–2046 CrossRef.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1–10 RSC.
- L. Thomas, F. Lionti, R. Ballou, D. Gatteschi, R. Sessoli and B. Barbara, Nature, 1996, 383, 145–147 CrossRef CAS.
- W. Wernsdorfer and R. Sessoli, Science, 1999, 284, 133–135 CrossRef CAS.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, New York, 2006 Search PubMed.
- J. M. Zadrozny, J. Liu, N. A. Piro, C. J. Chang, S. Hill and J. R. Long, Chem. Commun., 2012, 48, 3927–3929 RSC.
- J. Vallejo, I. Castro, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, G. De Munno, W. Wernsdorfer and E. Pardo, J. Am. Chem. Soc., 2012, 134, 15704–15707 CrossRef CAS PubMed.
- E. Colacio, J. Ruiz, E. Ruiz, E. Cremades, J. Krzystek, S. Carretta, J. Cano, T. Guidi, W. Wernsdorfer and E. K. Brechin, Angew. Chem., Int. Ed., 2013, 52, 9130–9134 CrossRef CAS PubMed.
- K. S. Pedersen, M. Sigrist, M. A. Sørensen, A. L. Barra, T. Weyhermueller, S. Piligkos, C. A. Thuesen, M. G. Vinum, H. Mutka, H. Weihe, R. Clérac and J. Bendix, Angew. Chem., Int. Ed., 2014, 53, 1351–1354 CrossRef CAS PubMed.
- H. Miyasaka, R. Clérac, W. Wernsdorfer, L. Lecren, C. Bonhomme, K. Sugiura and M. Yamashita, Angew. Chem., Int. Ed., 2004, 43, 2801–2805 CrossRef CAS PubMed.
- J. Villain, F. Hartman-Boutron, R. Sessoli and A. Rettori, Europhys. Lett., 1994, 27, 159–164 CrossRef CAS.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS PubMed.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clérac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926–4929 CrossRef CAS PubMed.
- O. Waldmann, A. M. Ako, H. U. Güdel and A. K. Powell, Inorg. Chem., 2008, 47, 3486–3488 CrossRef CAS PubMed.
- C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 2754–2755 CrossRef CAS PubMed.
- C. J. Milios, R. Inglis, A. Vinslava, R. Bagai, W. Wernsdorfer, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 12505–12511 CrossRef CAS PubMed.
- O. Waldmann, Inorg. Chem., 2007, 46, 10035–10037 CrossRef CAS PubMed.
- F. Neese and D. A. Pantazis, Faraday Discuss., 2011, 148, 229–238 RSC.
- E. Ruiz, J. Cirera, J. Cano, S. Alvarez, C. Loose and J. Kortus, Chem. Commun., 2008, 52–54 RSC.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- L. M. C. Beltran and J. R. Long, Acc. Chem. Res., 2005, 38, 325–334 CrossRef CAS PubMed.
- F. Allen, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2002, 58, 380–388 CrossRef PubMed.
- M. Shatruk, C. Avendano and K. R. Dunbar, in Prog. Inorg. Chem., ed. K. D. Karlin, 2009, vol. 56, pp. 155–334 Search PubMed.
- J. L. Heinrich, P. A. Berseth and J. R. Long, Chem. Commun., 1998, 1231–1232 RSC.
- M. Affronte, S. Carretta, G. A. Timco and R. E. P. Winpenny, Chem. Commun., 2007, 1789–1797 RSC.
- R. E. P. Winpenny, Adv. Inorg. Chem., 2001, 52, 1–111 CrossRef CAS.
- G. Aromi, S. M. J. Aubin, M. A. Bolcar, G. Christou, H. J. Eppley, K. Folting, D. N. Hendrickson, J. C. Huffman, R. C. Squire, H. L. Tsai, S. Wang and M. W. Wemple, Polyhedron, 1998, 17, 3005–3020 CrossRef CAS.
- P. Zhang, Y.-N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef CAS PubMed.
- G. E. Kostakis, A. M. Ako and A. K. Powell, Chem. Soc. Rev., 2010, 39, 2238–2271 RSC.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760–1763 CrossRef CAS.
- R. Clérac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837–12844 CrossRef PubMed.
- H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420–3437 CrossRef CAS PubMed.
- H. Miyasaka and R. Clérac, Bull. Chem. Soc. Jpn., 2005, 78, 1725–1748 CrossRef CAS.
- H. Miyasaka, R. Clérac, K. Mizushima, K. Sugiura, M. Yamashita, W. Wernsdorfer and C. Coulon, Inorg. Chem., 2003, 42, 8203–8213 CrossRef CAS PubMed.
- M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K. Sugiura, M. Yamashita, C. Coulon and R. Clérac, J. Am. Chem. Soc., 2005, 127, 3090–3099 CrossRef CAS PubMed.
- H. Miyasaka, A. Saitoh, M. Yamashita and R. Clérac, Dalton Trans., 2008, 2422–2427 RSC.
- W. R. Entley and G. S. Girolami, Science, 1995, 268, 397–400 CAS.
- M. Verdaguer, A. Bleuzen, V. Marvaud, J. Vaissermann, M. Seuleiman, C. Desplanches, A. Scuiller, C. Train, R. Garde, G. Gelly, C. Lomenech, I. Rosenman, P. Veillet, C. Cartier and F. Villain, Coord. Chem. Rev., 1999, 192, 1023–1047 CrossRef.
- J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563–570 CrossRef CAS PubMed.
- H. Weihe and H. U. Güdel, Comments Inorg. Chem., 2000, 22, 75–103 CrossRef CAS.
- D. Visinescu, C. Desplanches, I. Imaz, V. Bahers, R. Pradhan, F. A. Villamena, P. Guionneau and J. P. Sutter, J. Am. Chem. Soc., 2006, 128, 10202–10212 CrossRef CAS PubMed.
- D. E. Freedman, D. M. Jenkins and J. R. Long, Chem. Commun., 2009, 4829–4831 RSC.
- T. D. Harris, C. Coulon, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2011, 133, 123–130 CrossRef CAS PubMed.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CAS.
- F. Tuyeras, A. Scuiller, C. Duhayon, M. Hernandez-Molina, F. F. de Biani, M. Verdaguer, T. Mallah, W. Wernsdorfer and V. Marvaud, Inorg. Chim. Acta, 2008, 361, 3505–3518 CrossRef CAS PubMed.
- V. Marvaud, C. Decroix, A. Scuiller, C. Guyard-Duhayon, J. Vaissermann, F. Gonnet and M. Verdaguer, Chem.–Eur. J., 2003, 9, 1677–1691 CrossRef CAS PubMed.
- J. N. Rebilly and T. Mallah, in Single-Molecule Magnets and Related Phenomena, ed. R. Winpenny, 2006, pp. 103–131 Search PubMed.
- A. G. Sharpe, The Chemistry of Cyano Complexes of the Transition Metals, Academic Press, New York, 1976 Search PubMed.
- W. R. Entley, C. R. Treadway, S. R. Wilson and G. S. Girolami, J. Am. Chem. Soc., 1997, 119, 6251–6258 CrossRef CAS.
- K. J. Nelson, I. D. Giles, S. A. Troff, A. M. Arif and J. S. Miller, Inorg. Chem., 2006, 45, 8922–8929 CrossRef CAS PubMed.
- L. G. Beauvais and J. R. Long, J. Am. Chem. Soc., 2002, 124, 2110–2111 CrossRef CAS PubMed.
- J. Bendix, P. Steenberg and I. Søtofte, Inorg. Chem., 2003, 42, 4510–4512 CrossRef CAS PubMed.
- P. Albores, L. D. Slep, L. M. Baraldo, R. Baggio, M. T. Garland and E. Rentschler, Inorg. Chem., 2006, 45, 2361–2363 CrossRef CAS PubMed.
- T. L. Pappenhagen and D. W. Margerum, J. Am. Chem. Soc., 1985, 107, 4576–4577 CrossRef CAS.
- J. B. Goodenough, Magnetism and the Chemical Bond, Interscience, New York, 1963 Search PubMed.
- T. Mallah, C. Auberger, M. Verdaguer and P. Veillet, J. Chem. Soc., Chem. Commun., 1995, 61–62 RSC.
- C. P. Berlinguette, D. Vaughn, C. Canada-Vilalta, J. R. Galan-Mascaros and K. R. Dunbar, Angew. Chem., Int. Ed., 2003, 42, 1523–1526 CrossRef CAS PubMed.
- K. E. Funck, M. G. Hilfiger, C. P. Berlinguette, M. Shatruk, W. Wernsdorfer and K. R. Dunbar, Inorg. Chem., 2009, 48, 3438–3452 CrossRef CAS PubMed.
- A. V. Palii, S. M. Ostrovsky, S. I. Klokishner, B. S. Tsukerblat, C. P. Berlinguette, K. R. Dunbar and J. R. Galan-Mascaros, J. Am. Chem. Soc., 2004, 126, 16860–16867 CrossRef CAS PubMed.
- A. Palii, S. M. Ostrovsky, S. I. Klokishner, B. S. Tsukerblat and K. R. Dunbar, ChemPhysChem, 2006, 7, 871–879 CrossRef CAS PubMed.
- A. Palii, B. Tsukerblat, S. Klokishner, K. R. Dunbar, J. M. Clemente-Juan and E. Coronado, Chem. Soc. Rev., 2011, 40, 3130–3156 RSC.
- M. G. Hilfiger, M. Chen, T. V. Brinzari, T. M. Nocera, M. Shatruk, D. T. Petasis, J. L. Musfeldt, C. Achim and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 1410–1413 CrossRef CAS PubMed.
- P. L. W. Tregenna-Piggott, D. Sheptyakov, L. Keller, S. I. Klokishner, S. M. Ostrovsky, A. V. Palii, O. S. Reu, J. Bendix, T. Brock-Nannestad, K. Pedersen, H. Weihe and H. Mutka, Inorg. Chem., 2009, 48, 128–137 CrossRef CAS PubMed.
- H. Miyasaka, H. Ieda, N. Matsumoto, N. Re, R. Crescenzi and C. Floriani, Inorg. Chem., 1998, 37, 255–263 CrossRef CAS.
- H. J. Choi, J. J. Sokol and J. R. Long, Inorg. Chem., 2004, 43, 1606–1608 CrossRef CAS PubMed.
- H. J. Choi, J. J. Sokol and J. R. Long, J. Phys. Chem. Solids, 2004, 65, 839–844 CrossRef CAS PubMed.
- H. Miyasaka, A. Saitoh and S. Abe, Coord. Chem. Rev., 2007, 251, 2622–2664 CrossRef CAS PubMed.
- J. Dreiser, A. Schnegg, K. Holldack, K. S. Pedersen, M. Schau-Magnussen, J. Nehrkorn, P. Tregenna-Piggott, H. Mutka, H. Weihe, J. Bendix and O. Waldmann, Chem.–Eur. J., 2011, 17, 7492–7498 CrossRef CAS PubMed.
- K. S. Pedersen, M. Schau-Magnussen, J. Bendix, H. Weihe, A. V. Palii, S. I. Klokishner, S. Ostrovsky, O. S. Reu, H. Mutka and P. L. W. Tregenna-Piggott, Chem.–Eur. J., 2010, 16, 13458–13464 CrossRef CAS PubMed.
- K. S. Pedersen, J. Dreiser, J. Nehrkorn, M. Gysler, M. Schau-Magnussen, A. Schnegg, K. Holldack, R. Bittl, S. Piligkos, H. Weihe, P. Tregenna-Piggott, O. Waldmann and J. Bendix, Chem. Commun., 2011, 47, 6918–6920 RSC.
- K. S. Pedersen, M. Sigrist, H. Weihe, P. L. W. Tregenna-Piggott, M. Schau-Magnussen, J. Dreiser, H. Mutka, A.-L. Barra and J. Bendix, Inorg. Chem. Commun., 2012, 24, 24–28 CrossRef CAS PubMed.
- A. Palii, B. Tsukerblat, J. M. Clemente-Juan and E. Coronado, Int. Rev. Phys. Chem., 2010, 29, 135–230 CrossRef CAS.
- V. S. Mironov, Dokl. Phys. Chem., 2006, 408, 130–136 CrossRef CAS.
- A. V. Palii, O. S. Reu, S. M. Ostrovsky, S. I. Klokishner, B. S. Tsukerblat, M. Hilfiger, M. Shatruk, A. Prosvirin and K. R. Dunbar, J. Phys. Chem. A, 2009, 113, 6886–6890 CrossRef CAS PubMed.
- J. S. Griffith, The Theory of Transition Metal Ions, Cambridge University Press, Cambridge, 1961 Search PubMed.
- J. Dreiser, K. S. Pedersen, A. Schnegg, K. Holldack, J. Nehrkorn, M. Sigrist, P. Tregenna-Piggott, H. Mutka, H. Weihe, V. S. Mironov, J. Bendix and O. Waldmann, Chem.–Eur. J., 2013, 19, 3693–3701 CrossRef CAS PubMed.
- J. Larionova, B. Mombelli, J. Sanchiz and O. Kahn, Inorg. Chem., 1998, 37, 679–684 CrossRef CAS.
- J. Bendix, M. Brorson and C. E. Schäffer, Inorg. Chem., 1993, 32, 2838–2849 CrossRef CAS.
- M. Atanasov, C. Busche, P. Comba, F. El Hallak, B. Martin, G. Rajaraman, J. van Slageren and H. Wadepohl, Inorg. Chem., 2008, 47, 8112–8125 CrossRef CAS PubMed.
- X.-Y. Wang, C. Avendano and K. R. Dunbar, Chem. Soc. Rev., 2011, 40, 3213–3238 RSC.
- H. Miyasaka, H. Takahashi, T. Madanbashi, K. Sugiura, R. Clérac and H. Nojiri, Inorg. Chem., 2005, 44, 5969–5971 CrossRef CAS PubMed.
- Y.-Z. Zhang, B.-W. Wang, O. Sato and S. Gao, Chem. Commun., 2010, 46, 6959–6961 RSC.
- T. Glaser, M. Heidemeier, T. Weyhermüller, R.-D. Hoffmann, H. Rupp and P. Müller, Angew. Chem., Int. Ed., 2006, 45, 6033–6037 CrossRef CAS PubMed.
- T. Glaser, Chem. Commun., 2011, 47, 116–130 RSC.
- V. Hoeke, E. Krickemeyer, M. Heidemeier, H. Theil, A. Stammler, H. Bögge, T. Weyhermüller, J. Schnack and T. Glaser, Eur. J. Inorg. Chem., 2013, 4398–4409 CrossRef CAS.
- V. Hoeke, A. Stammler, H. Bögge, J. Schnack and T. Glaser, Inorg. Chem., 2013, 53, 257–268 CrossRef PubMed.
- V. Hoeke, K. Gieb, P. Müller, L. Ungur, L. Chibotaru, M. Heidemeier, E. Krickemeyer, A. Stammler, H. Bögge, C. Schröder, J. Schnack and T. Glaser, Chem. Sci., 2012, 3, 2868–2882 RSC.
- V. Hoeke, M. Heidemeier, E. Krickemeyer, A. Stammler, H. Bögge, J. Schnack and T. Glaser, Dalton Trans., 2012, 41, 12942–12959 RSC.
- D. E. Freedman, D. M. Jenkins, A. T. Iavarone and J. R. Long, J. Am. Chem. Soc., 2008, 130, 2884–2885 CrossRef CAS PubMed.
- J. M. Zadrozny, D. E. Freedman, D. M. Jenkins, T. D. Harris, A. T. Iavarone, C. Mathonière, R. Clérac and J. R. Long, Inorg. Chem., 2010, 49, 8886–8896 CrossRef CAS PubMed.
- M. V. Bennett and J. R. Long, J. Am. Chem. Soc., 2003, 125, 2394–2395 CrossRef CAS PubMed.
- V. S. Mironov, L. F. Chibotaru and A. Ceulemans, J. Am. Chem. Soc., 2003, 125, 9750–9760 CrossRef CAS PubMed.
- X.-Y. Wang, A. V. Prosvirin and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 5081–5084 CrossRef CAS PubMed.
- K. Qian, X.-C. Huang, C. Zhou, X.-Z. You, X.-Y. Wang and K. R. Dunbar, J. Am. Chem. Soc., 2013, 135, 13302–13305 CrossRef CAS PubMed.
- P. Przychodzeń, T. Korzeniak, R. Podgajny and B. Sieklucka, Coord. Chem. Rev., 2006, 250, 2234–2260 CrossRef PubMed.
- B. Nowicka, T. Korzeniak, O. Stefańczyk, D. Pinkowicz, S. Chorży, R. Podgajny and B. Sieklucka, Coord. Chem. Rev., 2012, 256, 1946–1971 CrossRef CAS PubMed.
- B. Sieklucka, R. Podgajny, P. Przychodzeń and T. Korzeniak, Coord. Chem. Rev., 2005, 249, 2203–2221 CrossRef CAS PubMed.
- B. Sieklucka, R. Podgajny, D. Pinkowicz, B. Nowicka, T. Korzeniak, M. Balanda, T. Wasiutynski, R. Pelka, M. Makarewicz, M. Czapla, M. Rams, B. Gawel and W. Lasocha, CrystEngComm, 2009, 11, 2032–2039 RSC.
- M. G. Hilfiger, H. Zhao, A. Prosvirin, W. Wernsdorfer and K. R. Dunbar, Dalton Trans., 2009, 5155–5163 RSC.
- J. H. Lim, H. S. Yoo, J. H. Yoon, E. K. Koh, H. C. Kim and C. S. Hong, Polyhedron, 2008, 27, 299–303 CrossRef CAS PubMed.
- J. H. Lim, J. H. Yoon, H. C. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2006, 45, 7424–7426 CrossRef CAS PubMed.
- D. E. Freedman, M. V. Bennett and J. R. Long, Dalton Trans., 2006, 2829–2834 RSC.
- J.-P. Sutter, S. Dhers, J.-P. Costes and C. Duhayon, C. R. Chim., 2008, 11, 1200–1206 CrossRef CAS PubMed.
- J. Long, L.-M. Chamoreau and V. Marvaud, Dalton Trans., 2010, 39, 2188–2190 RSC.
- M. Kozieł, R. Pełka, M. Rams, W. Nitek and B. Sieklucka, Inorg. Chem., 2010, 49, 4268–4277 CrossRef PubMed.
- S. Dhers, S. Sahoo, J.-P. Costes, C. Duhayon, S. Ramasesha and J.-P. Sutter, CrystEngComm, 2009, 11, 2078–2083 RSC.
- J.-P. Sutter, S. Dhers, R. Rajamani, S. Ramasesha, J.-P. Costes, C. Duhayon and L. Vendier, Inorg. Chem., 2009, 48, 5820–5828 CrossRef CAS PubMed.
- D. Visinescu, I.-R. Jeon, A. M. Madalan, M.-G. Alexandru, B. Jurca, C. Mathonière, R. Clérac and M. Andruh, Dalton Trans., 2012, 41, 13578–13581 RSC.
- J.-P. Costes, F. Dahan, A. Dupuis and J.-P. Laurent, Inorg. Chem., 1996, 35, 2400–2402 CrossRef CAS.
- M. Andruh, Chem. Commun., 2011, 47, 3025–3042 RSC.
- S. Wang, X. H. Ding, Y. H. Li and W. Huang, Coord. Chem. Rev., 2012, 256, 439–464 CrossRef CAS PubMed.
- S. Wang, X. H. Ding, J. L. Zuo, X. Z. You and W. Huang, Coord. Chem. Rev., 2011, 255, 1713–1732 CrossRef CAS PubMed.
- J. J. Sokol, A. G. Hee and J. R. Long, J. Am. Chem. Soc., 2002, 124, 7656–7657 CrossRef CAS PubMed.
- J. L. Heinrich, J. J. Sokol, A. G. Hee and J. R. Long, J. Solid State Chem., 2001, 159, 293–301 CrossRef CAS.
- B. N. Figgis, Trans. Faraday Soc., 1960, 56, 1553–1558 RSC.
- T. D. Harris, M. V. Bennett, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2010, 132, 3980–3988 CrossRef CAS PubMed.
- X. W. Feng, J. J. Liu, T. D. Harris, S. Hill and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7521–7529 CrossRef CAS PubMed.
- X. Feng, T. David Harris and J. R. Long, Chem. Sci., 2011, 2, 1688–1694 RSC.
- T. D. Harris, H. S. Soo, C. J. Chang and J. R. Long, Inorg. Chim. Acta, 2011, 369, 91–96 CrossRef CAS PubMed.
- L. M. Toma, L. D. Toma, F. S. Delgado, C. Ruiz-Perez, J. Sletten, J. Cano, J. M. Clemente-Juan, F. Lloret and M. Julve, Coord. Chem. Rev., 2006, 250, 2176–2193 CrossRef CAS PubMed.
- W. F. Yeung, P. H. Lau, T. C. Lau, H. Y. Wei, H. L. Sun, S. Gao, Z. D. Chen and W. T. Wong, Inorg. Chem., 2005, 44, 6579–6590 CrossRef CAS PubMed.
- J. F. Guo, W. F. Yeung, P. H. Lau, X. T. Wang, S. Gao, W. T. Wong, S. S. Y. Chui, C. M. Che, W. Y. Wong and T. C. Lau, Inorg. Chem., 2010, 49, 1607–1614 CrossRef CAS PubMed.
- J. Xiang, L. H. Jia, W. L. Man, K. Qian, S. M. Yiu, G. H. Lee, S. M. Peng, S. Gao and T. C. Lau, Chem. Commun., 2011, 47, 8694–8696 RSC.
- Z. H. Ni, H. Z. Kou, L. F. Zhang, C. H. Ge, A. L. Cui, R. J. Wang, Y. D. Li and O. Sato, Angew. Chem., Int. Ed., 2005, 44, 7742–7745 CrossRef CAS PubMed.
- Z. H. Ni, L. F. Zhang, V. Tangoulis, W. Wernsdorfer, A. L. Cui, O. Sato and H. Z. Kou, Inorg. Chem., 2007, 46, 6029–6037 CrossRef CAS PubMed.
- C. F. Wang, J. L. Zuo, B. M. Bartlett, Y. Song, J. R. Long and X. Z. You, J. Am. Chem. Soc., 2006, 128, 7162–7163 CrossRef CAS PubMed.
- S. Wang, J. L. Zuo, H. C. Zhou, H. J. Choi, Y. X. Ke, J. R. Long and X. Z. You, Angew. Chem., Int. Ed., 2004, 43, 5940–5943 CrossRef CAS PubMed.
- D. F. Li, S. Parkin, G. B. Wang, G. T. Yee, A. V. Prosvirin and S. M. Holmes, Inorg. Chem., 2005, 44, 4903–4905 CrossRef CAS PubMed.
- C. F. Wang, W. Liu, Y. Song, X. H. Zhou, J. L. Zuo and X. Z. You, Eur. J. Inorg. Chem., 2008, 717–727 CrossRef.
- D. F. Li, R. Clérac, S. Parkin, G. B. Wang, G. T. Yee and S. M. Holmes, Inorg. Chem., 2006, 45, 5251–5253 CrossRef CAS PubMed.
- D. Y. Wu, Y. J. Zhang, W. Huang and O. Sato, Dalton Trans., 2010, 39, 5500–5503 RSC.
- Y. Zhang, U. P. Mallik, N. Rath, G. T. Yee, R. Clérac and S. M. Holmes, Chem. Commun., 2010, 46, 4953–4955 RSC.
- Y. Z. Zhang, U. P. Mallik, R. Clérac, N. P. Rath and S. M. Holmes, Chem. Commun., 2011, 47, 7194–7196 RSC.
- Y. Z. Zhang, U. P. Mallik, N. P. Rath, R. Clérac and S. M. Holmes, Inorg. Chem., 2011, 50, 10537–10539 CrossRef CAS PubMed.
- B. M. Bartlett, T. D. Harris, M. W. DeGroot and J. R. Long, Z. Anorg. Allg. Chem., 2007, 633, 2380–2385 CrossRef CAS.
- E. J. Schelter, A. V. Prosvirin and K. R. Dunbar, J. Am. Chem. Soc., 2004, 126, 15004–15005 CrossRef CAS PubMed.
- A. V. Palii, S. M. Ostrovsky, S. I. Klokishner, B. S. Tsukerblat, E. J. Schelter, A. Prosvirin and K. R. Dunbar, Inorg. Chim. Acta, 2007, 360, 3915–3924 CrossRef CAS PubMed.
- F. Karadas, C. Avendano, M. G. Hilfiger, A. V. Prosvirin and K. R. Dunbar, Dalton Trans., 2010, 39, 4968–4977 RSC.
- E. J. Schelter, F. Karadas, C. Avendano, A. V. Prosvirin, W. Wernsdorfer and K. R. Dunbar, J. Am. Chem. Soc., 2007, 129, 8139–8149 CrossRef CAS PubMed.
- K. R. Dunbar, E. J. Schelter, A. V. Palii, S. M. Ostrovsky, V. Y. Mirovitskii, J. M. Hudson, M. A. Omary, S. I. Klokishner and B. S. Tsukerblat, J. Phys. Chem. A, 2003, 107, 11102–11111 CrossRef CAS.
- C. G. F. von Richthofen, A. Stammler, H. Bogge, M. W. DeGroot, J. R. Long and T. Glaser, Inorg. Chem., 2009, 48, 10165–10176 CrossRef CAS PubMed.
- P. J. Ferko and S. M. Holmes, Curr. Inorg. Chem., 2013, 3, 172–193 CrossRef CAS.
- D. F. Li, S. Parkin, G. B. Wang, G. T. Yee, R. Clérac, W. Wernsdorfer and S. M. Holmes, J. Am. Chem. Soc., 2006, 128, 4214–4215 CrossRef CAS PubMed.
- Z. G. Gu, W. Liu, Q. F. Yang, X. H. Zhou, J. L. Zuo and X. Z. You, Inorg. Chem., 2007, 46, 3236–3244 CrossRef CAS PubMed.
- D. Li, S. Parkin, R. Clérac and S. M. Holmes, Inorg. Chem., 2006, 45, 7569–7571 CrossRef CAS PubMed.
- K. Mitsumoto, H. Nishikawa, G. N. Newton and H. Oshio, Dalton Trans., 2012, 41, 13601–13608 RSC.
- J. Y. Yang, M. P. Shores, J. J. Sokol and J. R. Long, Inorg. Chem., 2003, 42, 1403–1419 CrossRef CAS PubMed.
- D. Li, R. Clérac, O. Roubeau, E. Harté, C. Mathonière, R. Le Bris and S. M. Holmes, J. Am. Chem. Soc., 2007, 130, 252–258 CrossRef PubMed.
- N. Hoshino, Y. Sekine, M. Nihei and H. Oshio, Chem. Commun., 2010, 46, 6117–6119 RSC.
- Y.-Z. Zhang, U. P. Mallik, R. Clérac, N. P. Rath and S. M. Holmes, Polyhedron, 2013, 52, 115–121 CrossRef CAS PubMed.
- Y.-H. Peng, Y.-F. Meng, L. Hu, Q.-X. Li, Y.-Z. Li, J.-L. Zuo and X.-Z. You, Inorg. Chem., 2010, 49, 1905–1912 CrossRef CAS PubMed.
- W. Liu, C.-F. Wang, Y.-Z. Li, J.-L. Zuo and X.-Z. You, Inorg. Chem., 2006, 45, 10058–10065 CrossRef CAS PubMed.
- L.-C. Kang, M.-X. Yao, X. Chen, Y.-Z. Li, Y. Song, J.-L. Zuo and X.-Z. You, Dalton Trans., 2011, 40, 2204–2212 RSC.
- D. Li, R. Clérac, G. Wang, G. T. Yee and S. M. Holmes, Eur. J. Inorg. Chem., 2007, 2007, 1341–1346 CrossRef.
- Y.-Z. Zhang, U. P. Mallik, R. Clérac, N. P. Rath and S. M. Holmes, Chem. Commun., 2011, 47, 7194–7196 RSC.
- Y. Z. Zhang, D. F. Li, R. Clérac, M. Kalisz, C. Mathonière and S. M. Holmes, Angew. Chem., Int. Ed., 2010, 49, 3752–3756 CrossRef CAS PubMed.
- D. Siretanu, D. F. Li, L. Buisson, D. M. Bassani, S. M. Holmes, C. Mathonière and R. Clérac, Chem.–Eur. J., 2011, 17, 11704–11708 CrossRef CAS PubMed.
- G. N. Newton, M. Nihei and H. Oshio, Eur. J. Inorg. Chem., 2011, 3031–3042 CrossRef CAS.
- J. Mercurol, Y. L. Li, E. Pardo, O. Risset, M. Seuleiman, H. Rousseliere, R. Lescouezec and M. Julve, Chem. Commun., 2010, 46, 8995–8997 RSC.
- A. Mondal, Y. Li, M. Seuleiman, M. Julve, L. Toupet, M. Buron-Le Cointe and R. Lescouëzec, J. Am. Chem. Soc., 2013, 135, 1653–1656 CrossRef CAS PubMed.
- M. Nihei, Y. Sekine, N. Suganami and H. Oshio, Chem. Lett., 2010, 39, 978–979 CrossRef CAS.
- M. Nihei, Y. Sekine, N. Suganami, K. Nakazawa, A. Nakao, H. Nakao, Y. Murakami and H. Oshio, J. Am. Chem. Soc., 2011, 133, 3592–3600 CrossRef CAS PubMed.
- M. Nihei, Y. Okamoto, Y. Sekine, N. Hoshino, T. Shiga, I. P.-C. Liu and H. Oshio, Angew. Chem., Int. Ed., 2012, 51, 6361–6364 CrossRef CAS PubMed.
- Z.-H. Ni, H.-Z. Kou, L.-F. Zhang, W.-W. Ni, Y.-B. Jiang, A.-L. Cui, J. Ribas and O. Sato, Inorg. Chem., 2005, 44, 9631–9633 CrossRef CAS PubMed.
- J. I. Kim, H. S. Yoo, E. K. Koh and C. S. Hong, Inorg. Chem., 2007, 46, 10461–10463 CrossRef CAS PubMed.
- J. I. Kim, J. H. Yoon, H. Y. Kwak, E. K. Koh and C. S. Hong, Eur. J. Inorg. Chem., 2008, 2008, 2756–2763 CrossRef.
- J. I. Kim, H. Y. Kwak, J. H. Yoon, D. W. Ryu, I. Y. Yoo, N. Yang, B. K. Cho, J.-G. Park, H. Lee and C. S. Hong, Inorg. Chem., 2009, 48, 2956–2966 CrossRef CAS PubMed.
- M. Atanasov, P. Comba and S. Helmle, Inorg. Chem., 2012, 51, 9357–9368 CrossRef CAS PubMed.
- A. Panja, P. Guionneau, I.-R. Jeon, S. M. Holmes, R. Clérac and C. Mathonière, Inorg. Chem., 2012, 51, 12350–12359 CrossRef CAS PubMed.
- R. Lescouezec, J. Vaissermann, L. M. Toma, R. Carrasco, F. Lloret and M. Julve, Inorg. Chem., 2004, 43, 2234–2236 CrossRef PubMed.
- T. Senapati, C. Pichón, R. Ababei, C. Mathonière and R. Clérac, Inorg. Chem., 2012, 51, 3796–3812 CrossRef CAS PubMed.
- S. A. Sulway, R. A. Layfield, F. Tuna, W. Wernsdorfer and R. E. P. Winpenny, Chem. Commun., 2012, 48, 1508–1510 RSC.
- M. Mousavi, V. Bereau, C. Desplanches, C. Duhayon and J. P. Sutter, Chem. Commun., 2010, 46, 7519–7521 RSC.
- R. González, A. Acosta, R. Chiozzone, C. Kremer, D. Armentano, G. De Munno, M. Julve, F. Lloret and J. Faus, Inorg. Chem., 2012, 51, 5737–5747 CrossRef PubMed.
- J. Xiang, L. H. Jia, W. L. Man, K. Qian, G. H. Lee, S. M. Peng, S. M. Yiu, S. Gao and T. C. Lau, Dalton Trans., 2012, 41, 5794–5798 RSC.
- F. Pointillart, K. Bernot, R. Sessoli and D. Gatteschi, Inorg. Chem., 2010, 49, 4355–4361 CrossRef CAS PubMed.
- K. S. Pedersen, J. Dreiser, M. Schau-Magnussen, C. A. Thuesen, H. Weihe and J. Bendix, Polyhedron, 2012, 46, 47–52 CrossRef CAS PubMed.
- C. A. Thuesen, K. S. Pedersen, M. Schau-Magnussen, M. Evangelisti, J. Vibenholt, S. Piligkos, H. Weihe and J. Bendix, Dalton Trans., 2012, 41, 11284–11292 RSC.
- T. Birk, K. S. Pedersen, C. A. Thuesen, T. Weyhermueller, M. Schau-Magnussen, S. Piligkos, H. Weihe, S. Mossin, M. Evangelisti and J. Bendix, Inorg. Chem., 2012, 51, 5435–5443 CrossRef CAS PubMed.
- J. Dreiser, K. S. Pedersen, C. Piamonteze, S. Rusponi, Z. Salman, M. E. Ali, M. Schau-Magnussen, C. A. Thuesen, S. Piligkos, H. Weihe, H. Mutka, O. Waldmann, P. Oppeneer, J. Bendix, F. Nolting and H. Brune, Chem. Sci., 2012, 3, 1024–1032 RSC.
- T. Birk, K. S. Pedersen, S. Piligkos, C. A. Thuesen, H. Weihe and J. Bendix, Inorg. Chem., 2011, 50, 5312–5314 CrossRef CAS PubMed.
- S. K. Singh, K. S. Pedersen, M. Sigrist, C. A. Thuesen, M. Schau-Magnussen, H. Mutka, S. Piligkos, H. Weihe, G. Rajaraman and J. Bendix, Chem. Commun., 2013, 49, 5583–5585 RSC.
- K. S. Pedersen, G. Lorusso, J. J. Morales, T. Weyhermüller, S. Piligkos, S. K. Singh, D. Larsen, M. Schau-Magnussen, G. Rajaraman, M. Evangelisti and J. Bendix, Angew. Chem., Int. Ed., 2014, 53, 2394–2397 CrossRef CAS PubMed.
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460–1499 RSC.
- H. Tamaki, Z. J. Zhong, N. Matsumoto, S. Kida, M. Koikawa, N. Achiwa, Y. Hashimoto and H. Okawa, J. Am. Chem. Soc., 1992, 114, 6974–6979 CrossRef CAS.
- R. Chiozzone, R. González, C. Kremer, G. De Munno, J. Cano, F. Lloret, M. Julve and J. Faus, Inorg. Chem., 1999, 38, 4745–4752 CrossRef CAS PubMed.
- J. Martinez-Lillo, D. Armentano, G. De Munno, W. Wernsdorfer, M. Julve, F. Lloret and J. Faus, J. Am. Chem. Soc., 2006, 128, 14218–14219 CrossRef CAS PubMed.
- J. Martinez-Lillo, D. Armentano, G. De Munno, W. Wernsdorfer, J. M. Clemente-Juan, J. Krzystek, F. Lloret, M. Julve and J. Faus, Inorg. Chem., 2009, 48, 3027–3038 CrossRef CAS PubMed.
- J. Martínez-Lillo, T. F. Mastropietro, E. Lhotel, C. Paulsen, J. Cano, G. De Munno, J. Faus, F. Lloret, M. Julve, S. Nellutla and J. Krzystek, J. Am. Chem. Soc., 2013, 135, 13737–13748 CrossRef PubMed.
- J. Martinez-Lillo, L. Canadillas-Delgado, J. Cano, F. Lloret, M. Julve and J. Faus, Chem. Commun., 2012, 48, 9242–9244 RSC.
- J. Vallejo, I. Castro, J. Ferrando-Soria, M. D. Deniz-Hernandez, C. Ruiz-Perez, F. Lloret, M. Julve, R. Ruiz-Garcia and J. Cano, Inorg. Chem., 2011, 50, 2073–2075 CrossRef CAS PubMed.
- B. J. Kennedy and K. S. Murray, Inorg. Chem., 1985, 24, 1552–1557 CrossRef CAS.
- Z. L. Lu, M. Yuan, F. Pan, S. Gao, D. Q. Zhang and D. B. Zhu, Inorg. Chem., 2006, 45, 3538–3548 CrossRef PubMed.
- L. Lecren, W. Wernsdorfer, Y. G. Li, A. Vindigni, H. Miyasaka and R. Clérac, J. Am. Chem. Soc., 2007, 129, 5045–5051 CrossRef CAS PubMed.
- R. Ababei, C. Pichon, O. Roubeau, Y.-G. Li, N. Bréfuel, L. Buisson, P. Guionneau, C. Mathonière and R. Clérac, J. Am. Chem. Soc., 2013, 135, 14840–14853 CrossRef CAS PubMed.
- R. Ababei, Y. G. Li, O. Roubeau, M. Kalisz, N. Bréfuel, C. Coulon, E. Harté, X. T. Liu, C. Mathonière and R. Clérac, New J. Chem., 2009, 33, 1237–1248 RSC.
- H. Miyasaka, N. Matsumoto, H. Okawa, N. Re, E. Gallo and C. Floriani, J. Am. Chem. Soc., 1996, 118, 981–994 CrossRef CAS.
- W.-W. Ni, Z.-H. Ni, A.-L. Cui, X. Liang and H.-Z. Kou, Inorg. Chem., 2007, 46, 22–33 CrossRef CAS PubMed.
- X. T. Liu, O. Roubeau and R. Clérac, C. R. Chim., 2008, 11, 1182–1191 CrossRef CAS PubMed.
- J. H. Yoon, J. H. Lim, H. C. Kim and C. S. Hong, Inorg. Chem., 2006, 45, 9613–9615 CrossRef CAS PubMed.
- H. Miyasaka, T. Nezu, K. Sugimoto, K.-i. Sugiura, M. Yamashita and R. Clérac, Inorg. Chem., 2004, 43, 5486–5488 CrossRef CAS PubMed.
- H. Miyasaka, T. Nezu, K. Sugimoto, K. Sugiura, M. Yamashita and R. Clérac, Chem.–Eur. J., 2005, 11, 1592–1602 CrossRef CAS PubMed.
- C. Kachi-Terajima, H. Miyasaka, A. Saitoh, N. Shirakawa, M. Yamashita and R. Clérac, Inorg. Chem., 2007, 46, 5861–5872 CrossRef CAS PubMed.
- C. Kachi-Terajima, H. Miyasaka, K.-i. Sugiura, R. Clérac and H. Nojiri, Inorg. Chem., 2006, 45, 4381–4390 CrossRef CAS PubMed.
- J. Cirera, E. Ruiz, S. Alvarez, F. Neese and J. Kortus, Chem.–Eur. J., 2009, 15, 4078–4087 CrossRef CAS PubMed.
- Y.-Y. Zhu, C. Cui, Y.-Q. Zhang, J.-H. Jia, X. Guo, C. Gao, K. Qian, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Chem. Sci., 2013, 4, 1802–1806 RSC.
- R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS PubMed.
- G. Cucinotta, M. Perfetti, J. Luzon, M. Etienne, P. E. Car, A. Caneschi, G. Calvez, K. Bernot and R. Sessoli, Angew. Chem., Int. Ed., 2012, 51, 1606–1610 CrossRef CAS PubMed.
- N. F. Chilton, D. Collison, E. J. McInnes, R. E. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 Search PubMed.
- D. Aravena and E. Ruiz, Inorg. Chem., 2013, 52, 13770–13778 CrossRef CAS PubMed.
- M. Holynska, D. Premuzic, I. R. Jeon, W. Wernsdorfer, R. Clérac and S. Dehnen, Chem.–Eur. J., 2011, 17, 9605–9610 CrossRef CAS PubMed.
- R. J. Blagg, C. A. Muryn, E. J. L. McInnes, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530–6533 CrossRef CAS PubMed.
- R. J. Blagg, F. Tuna, E. J. L. McInnes and R. E. P. Winpenny, Chem. Commun., 2011, 47, 10587–10589 RSC.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- J. Luzon and R. Sessoli, Dalton Trans., 2012, 41, 13556–13567 RSC.
- F. Habib, P. H. Lin, J. Long, I. Korobkov, W. Wernsdorfer and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 8830–8833 CrossRef CAS PubMed.
- A. M. Madalan, N. Avarvari, M. Fourmigue, R. Clérac, L. F. Chibotaru, S. Clima and M. Andruh, Inorg. Chem., 2008, 47, 940–950 CrossRef CAS PubMed.
- M. A. Palacios, A. J. Mota, J. Ruiz, M. M. Hänninen, R. Sillanpää and E. Colacio, Inorg. Chem., 2012, 51, 7010–7012 CrossRef CAS PubMed.
- G. Poneti, K. Bernot, L. Bogani, A. Caneschi, R. Sessoli, W. Wernsdorfer and D. Gatteschi, Chem. Commun., 2007, 1807–1809 RSC.
- K. Bernot, F. Pointillart, P. Rosa, M. Etienne, R. Sessoli and D. Gatteschi, Chem. Commun., 2010, 46, 6458–6460 RSC.
- X. Yi, K. Bernot, F. Pointillart, G. Poneti, G. Calvez, C. Daiguebonne, O. Guillou and R. Sessoli, Chem.–Eur. J., 2012, 18, 11379–11387 CrossRef CAS PubMed.
- Y. Ma, G.-F. Xu, X. Yang, L.-C. Li, J. Tang, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem. Commun., 2010, 46, 8264–8266 RSC.
- F. Mori, T. Ishida and T. Nogami, Polyhedron, 2005, 24, 2588–2592 CrossRef CAS PubMed.
- F. Mori, T. Nyui, T. Ishida, T. Nogami, K.-Y. Choi and H. Nojiri, J. Am. Chem. Soc., 2006, 128, 1440–1441 CrossRef CAS PubMed.
- T. Yamaguchi, Y. Sunatsuki, H. Ishida, M. Kojima, H. Akashi, N. Re, N. Matsumoto, A. Pochaba and J. Mroziński, Inorg. Chem., 2008, 47, 5736–5745 CrossRef CAS PubMed.
- J.-P. Costes, S. Shova and W. Wernsdorfer, Dalton Trans., 2008, 1843–1849 RSC.
- M. Ferbinteanu, T. Kajiwara, K. Y. Choi, H. Nojiri, A. Nakamoto, N. Kojima, F. Cimpoesu, Y. Fujimura, S. Takaishi and M. Yamashita, J. Am. Chem. Soc., 2006, 128, 9008–9009 CrossRef CAS PubMed.
- F. Pointillart, K. Bernot, R. Sessoli and D. Gatteschi, Chem.–Eur. J., 2007, 13, 1602–1609 CrossRef PubMed.
- G.-F. Xu, P. Gamez, J. Tang, R. Clérac, Y.-N. Guo and Y. Guo, Inorg. Chem., 2012, 51, 5693–5698 CrossRef CAS PubMed.
- G.-F. Xu, Q.-L. Wang, P. Gamez, Y. Ma, R. Clérac, J. Tang, S.-P. Yan, P. Cheng and D.-Z. Liao, Chem. Commun., 2010, 46, 1506–1508 RSC.
- A. McRobbie, A. R. Sarwar, S. Yeninas, H. Nowell, M. L. Baker, D. Allan, M. Luban, C. A. Muryn, R. G. Pritchard, R. Prozorov, G. A. Timco, F. Tuna, G. F. S. Whitehead and R. E. P. Winpenny, Chem. Commun., 2011, 47, 6251–6253 RSC.
- J. D. Rinehart, T. D. Harris, S. A. Kozimor, B. M. Bartlett and J. R. Long, Inorg. Chem., 2009, 48, 3382–3395 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Dalton Trans., 2012, 41, 13572–13574 RSC.
- J. D. Rinehart and J. R. Long, J. Am. Chem. Soc., 2009, 131, 12558–12559 CrossRef CAS PubMed.
- M. A. Antunes, L. C. J. Pereira, I. C. Santos, M. Mazzanti, J. Marçalo and M. Almeida, Inorg. Chem., 2011, 50, 9915–9917 CrossRef CAS PubMed.
- J. D. Rinehart, K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7572–7573 CrossRef CAS PubMed.
- J. T. Coutinho, M. A. Antunes, L. C. J. Pereira, H. Bolvin, J. Marcalo, M. Mazzanti and M. Almeida, Dalton Trans., 2012, 41, 13568–13571 RSC.
- F. Moro, D. P. Mills, S. T. Liddle and J. van Slageren, Angew. Chem., Int. Ed., 2013, 52, 3430–3433 CrossRef CAS PubMed.
- N. Magnani, C. Apostolidis, A. Morgenstern, E. Colineau, J.-C. Griveau, H. Bolvin, O. Walter and R. Caciuffo, Angew. Chem., Int. Ed., 2011, 50, 1696–1698 CrossRef CAS PubMed.
- D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454–460 CAS.
- V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J.-C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- S. M. Cornet, L. J. L. Haller, M. J. Sarsfield, D. Collison, M. Helliwell, I. May and N. Kaltsoyannis, Chem. Commun., 2009, 917–919 RSC.
Footnotes |
| † Dedicated to Professor Marius Andruh on the occasion of his 60th birthday. |
| ‡ In this report, a magnet is defined as a system exhibiting magnetic bistability, i.e. an M vs. H hysteresis loop. |
§ Alternative Hamiltonian conventions (as a matter of personal taste) such as 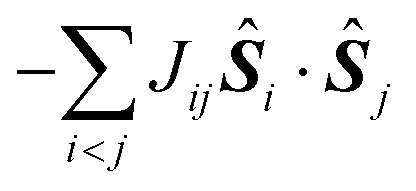 or or 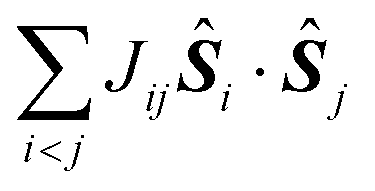 are often found in the literature and a special attention to the employed definition should be given when comparing parameter values. Throughout this Feature article, we will consistently adopt the definition given in eqn (1). are often found in the literature and a special attention to the employed definition should be given when comparing parameter values. Throughout this Feature article, we will consistently adopt the definition given in eqn (1). |
| ¶ It is worth noting that in most of the cases, τ0 is not easy to estimate accurately for a few reasons: (i) it is not always experimentally possible to obtain a clear (i.e. linear) thermally activated behavior of the relaxation time over many decades of time (that requires different experimental setups); there is often some kind of curvature in ln(τ) vs. T−1 (likely originating from additional relaxation mechanisms) if the measurements cannot reach sufficiently high temperatures; (ii) τ0 is also strongly influenced by the “bath”, i.e. the environment, in which the magnetization of the SMMs slowly relaxes. For example, weak magnetic coupling between SMMs influence τ0 as illustrated in chains of SMMs or SCM systems in which τ0 is also thermally activated and function of the intra-chain interactions; (iii) in some systems, which display a very broad spectrum of energy, multiple relaxation processes can be explored increasing the temperature and thus τ0 might change depending of the relaxation process explored. Quite generally, τ0 should be of the order of 10−10 to 10−12 s to be compatible with typical vibrations of the network that govern the reversal of magnetization. In many SMM examples, τ0 is reported to be larger and sometimes much larger than 10−10 s, suggesting that additional effects are indeed hidden in this parameter. |
| || In this context, the experimental estimation of Δeff by assuming a pure thermally activated process at the highest available temperatures may be inappropriate in some cases as recently demonstrated independently by Sorace, Dreiser and co-workers.37,38 |
| This journal is © The Royal Society of Chemistry 2014 |