C–H nitrogenation and oxygenation by ruthenium catalysis
Vedhagiri S.
Thirunavukkarasu
,
Sergei I.
Kozhushkov
and
Lutz
Ackermann
*
Institut fuer Organische und Biomolekulare Chemie, Georg-August-Universitaet, Tammannstrasse 2, 37077 Goettingen, Germany. E-mail: lutz.ackermann@chemie.uni-goettingen.de; Fax: +49-551-39-6777; Tel: +49-551-39-3202
First published on 25th October 2013
Abstract
Remarkable recent progress has been accomplished in direct C–H functionalizations for the formation of C–N and C–O bonds through the use of readily accessible ruthenium catalysts. Particularly, ruthenium(II) complexes allowed for challenging direct C(sp2)–H hydroxylation of arenes. These catalysts set the stage for step-economical C–H functionalization with electron-rich as well as electron-deficient (hetero)arenes and, therefore, provided versatile access to diversely decorated phenols. While a number of synthetically useful protocols for ruthenium-catalyzed C(sp3)–H bond nitrogenation have been elaborated, the analogous transformations of more stable C(sp2)–H bonds were very recently achieved.
 Vedhagiri S. Thirunavukkarasu | Vedhagiri S. Thirunavukkarasu was born in 1983 in Tamil Nadu, India. He studied chemistry at Presidency College in Chennai, India, where he obtained his master's degree at the University of Madras, India. In 2007, he joined the research group of Chien-Hong Cheng (National Tsing Hua University in Hsinchu, Taiwan) and obtained his PhD degree in 2011. Since 2011, he has been working in the research group of Prof. Dr Lutz Ackermann (Georg-August-Universität Göttingen, Germany) as an Alexander von Humboldt Research Fellow. |
 Sergei I. Kozhushkov | Sergei I. Kozhushkov was born in 1956 in Kharkov, USSR. He studied chemistry at Lomonosov Moscow State University, where he obtained his doctoral degree in 1983 under the supervision of Professor N. S. Zefirov and performed his “Habilitation” in 1998. From 1983 to 1991, he worked at Moscow State University and then at Zelinsky Institute of Organic Chemistry. In 1991, he joined the research group of Professor A. de Meijere (Georg-August-Universität Göttingen, Germany) as an Alexander von Humboldt Research Fellow. Since 2001 he has held a permanent position as a Senior Scientist at the Georg-August-University of Göttingen. Since 2007, he has been working in the research group of Professor L. Ackermann (Georg-August-Universität Göttingen, Germany). |
 Lutz Ackermann | Lutz Ackermann (1972) studied Chemistry at the Christian-Albrechts-University Kiel, Germany, and received his PhD from the University of Dortmund in 2001 for research under the supervision of Alois Fürstner at the Max-Plank-Institut für Kohlenforschung in Mülheim/Ruhr. He was a postdoctoral coworker in the laboratories of Robert G. Bergman at the University of California at Berkeley before initiating his independent career in 2003 at the Ludwig-Maximilians-University München supported by the Emmy Noether-programme of the DFG. In 2007, he was appointed as Full Professor at the Georg-August-University Göttingen. His recent awards and distinctions include a JSPS visiting professor fellowship (2009), an AstraZeneca Excellence in Chemistry Award (2011) and an ERC Grant (2012) as well as visiting professorships at the Universita degli Studi di Milano, Italy (2007), and the University of Wisconsin at Madison (2008). The development of novel concepts for sustainable catalysis, with a particular focus on C–H functionalizations, and their applications to organic synthesis constitute his major current research interests. |
1. Introduction
Oxygenated and nitrogenated aromatic molecules are key intermediates in organic synthesis and constitute important structural motifs of useful pharmaceuticals, agrochemicals, polymers, and biologically active compounds (Scheme 1).1 The practical importance of substituted anilines and phenols in these applied areas has resulted in a continued strong demand for versatile methods for their preparation. The direct catalytic transformation of otherwise unreactive C(sp2)–H or C(sp3)–H bonds2 into C–N or C–O bonds3 represents an environmentally benign as well as economically attractive strategy (Scheme 2). This approach compares favorably to classical protocols with respect to the overall minimization of by-product formation (atom-economy), and the optimization of the required reaction steps (step-economy).4 For instance, the direct C–H amination represents an appealing alternative to the useful palladium- or copper-catalyzed amination of organic electrophiles5 or to other indirect methods.6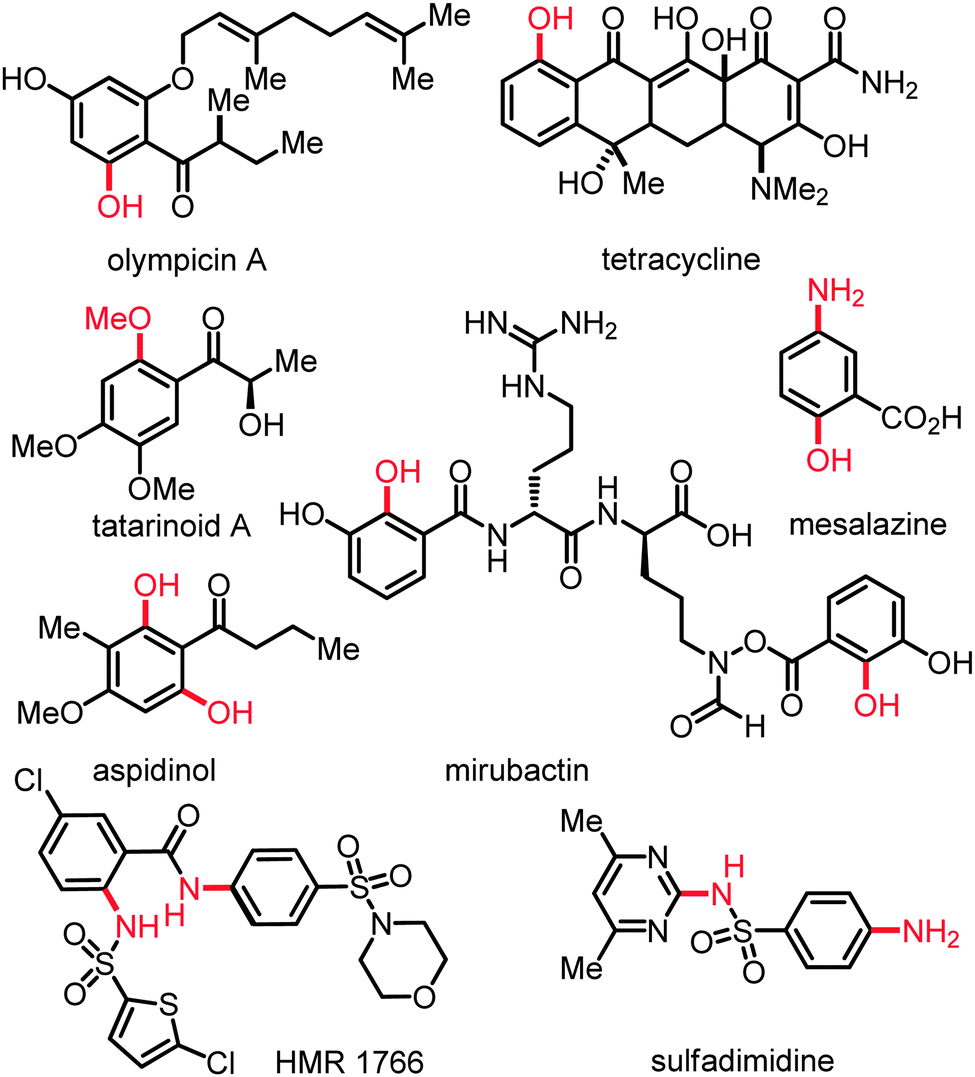 | ||
| Scheme 1 Selected bioactive compounds based on phenols or anilines. | ||
 | ||
| Scheme 2 C–H activation-based direct aniline and phenol syntheses. | ||
Since the early discoveries reported by Breslow and Gellman7a,b as well as Fujiwara,7c a plethora of synthetically useful protocols for catalyzed direct nitrogenation and oxygenation8 of alkanes and arenes has been devised, mostly using palladium,9,10 rhodium,11 copper12 or iron13 catalysts. In stark contrast, readily available ruthenium complexes14 have until recently been underdeveloped as catalysts for C–H bond nitrogenation and oxygenation. This feature article summarizes the recent rapid development of ruthenium-catalyzed chelation-assisted direct C(sp3)–H and C(sp2)–H bond nitrogenation and oxygenation up to autumn 2013, with a particular focus on the recent progress.
2. Direct C–H nitrogenation
Significant advances in the development of ruthenium-catalyzed C–H bond nitrogenation were achieved over the past two decades by Che, Du Bois and Blakey as well as Cenini, Gallo and Ragaini among others.15–18 Amidation of C(sp3)–H bonds with ruthenium catalysts has been studied extensively and developed to a level of efficiency that proved suitable for its application to complex molecule syntheses. In contrast, only a few selected reports on ruthenium-catalyzed amination and amidation of more stable C(sp2)–H bonds are available as of yet.2.1 Amidation of C(sp3)–H bonds
Several elegant protocols for the ruthenium-catalyzed intermolecular amidation of C(sp3)–H bonds have been established by Che and co-workers.15 Thus, amidation of aliphatic, benzylic and allylic C–H bonds proved to be viable with iminoiodinane RSO2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) IPh as the nitrene source, which could be conveniently generated in situ from RSO2NH2 and (diacetoxyiodo)benzene, PhI(OAc)2. These reactions were conducted with both achiral and chiral ruthenium porphyrin complexes (Scheme 3). In the latter cases, only moderate to low yields and enantioselectivities were obtained.
IPh as the nitrene source, which could be conveniently generated in situ from RSO2NH2 and (diacetoxyiodo)benzene, PhI(OAc)2. These reactions were conducted with both achiral and chiral ruthenium porphyrin complexes (Scheme 3). In the latter cases, only moderate to low yields and enantioselectivities were obtained.
 | ||
| Scheme 3 Intermolecular amidation of aliphatic and benzylic C(sp3)–H bonds; conditions: cat. [Ru], CH2Cl2, 40 °C, 2 h. | ||
The entropically favored intramolecular amidation of C(sp3)–H bonds is characterized by improved efficacies, as well as excellent diastereo- and remarkable enantioselectivities. Several efficient protocols were elaborated for the catalytic amidation of benzylic and allylic C–H bonds in substrates 1 to form five- and six-membered heterocycles 2. Indeed, the rational design of different types of complexes highlighted different ruthenium catalysts to be suitable in the presence of weak bases or molecular sieves, such as ruthenium porphyrins,16 cationic ruthenium(II)-pybox catalysts, as for instance complex 3,17 or the mixed-valent paddlewheel ruthenium complexes tetrakis(2-oxypyridinato)diruthenium(II,III) chloride, [Ru2(hp)4Cl]18 (Scheme 4).
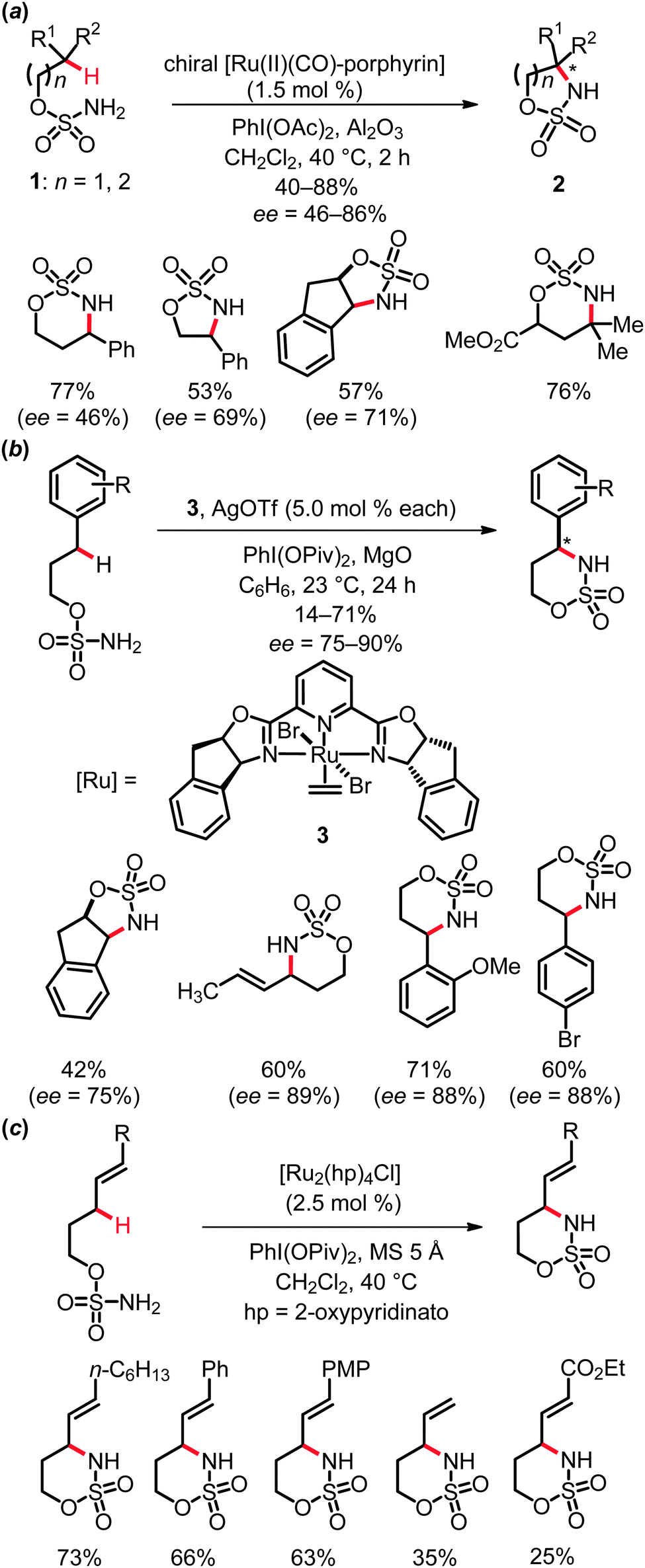 | ||
| Scheme 4 Intramolecular amidation of C(sp3)–H bonds. | ||
Notably, ruthenium catalysts allowed for intramolecular amidation of allylic C–H bonds, while related rhodium complexes led to a different chemoselectivity, namely aziridination of the double bond.11a,18 Mechanistically, the key step of the ruthenium-catalyzed direct amidation was found to be the insertion of ruthenium nitrenoids into C(sp3)–H bonds, which therefore renders potential transformations of more stable C(sp2)–H bonds significantly more challenging. Most probably, the amidation reaction initiates from iminoiodinane 4via treatment of substrate 1 with (diacetoxyiodo)benzene PhI(OAc)2.10a Coordination of compound 4 to the ruthenium complex gives rise to intermediate A and subsequently generates the active ruthenium nitrenoid intermediate B. Finally, the outer-sphere C–H bond functionalization step furnishes amide C, which thereafter affords the heterocyclic product 2, regenerating the active catalyst (Scheme 5).
 | ||
| Scheme 5 Mechanistic rationalization of ruthenium-catalyzed intramolecular amidation of C(sp3)–H bonds. | ||
The exact working mode of the elementary transformation B → C does not only depend on the nature of the transition metal, but also on the specific coordination chemistry of the ruthenium complex. Both computational and experimental studies on [Ru2(hp)4Cl]-promoted amidation [kinetic isotope effect (KIE) kH/kD ≈ 4.9]18 disclosed a stepwise C–H insertion with the formation of a short-lived diradical species, which is in contrast to the concerted insertion process postulated for rhodium catalysis (KIE ≈ 2.6). However, the two pathways of C–H bond insertion and hydrogen atom transfer were both proposed on the basis of computational studies (DFT) of reactions catalyzed by cationic ruthenium(II)-pybox complexes.17b These studies demonstrated the importance of ligand acceleration and substrate structure in these amidations.
2.2 Amination and amidation of C(sp2)–H bonds
Given the outer-sphere reaction mode of the above mentioned C–H bond nitrogenations, only relatively few examples of related C(sp2)–H bond aminations and amidations are available. Yet, the versatility and mechanistic diversity of these C–N bond forming processes render them especially attractive for theoreticians and synthetically oriented chemists. These transformations can be categorized into two distinct modes of action. First, direct functionalizations were viable by the insertion of ruthenium nitrenoids into unactivated C–H bonds. Second, approaches exploiting dehydrogenative19 aminations by C–H/N–H bond activation were realized. As an example of the former transformations, the intramolecular amination reactions of ortho-aryl phenylazides 5 and 1-azido-1,3-butadienes 7 furnished carbazoles 6 and pyrroles 8, respectively. In this transformation the active catalyst was generated from the user-friendly, inexpensive [RuCl3·nH2O], which effectively facilitated the C–H bond aminations (Scheme 6).20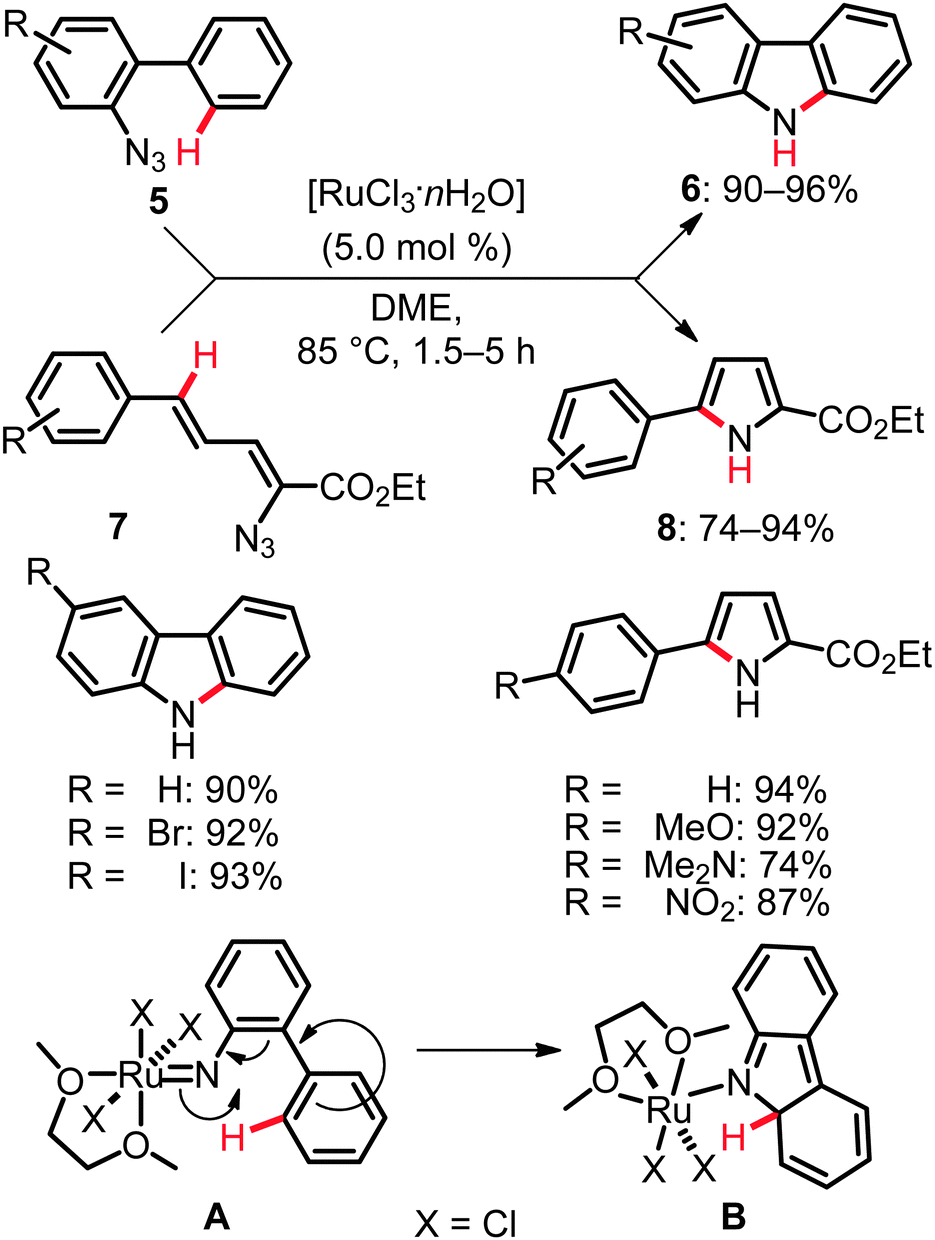 | ||
| Scheme 6 Intramolecular ruthenium-catalyzed amination of C(sp3)–H bonds. | ||
Computational studies on these aminations20,21 indicated that (i) the ruthenium species exhibited a higher activity than typical iridium or rhodium complexes, (ii) these catalytic reactions formally invoked a Ru(III)/Ru(V) catalytic cycle and (iii) a two-step process including formal electrocyclization A → B (Scheme 6) was involved in the catalytic amination. The latter hypothesis was further experimentally supported.20
In contrast to these intramolecular reactions, cationic ruthenium(II) complexes derived from [RuCl2(p-cymene)]2 proved to be most effective for intermolecular nitrogenation of C(sp2)–H bonds with an excellent site- and chemo-selectivity, along with a remarkably broad substrate scope. Thus, the catalytic system consisting of [RuCl2(p-cymene)]2, AgSbF6 and KOAc appeared to be appropriate for intermolecular ortho-amidations through direct C(sp2)–H bond nitrogenation on N-benzoylated sulfoximines 9 with sulfonyl azides (Scheme 7).22 These transformations displayed an excellent tolerance of various functional groups and provided an expedient approach to anthranilic acid derivatives through simple base-mediated hydrolysis of thus obtained amidation products 10. Likely, KOAc facilitated the formation of a cationic ruthenium(II) carboxylate catalyst.
 | ||
| Scheme 7 Ruthenium-catalyzed amidation of N-benzoylated sulfoximines 9. | ||
The power of this strategy was elegantly highlighted by the synthesis of HMR 1766 (Scheme 1), which targets deficient NO signaling in hypertension, peripheral, and coronary artery disease as well as heart failure.
Comparable catalytic conditions were utilized by Chang and co-workers in independent studies on ruthenium-catalyzed C–H bond amidations on arenes bearing synthetically useful directing groups,23 such as amides (11) or ketones (12) (Scheme 8).24
 | ||
| Scheme 8 Intermolecular C–H amidation of benzamides 11 and ketones 12. | ||
A wide range of benzamides 11 and aryl ketone 12 was readily amidated at the ortho-position by using sulfonyl azides with excellent catalytic efficacy and selectivity. It is particularly noteworthy that such a facile nitrogenation has not been reported for rhodium or palladium catalysis. The practical importance of the thus obtained products 13 and 14 was showcased by the preparation of a wide range of heterocycles with potential biological activities.
As to the catalysts working mode, detailed mechanistic studies revealed KIE for these amidations of kH/kD ≈ 5.9 for the amides 11 and kH/kD ≈ 2.7 for the phenones 12.24 These findings indicated the initial C–H bond activation to occur via an electrophilic-type metalation in an irreversible fashion and, thus, to be kinetically relevant. In contrast, in the related ruthenium(II)-catalyzed intermolecular ortho-C–H amidation of phenones 12 in the presence of substoichiometric quantities of Cu(OAc)2·H2O (Scheme 9)25 the C–H bond metalation was suggested to be reversible in nature. This observation can, for example, be rationalized in terms of heterobimetallic cooperative26 catalysis being of importance in the latter case.25a
 | ||
| Scheme 9 Ruthenium-catalyzed C–H amidation of aromatic ketones 12. | ||
Moreover, valuable heteroaromatic groups allowed for chelation-assisted amidation of arenes with various alkyl and aryl sulfonyl azides, thereby setting the stage for C–N bond formations on pyrazolyl-, pyrimidyl- or pyridyl-substituted arenes and heteroarenes 15 in good to excellent yields (Scheme 10).27 Intermolecular competition experiments with isotopically labeled starting materials were indicative of a reversible ruthenation event with a KIE of kH/kD ≈ 1.3. Hence, the C–H bond activation likely occurred here by a reversible electrophilic-type metalation and is not involved in the rate limiting step. The proposed working mode postulated for this catalytic system is depicted in Scheme 11.27a
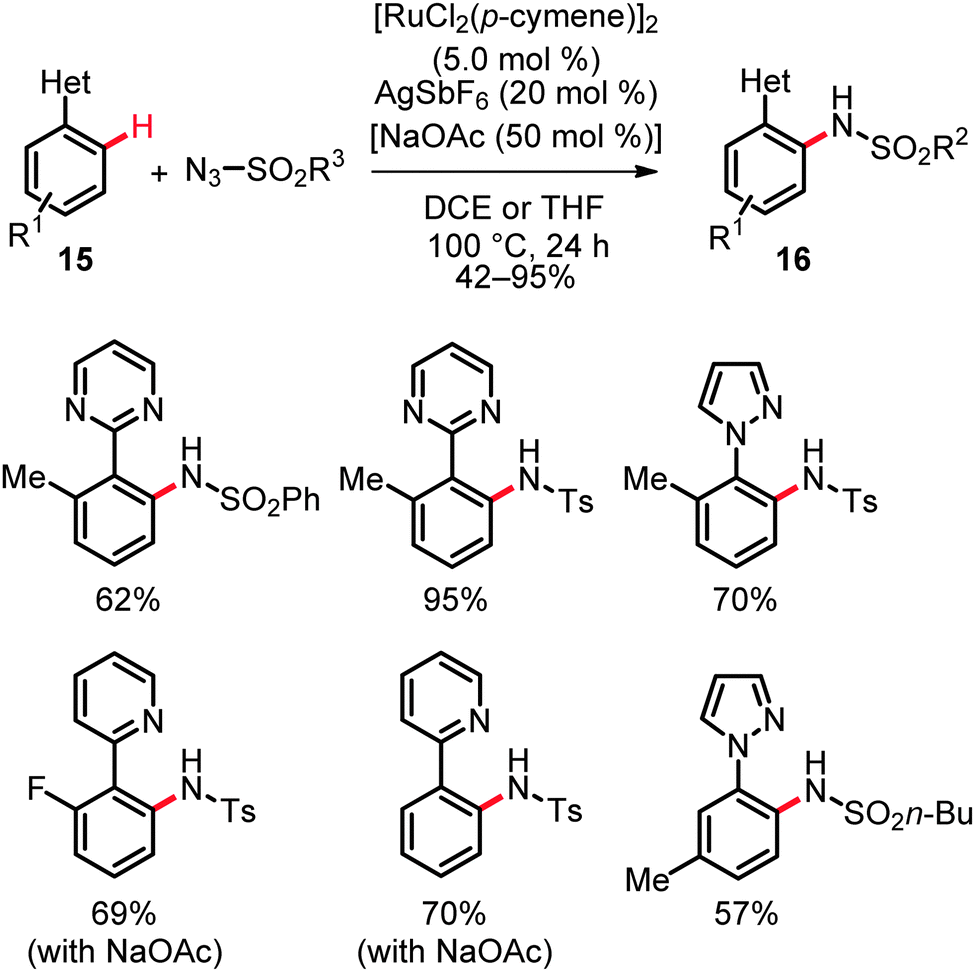 | ||
| Scheme 10 C–H amidation of substrates 15. | ||
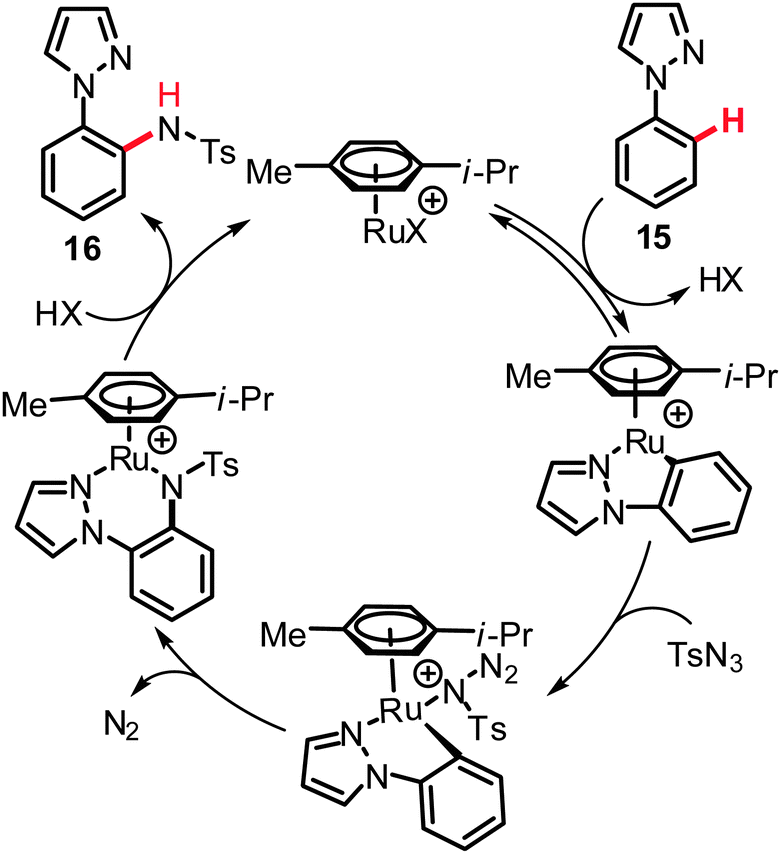 | ||
| Scheme 11 Proposed mechanism of the intermolecular C(sp2)–H amidations (X = Cl or OAc). | ||
The second approach, namely ruthenium-catalyzed dehydrogenative amination by C–H/N–H bond functionalization, was accomplished with unsaturated hydrazones 17 to furnish tri- and tetrasubstituted pyrazoles 1828 (Scheme 12a) and for the dehydrogenative homo-coupling of carbazoles 19 (Scheme 12b).29 These oxidative couplings proved viable with oxygen or air, respectively, as the sacrificial oxidants. This strategy was found to be generally useful and demonstrated a broad substrate scope and a considerable tolerance of important functional groups.
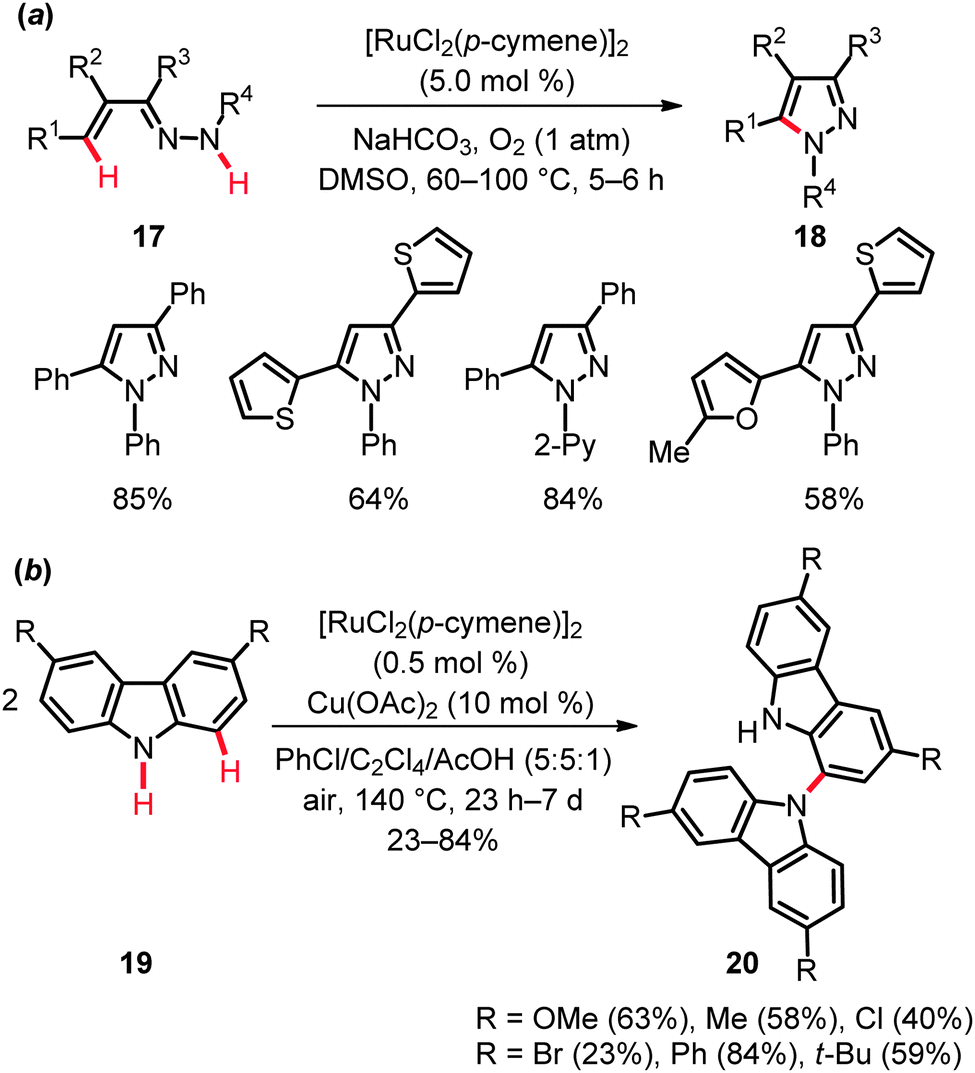 | ||
| Scheme 12 Ruthenium-catalyzed dehydrogenative amination. | ||
The KIE value of kH/kD ≈ 2.4 obtained for the pyrazole formation suggested the C–H metalation in substrate 17 to be kinetically relevant.28 In contrast, preliminary mechanistic investigations on the oxidative carbazole homo-coupling by Patureau illustrated the initial C–H activation in carbazole 19 not to be rate limiting (cf.Schemes 8–11). Notably, both ruthenium and copper complexes were found to be mandatory for the C–H bond activation step.29
3. Direct C–H oxygenation
During the past few years, several ruthenium-catalyzed hydroxylations of C–H bonds were reported by inter alia Rao, Du Bois and our research group. Contrary to the corresponding nitrogenations, solely tertiary C(sp3)–H bonds and C(sp2)–H bonds in (hetero)arenes were thus far hydroxylated. The limitation to tertiary C(sp3)–H bonds is likely due to the inherently high catalytic activity of ruthenium complexes as to undesired overoxidation to the corresponding carbonyl compounds.30,313.1 Hydroxylation of C(sp3)–H bonds
Early RuO4-mediated hydroxylation of unactivated tertiary C(sp3)–H bonds in hydrocarbons as well as mechanistic aspects of these oxygenations were studied in great detail by Bakke and coworkers among others.31 Yet, novel ruthenium catalysts recently disclosed by Du Bois and coworkers32a allowed them to significantly improve the substrate scope of the hydroxylation. Indeed, the use of catalytic quantities of [RuCl3·nH2O], along with pyridine as the additive and KBrO3 as the stoichiometric oxidant, resulted in the development of a practical, user-friendly protocol that proved applicable to variously substituted substrates 21 (Scheme 13). This reaction not only afforded hydroxylated esters, epoxides, sulfones, oxazolidinones, carbamates and sulfamates 22 in yields generally exceeding 50%, but also constituted a highly convenient method for 18O-label incorporation.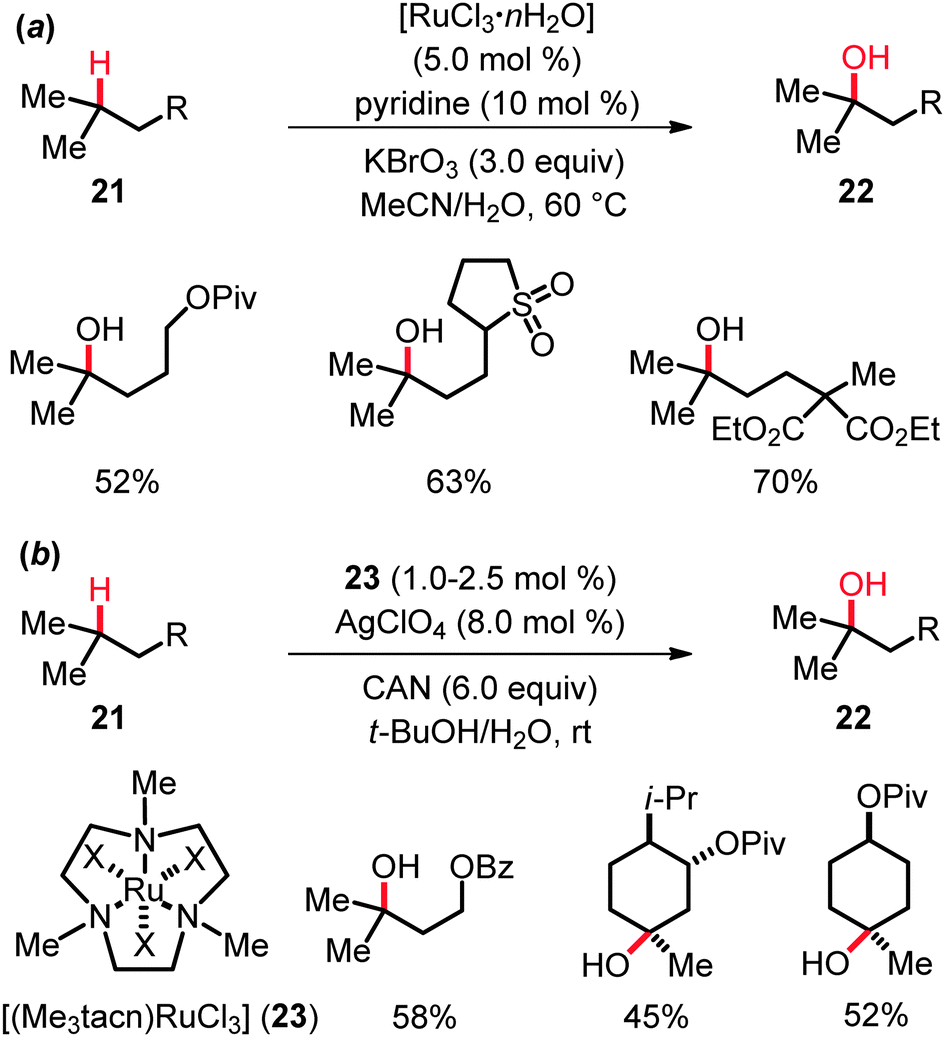 | ||
| Scheme 13 Ruthenium-catalyzed hydroxylation of C(sp3)–H bonds. | ||
However, the coordination of RuCl3 by trimethyltriazacyclononane, as found in [(Me3tacn)RuCl3] (23), in combination with AgClO4 as the additive and CAN as the oxidant, improved the yield and allowed for a reduction of the catalyst loading as well as of the reaction temperature (Scheme 13).32b Interestingly, the KIE value of kH/kD ≈ 6.7 obtained in experiments with isotopically labeled starting materials 21 led the authors to propose a two-step mechanism, involving hydrogen atom abstraction followed by a fast, solvent-caged radical rebound. This mechanistic rationale is related to the mode of action in ruthenium-catalyzed intramolecular amidations (cf.Scheme 5), but differs from the generally accepted concerted asynchronous (3+2) pathway previously postulated for C–H hydroxylations catalyzed with RuO4 (KIE ≈ 5–7).31
3.2 Hydroxylation of C(sp2)–H bonds
The past two years have witnessed a tremendous development in the direct hydroxylation of C(sp2)–H bonds in arenes and heteroarenes with readily accessible ruthenium catalysts (Scheme 14).33–42 While Rao and coworkers used the complex [RuCl2(p-cymene)]2 as the precatalyst and K2S2O8 or HIO3 as the oxidant,34 our group employed [RuCl2(p-cymene)]2, as well as the well-defined ruthenium(II) biscarboxylate complex [Ru(O2CMes)2(p-cymene)] (24),33 or inexpensive [RuCl3·nH2O] and (diacetoxyiodo)benzene [PhI(OAc)2] as the oxidant. For chelation-assisted C–O bond formation on arenes the solvent mixture comprising of TFA and TFAA turned out to be critical, which enabled successful hydroxylation of electron-deficient and electron-rich arenes as well as heteroarenes33–42 with weakly coordinating directing groups,23 such as esters or ketones.33–42 Thus, the ruthenium-catalyzed ortho-hydroxylation of benzoates 25 was found to be generally useful for the preparation of highly functionalized arenes, some of which are difficult to access via conventional approaches (Scheme 15).34 | ||
| Scheme 14 Overview of ruthenium-catalyzed direct hydroxylation of C(sp2)–H bonds. | ||
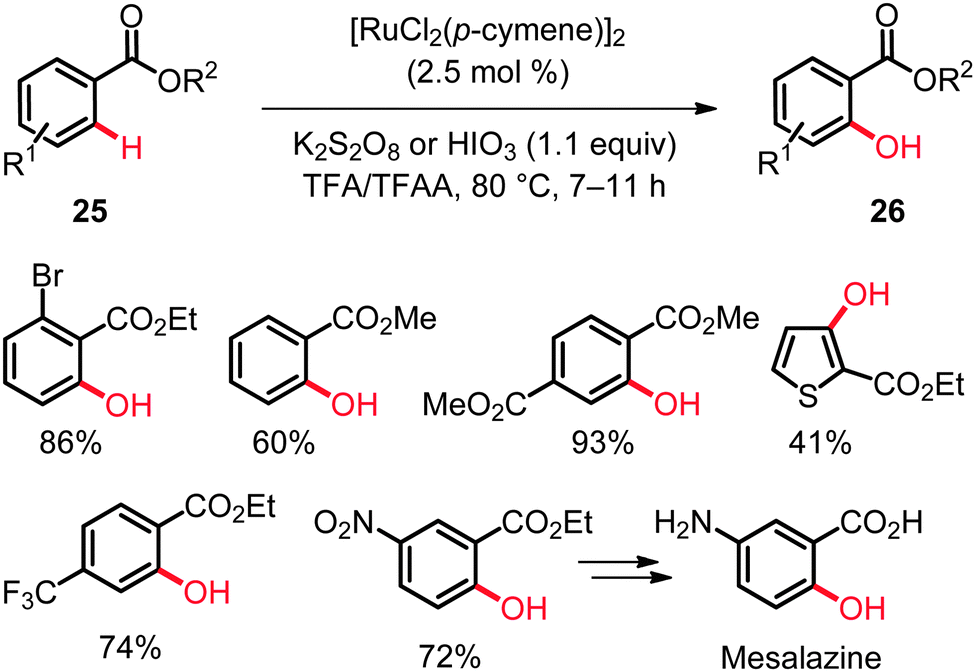 | ||
| Scheme 15 ortho-Hydroxylation of benzoates 25. | ||
The catalytic system exhibited a good functional group tolerance and delivered the products 26 in high yields. Notably, useful heteroarenes were also compatible with these catalytic reaction conditions. The KIE value of kH/kD ≈ 1.8 was suggestive of a kinetically relevant C–H metalation. These hydroxylations set the stage for a step-economical approach towards the synthesis of biologically important compounds, such as Mesalazine, an anti-inflammatory drug used for the treatment of inflammatory bowel disease. Indeed, ethyl 2-hydroxy-5-nitrobenzoate could be easily converted to Mesalazine by sequential hydrolysis and reduction.35
Studies on ruthenium-catalyzed C(sp2)–H bond oxygenation of arenes 11 bearing amide directing groups identified the user-friendly, inexpensive [RuCl3·nH2O] as a viable catalyst. PhI(OAc)2 proved to be an efficient oxidant, while O2, Cu(OAc)2·H2O, t-BuOOH, oxone, K2S2O8 or PhI(TFA)2 were found to be inferior. Interestingly, the most satisfactory results were obtained with a well-defined ruthenium(II) biscarboxylate complex [Ru(O2CMes)2(p-cymene)] (24) at a remarkably low catalyst loading of only 1.0 mol% (Scheme 16).36
 | ||
| Scheme 16 Oxidative hydroxylation of benzamides 11. | ||
Variously decorated N,N-disubstituted benzamides 11 bearing dimethylamino, diethylamino, diisopropylamino, pyrrolidinyl or morpholinyl moieties afforded the corresponding amidophenols 27 in good to excellent yields with the catalytic system. In contrast to palladium-catalyzed oxidative ortho-hydroxylation of benzamides with a rather limited substrate scope,9a,b the ruthenium-catalyzed reactions were compatible with methoxy, ester, nitro and fluoro substituents on the arenes. Thus, the reaction of 4-methoxy, 4-methoxycarbonyl and 4-fluorosubstituted benzamides 11 furnished the ortho-hydroxylated products 27 in high yields of up to 98%. The optimized catalyst displayed an excellent site-selectivity, as illustrated by the exclusive formation of 2-hydroxy-N,N-diisopropyl-5-nitrobenzamide (27a) as the sole product (Scheme 16). Mechanistic studies provided strong support for a reversible C–H bond metalation step.36
The ruthenium-catalyzed hydroxylation was not limited to relatively strongly coordinating directing groups. Indeed, weakly coordinating ketones in phenones 12 proved to be applicable exploiting the ruthenium complex 24 (Scheme 17).37 This unprecedented functionalization occurred with an excellent functional group tolerance and a broad substrate scope as well as high chemo- and site-selectivities.
 | ||
| Scheme 17 ortho-Hydroxylation of aromatic ketones 12. | ||
While acetophenone afforded a mixture of C(sp2)–H- and C(sp3)–H-functionalized products, benzophenone furnished mono- and dihydroxylated compounds in 85% yield. Variously substituted aromatic ketones 12 chemoselectively gave the corresponding phenol derivatives 28, while hydroxylation of electron-deficient substrates was less efficient. Similar ortho-hydroxylations could be performed with the inexpensive [RuCl3·nH2O] precatalyst and oxone or K2S2O8 as the terminal oxidant, albeit with a somewhat lower catalytic efficacy. The selective formation of methyl 3-hydroxy-4-pivaloylbenzoate (28a) in an intermolecular competition was of particular interest, since it revealed the relative directing group abilities.
Weinreb amides 29 constitute important structural motifs in a number of natural products and bioactive compounds and represent functional groups of key importance in synthetic organic chemistry. Thus, these functional groups are easily installed and can be chemoselectively transformed into the corresponding ketones and aldehydes.38 Unfortunately, amides 29 had been underutilized in ruthenium-catalyzed C–H bond functionalizations,14 and direct hydroxylations of aryl Weinreb amides had proven elusive until very recently. Yet, the ruthenium complex [RuCl2(p-cymene)]2, along with PhI(OAc)2 (1.0 equiv.) as the oxidant in TFA/TFAA, was found to enable unprecedented C–H bond oxygenations of aryl Weinreb amides 29 to give the desired products 30 with ample scope under exceedingly mild reaction conditions (Scheme 18).39 Mechanistic studies disclosed an irreversible, thus kinetically relevant C–H bond activation with a KIE of kH/kD ≈ 3.0. The step- and atom-economical syntheses of ortho-hydroxylated Weinreb amides 30 also provided access to valuable ortho-hydroxylated aldehydes. For example, o-hydroxy-N-methoxy-N-methylamide (30a) obtained via the ruthenium-catalyzed C–H bond oxygenation underwent a facile reduction to afford the corresponding ortho-hydroxyaldehyde (Scheme 18).39
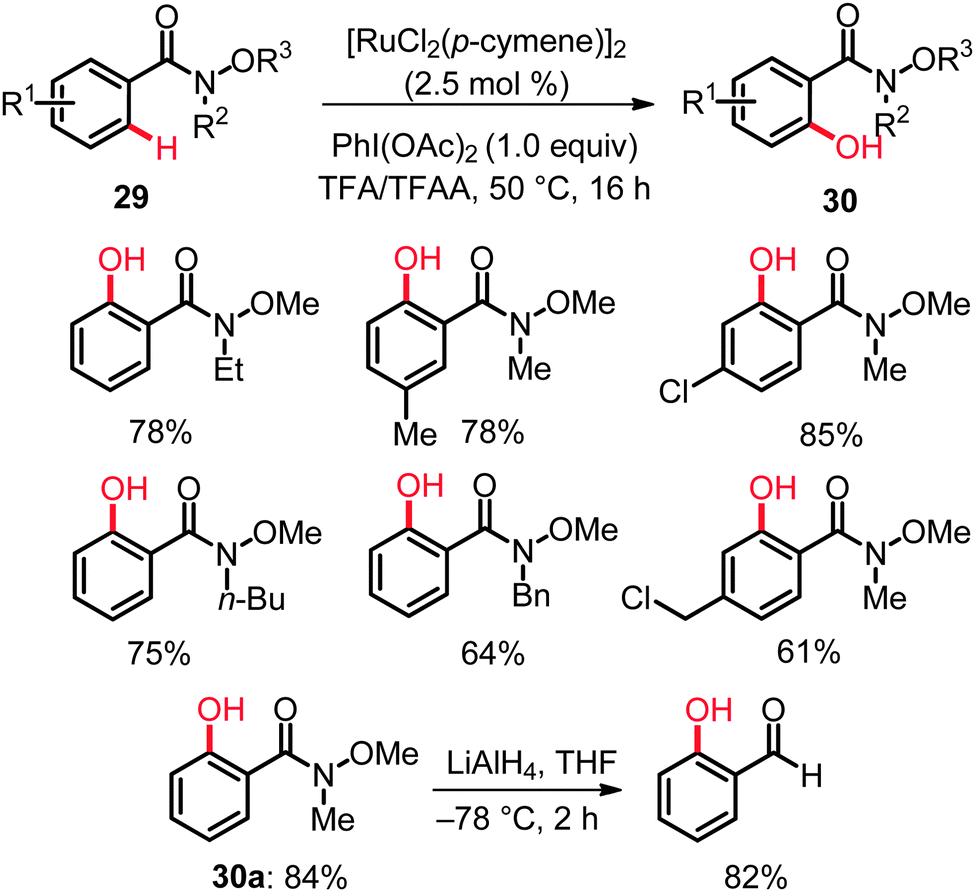 | ||
| Scheme 18 C–H hydroxylation of Weinreb amides 29. | ||
Whilst the previous studies had focused on the use of arenes bearing electron-withdrawing directing groups, we reported recently the first ruthenium-catalyzed C(sp2)–H bond oxygenation of phenol derivatives 31 applying [RuCl2(p-cymene)]2 as the catalyst and PhI(TFA)2 as the oxidant.40 Thus, direct hydroxylation of easily removable aryl carbamates 31 proceeded under mild reaction conditions with high catalytic efficacy and excellent chemoselectivity (Scheme 19).40,41 Studies with isotopically labeled substrates 31 disclosed a KIE of kH/kD ≈ 2.2, which is in accordance with a kinetically relevant C–H bond metalation step.40
 | ||
| Scheme 19 Direct ortho-hydroxylation of carbamates 31. | ||
It is noteworthy that the ruthenium(II) catalyst also allowed for the direct C–H bond functionalization of anisole derivatives 33 being devoid of Lewis-basic directing groups, occurring with a remarkable para-selectivity (Scheme 20).40
 | ||
| Scheme 20 para-Hydroxylation of anisoles 33. | ||
Ruthenium catalysis was not restricted to direct hydroxylation of aryl carbamates as electron-rich substrates. Hence, anilide-directed oxidative C–O bond formations were very recently reported by Rao employing K2S2O8 as the oxidant.41 In this transformation an efficient synthesis of mono- and dihydroxylated anilides 36 by C(sp2)–H bond oxygenation was accomplished (Scheme 21).42 The reaction featured excellent site-selectivities and a good functional group tolerance. As found for the aryl carbamates 31,40 the acylated amino moiety in product 36 constituted a removable directing group.43 Indeed, the 2-aminophenol 37a was obtained by simple hydrazinolysis (Scheme 21).42
 | ||
| Scheme 21 ortho-Hydroxylation of anilides 35. | ||
4. Conclusions
Recent years have witnessed tremendous progress in metal-catalyzed C–H bond functionalizations. Despite these advances, relatively inexpensive44 ruthenium complexes were until very recently not fully recognized as catalysts for direct amidations and hydroxylations through challenging functionalization of otherwise unreactive C(sp3)–H and C(sp2)–H bonds. These difficult oxidative C–H bond nitrogenations and oxygenations have proven to be widely applicable using versatile ruthenium complexes, with considerable progress being accomplished in the last two years. Notable features of the most user-friendly ruthenium catalysts include the remarkably broad substrate scope and the extraordinarily high chemo- and site-selectivity. The outstanding selectivity was among others reflected by the high functional group tolerance and catalytic activity, which compared, for example, favorably with rhodium catalysis in ortho-hydroxylation of arenes.41 Importantly, ruthenium-catalyzed nitrogenations and oxygenations enabled the challenging direct functionalization of unactivated C(sp3)–H bonds and were accomplished with substrates displaying only weakly coordinating directing groups, such as esters and ketones. Considering the practical importance of atom- and step-economical C–H bond amidations, aminations and hydroxylations for organic synthesis, material sciences or medicinal chemistry, significant further progress is expected in this rapidly evolving research area.Acknowledgements
L. A. sincerely thanks all his former and present co-workers involved in the C–H bond functionalization projects for their experimental work and their intellectual contributions. VST is indebted to the Humboldt Foundation for a research fellowship. The cited studies from our laboratories were partly funded by the European Research Council under the European Community 7th Framework Program and the Fonds der Chemischen Industrie.Notes and references
- (a) A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) The Chemistry of Anilines, ed. Z. Rappoport, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267–275 RSC; (d) S. A. Lawrence, Amines: Synthesis Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (e) The Chemistry of Phenols, ed. Z. Rappoport, Wiley-VCH, Weinheim, 2003 Search PubMed; (f) J. F. Hartwig, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-i. Negishi, Wiley-Interscience, New York, 2002, vol. 1, pp. 1051–1097 Search PubMed; (g) H. Fiegel, H. W. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H. J. Buysch, D. Garbe and W. Paulus, Phenol Derivatives in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, New York, 2002 Search PubMed; (h) J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, NewYork, 1996 Search PubMed.
- For selected reviews on C–H bond functionalization, see: (a) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed; (b) A. Sharma, D. Vacchani and E. Van der Eycken, Chem.–Eur. J., 2013, 19, 1158–1168 CrossRef CAS PubMed; (c) S. I. Kozhushkov, H. K. Potukuchi and L. Ackermann, Catal. Sci. Technol., 2013, 3, 562–571 RSC; (d) J. J. Mousseau and A. B. Charrette, Acc. Chem. Res., 2013, 46, 412–424 CrossRef CAS PubMed; (e) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253–3260 RSC; (f) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (g) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (h) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (i) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (j) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (k) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed; (l) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC; (m) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC; (n) Y. Nakao, Synthesis, 2011, 3209–3219 CrossRef CAS PubMed; (o) L. Ackermann and H. K. Potukuchi, Org. Biomol. Chem., 2010, 8, 4503–4513 RSC; (p) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (q) L. Ackermann, Chem. Commun., 2010, 46, 4866–4877 RSC; (r) M. Livendahl and A. M. Echavarren, Isr. J. Chem., 2010, 50, 630–651 CrossRef CAS; (s) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (t) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (u) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269–10310 CrossRef CAS PubMed; (v) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087–4109 CAS; (w) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673–714 CrossRef CAS; (x) L. Ackermann, Top. Organmet. Chem., 2007, 24, 35–60 CrossRef CAS; (y) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (z) D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35–41 Search PubMed; (a a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (a b) L. Ackermann, Synlett, 2007, 507–526 CrossRef CAS PubMed; (a c) T. Satoh and M. Miura, Chem. Lett., 2007, 200–205 CrossRef CAS.
- The values of C–H bond dissociation energies are equal to 438.9 and 472.4 kJ mol−1 for methane and benzene, respectively: S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CAS; (b) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed; (c) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- (a) N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC; (b) A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 1999, 219, 131–209 CrossRef; (c) J. F. Hartwig, Pure Appl. Chem., 1999, 71, 1416–1423 CrossRef.
- Selected reviews: (a) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed; (b) W. Song, S. I. Kozhushkov and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 6576–6578 CrossRef CAS PubMed; (c) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC.
- (a) R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400–1401 RSC; (b) R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728–6729 CrossRef CAS; (c) T. Jintoku, H. Taniguchi and Y. Fujiwara, Chem. Lett., 1987, 1865–1868 CrossRef CAS.
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- Palladium-catalyzed oxygenations, selected reviews: (a) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed . For selected recent contributions, see: ; (c) Y. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827–5831 CrossRef CAS PubMed; (d) J. B. Gary, A. K. Cook and M. S. Sanford, ACS Catal., 2013, 3, 700–703 CrossRef CAS; (e) F. A. Harraz, S. E. El-Hout, H. M. Killa and I. A. Ibrahim, J. Mol. Catal. A: Chem., 2013, 370, 182–188 CrossRef CAS PubMed; (f) A. Banerjee, A. Bera, S. Guin, S. K. Rout and B. K. Patel, Tetrahedron, 2013, 69, 2175–2183 CrossRef CAS PubMed; (g) P. Y. Choy and F. Y. Kwong, Org. Lett., 2013, 15, 270–273 CrossRef CAS PubMed; (h) F. Mo, L. J. Trzepkowski and G. Dong, Angew. Chem., Int. Ed., 2012, 51, 13075–13079 CrossRef CAS PubMed; (i) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070–13074 CrossRef CAS PubMed; (j) M. R. Yadav, R. K. Rit and A. K. Sahoo, Chem.–Eur. J., 2012, 18, 5541–5545 CrossRef CAS PubMed; (k) J. Piechowska, K. Huttunen, Z. Wróbel, H. Lemmetyinen, N. V. Tkachenko and D. T. Gryko, J. Phys. Chem. A, 2012, 116, 9614–9620 CrossRef CAS PubMed; (l) J. Piechowska and D. T. Gryko, J. Org. Chem., 2011, 76, 10220–10228 CrossRef CAS PubMed; (m) D. A. Alonso, C. Nájera, I. M. Pastor and M. Yus, Chem.–Eur. J., 2010, 16, 5274–5284 CrossRef CAS PubMed , and references cited therein.
- Palladium-catalyzed nitrogenations, selected reviews: see ref. 9b and (a) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061–5074 RSC For selected recent reports, see; (b) R. Shrestha, P. Mukherjee, Y. Tan, Z. C. Litman and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8480–8483 CrossRef CAS PubMed; (c) V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468–1471 CrossRef CAS PubMed; (d) K.-H. Ng, F.-N. Ng and W.-Y. Yu, Chem. Commun., 2012, 48, 11680–11682 RSC; (e) N. Cocherel, B. J. Lidster and M. L. Turner, PCT Int. Appl., WO2012076886 A3, 2012 Search PubMed; (f) B. Xiao, T.-J. Gong, J. Xu, Z.-J. Liu and L. Liu, J. Am. Chem. Soc., 2011, 133, 1466–1474 CrossRef CAS PubMed; (g) G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944–2947 CrossRef CAS PubMed; (h) X.-Y. Liu, P. Gao, Y.-W. Shen and Y.-M. Liang, Org. Lett., 2011, 13, 4196–4199 CrossRef CAS PubMed.
- For reviews on rhodium-catalyzed C–H nitrogenations, see ref. 10a and: (a) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed; (b) N. Boudet and S. B. Blakey, in Chiral Amine Synthesis, ed. T. C. Nugent, Wiley-VCH, Weinheim, 2010, pp. 337–396 Search PubMed; (c) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (d) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439–2463 CrossRef CAS PubMed; (e) J. Du Bois, in Modern Rhodium Catalyzed Organic Reactions, ed. P. A. Evans, Wiley, New York, 2005, pp. 379–416 Search PubMed; (f) H. M. L. Davies and M. S. Long, Angew. Chem., Int. Ed., 2005, 44, 3518–3520 CrossRef CAS PubMed; (g) P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2920 CrossRef PubMed Selected recent contributions: ; (h) J. Ryu, K. Shin, S. H. Park, J. Y. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 9904–9908 CrossRef CAS PubMed , and references cited therein; (i) J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, J. Am. Chem. Soc., 2012, 134, 9110–9113 CrossRef CAS PubMed; (j) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2012, 14, 656–659 CrossRef CAS PubMed; (k) K.-H. Ng, Z. Zhou and W.-Y. Yu, Org. Lett., 2012, 14, 272–275 CrossRef CAS PubMed; (l) R. P. Reddy and H. M. L. Davies, Org. Lett., 2006, 8, 5013–5016 CrossRef CAS PubMed.
- Reviews: see ref. 10a and: (a) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC; (b) A. Armstrong and J. C. Collins, Angew. Chem., Int. Ed., 2010, 49, 2282–2285 CrossRef CAS PubMed . Selected reports: ; (c) A. John and K. M. Nicholas, J. Org. Chem., 2011, 76, 4158–4162 CrossRef CAS PubMed; (d) L. Zhang, Z. Liu, H. Li, G. Fang, B.-D. Barry, T. A. Belay, X. Bi and Q. Liu, Org. Lett., 2011, 13, 6536–6539 CrossRef CAS PubMed; (e) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2860–2863 CrossRef CAS PubMed; (f) T. Kawano, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2010, 132, 6900–6901 CrossRef CAS PubMed.
- (a) Y. Liu, X. Guan, E. L.-M. Wong, P. Liu, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2013, 135, 7194–7204 CrossRef CAS PubMed; (b) M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721–9726 CrossRef CAS PubMed; (c) S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036–2039 CrossRef CAS PubMed; (d) T. Wang, W. Zhou, H. Yin, J.-A. Ma and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 10823–10826 CrossRef CAS PubMed; (e) C. Qin, W. Zhou, F. Chen, Y. Ou and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 12595–12599 CrossRef CAS PubMed; (f) Q. Xia, W. Chen and H. Qiu, J. Org. Chem., 2011, 76, 7577–7582 CrossRef CAS PubMed; (g) Z. Wang, Y. Zhang, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2008, 10, 1863–1866 CrossRef CAS PubMed.
- For recent reviews on ruthenium-catalyzed C–H bond functionalization, see: (a) L. Ackermann, Acc. Chem. Res., 2013, 46 DOI:10.1021/ar3002798; (b) B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744–5767 RSC; (c) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC; (d) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (e) L. Ackermann, Pure Appl. Chem., 2010, 82, 1403–1413 CrossRef CAS; (f) L. Ackermann and R. Vicente, Top. Curr. Chem., 2010, 292, 211–229 CrossRef CAS; (g) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077–1101 CrossRef CAS.
- (a) X.-G. Zhou, X.-Q. Yu, J.-S. Huang and C.-M. Che, Chem. Commun., 1999, 2377–2378 RSC; (b) X.-Q. Yu, J.-S. Huang, X.-G. Zhou and C.-M. Che, Org. Lett., 2000, 2, 2233–2236 CrossRef CAS PubMed; (c) J.-L. Liang, J.-S. Huang, X.-Q. Yu, N. Zhu and C.-M. Che, Chem.–Eur. J., 2002, 8, 1563–1572 CrossRef CAS; (d) J.-L. Liang, S.-X. Yuan, J.-S. Huang, W.-Y. Yu and C.-M. Che, Angew. Chem., Int. Ed., 2002, 41, 3465–3468 CrossRef CAS.
- (a) S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, N. Casati, P. Macchi and S. Cenini, Chem. Commun., 2009, 3952–3954 RSC; (b) T. Terada, T. Kurahashi and S. Matsubara, Heterocycles, 2012, 85, 2415–2419 CrossRef CAS; (c) D. Intrieri, A. Caselli, F. Ragaini, P. Macchi, N. Casati and E. Gallo, Eur. J. Inorg. Chem., 2012, 569–580 CrossRef CAS; (d) J. W. W. Chang and P. W. H. Chan, Angew. Chem., Int. Ed., 2008, 47, 1138–1140 CrossRef CAS PubMed; (e) L. He, P. W. H. Chan, W.-M. Tsui, W.-Y. Yu and C.-M. Che, Org. Lett., 2004, 6, 2405–2408 CrossRef CAS PubMed , and cited references.
- (a) E. Milczek, N. Boudet and S. Blakey, Angew. Chem., Int. Ed., 2008, 47, 6825–6828 CrossRef CAS PubMed; (b) D. G. Musaev and S. B. Blakey, Organometallics, 2012, 31, 4950–4961 CrossRef CAS; (c) J. L. Bon and S. B. Blakey, Heterocycles, 2012, 84, 1313–1323 CrossRef CAS PubMed.
- M. E. Harvey, D. G. Musaev and J. Du Bois, J. Am. Chem. Soc., 2011, 133, 17207–17216 CrossRef CAS PubMed.
- (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- W. G. Shou, J. Li, T. Guo, Z. Lin and G. Jia, Organometallics, 2009, 28, 6847–6854 CrossRef CAS.
- Q. Zhang, C. Wu, L. Zhou and J. Li, Organometallics, 2013, 32, 415–426 CrossRef CAS.
- M. R. Yadav, R. K. Rit and A. K. Sahoo, Org. Lett., 2013, 15, 1638–1641 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- J. Kim, J. Kim and S. Chang, Chem.–Eur. J., 2013, 19, 7328–7333 CrossRef CAS PubMed.
- (a) M. Bhanuchandra, M. R. Yadav, R. K. Rit, M. R. Kuram and A. K. Sahoo, Chem. Commun., 2013, 49, 5225–5227 RSC; (b) Q.-Z. Zheng, Y.-F. Liang, C. Qin and N. Jiao, Chem. Commun., 2013, 49, 5654–5656 RSC.
- Review: M. H. Pérez-Temprano, J. A. Casares and P. Espinet, Chem.–Eur. J., 2012, 18, 1864–1884 CrossRef PubMed.
- (a) V. S. Thirunavukkarasu, K. Raghuvanshi and L. Ackermann, Org. Lett., 2013, 15, 3286–3289 CrossRef CAS PubMed; (b) See ref. 24.
- J. Hu, S. Chen, Y. Sun, J. Yang and Y. Rao, Org. Lett., 2012, 14, 5030–5033 CrossRef CAS PubMed.
- M.-L. Louillat and F. W. Patureau, Org. Lett., 2013, 15, 164–167 CrossRef CAS PubMed.
- (a) M. S. Yusubov, V. N. Nemykin and V. V. Zhdankin, Tetrahedron, 2010, 66, 5745–5752 CrossRef CAS PubMed; (b) C. S. Yi, K.-H. Kwon and D. W. Lee, Org. Lett., 2009, 11, 1567–1569 CrossRef CAS PubMed; (c) C. Wang, K. V. Shalyaev, M. Bonchio, T. Carofiglio and J. T. Groves, Inorg. Chem., 2006, 45, 4769–4782 CrossRef CAS PubMed; (d) J. R. Bryant and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 10351–10361 CrossRef CAS PubMed.
- (a) J. M. Bakke and A. E. Frøhaug, J. Phys. Org. Chem., 1996, 9, 310–318 CrossRef CAS , and references cited therein; (b) M. Drees and T. Strassner, J. Org. Chem., 2006, 71, 1755–1760 CrossRef CAS PubMed.
- (a) E. McNeill and J. Du Bois, J. Am. Chem. Soc., 2010, 132, 10202–10204 CrossRef CAS PubMed; (b) E. McNeill and J. Du Bois, Chem. Sci., 2012, 3, 1810–1813 RSC.
- (a) L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed; (b) L. Ackermann, J. Pospech and H. K. Potukuchi, Org. Lett., 2012, 14, 2146–2149 CrossRef CAS PubMed.
- Y. Yang, Y. Lin and Y. Rao, Org. Lett., 2012, 14, 2874–2877 CrossRef CAS PubMed.
- G. Breviglieri, B. Giacomo, C. Sergio, A. Cinzia, E. Campanab and M. Panunzio, Molecules, 2001, 6, M260–M261 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu, J. Hubrich and L. Ackermann, Org. Lett., 2012, 14, 4210–4213 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu and L. Ackermann, Org. Lett., 2012, 14, 6206–6209 CrossRef CAS PubMed.
- Reviews: (a) S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 3707–3738 CAS; (b) M. Mentzel and H. M. R. Hoffmann, J. Prakt. Chem., 1997, 339, 517–524 CrossRef CAS.
- F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718–720 CrossRef CAS PubMed.
- W. Liu and L. Ackermann, Org. Lett., 2013, 15, 3484–3486 CrossRef CAS PubMed.
- For the comparison of the relative efficacies of PhI(OAc)2 and K2S2O8 as oxidants as well as of ruthenium and rhodium catalysts [RuCl2(p-cymene)]2 and [Rh(OAc)2]2 in ortho-hydroxylations, see also: G. Shan, X. Han, Y. Lin, S. Yu and Y. Rao, Org. Biomol. Chem., 2013, 11, 2318–2322 CAS.
- (a) X. Yang, G. Shan and Y. Rao, Org. Lett., 2013, 15, 2334–2337 CrossRef CAS PubMed ; see also:; (b) K. Padala and M. Jeganmohan, Chem. Commun., 2013, 49, 9651–9653 RSC.
- Review: C. Wang and Y. Huang, Synlett, 2013, 145–149 CAS.
- Thus, in September 2013, the prices of platinum, rhodium, gold, iridium, palladium and ruthenium were 1482, 1015, 1363, 800, 701 and 80 US$ per troy oz, respectively. See: http://taxfreegold.co.uk/preciousmetalpricesusdollars.html.
| This journal is © The Royal Society of Chemistry 2014 |