 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntriguing mechanistic labyrinths in gold(I) catalysis
Carla
Obradors
a and
Antonio M.
Echavarren
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: aechavarren@iciq.es
bDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, C/Marcel. li Domingo s/n, 43007 Tarragona, Spain
First published on 17th October 2013
Abstract
Many mechanistically intriguing reactions have been developed in the last decade using gold(I) as catalyst. Here we review the main mechanistic proposals in gold-catalysed activation of alkynes and allenes, in which this metal plays a central role by stabilising a variety of complex cationic intermediates.
 Carla Obradors | Carla Obradors was born in Manresa (Barcelona) in 1987. She graduated in Chemistry at Universitat Autònoma de Barcelona in 2010. Meanwhile, she also worked at Parc Científic de Barcelona and Institut de Química Avançada de Catalunya (CSIC). In 2011, she was awarded the Master of Synthesis and Catalysis Extraordinary Prize at Universitat Rovira i Virgili (Tarragona). Since 2011, she has been carrying out her PhD studies under the supervision of Prof. Echavarren at the Institute of Chemical Research of Catalonia (ICIQ). |
 Antonio M. Echavarren | Antonio M. Echavarren obtained his PhD at the Universidad Autónoma de Madrid (UAM) in 1982. After postdoctoral stays in Boston College and Colorado State University, he joined the CSIC (Institute of Organic Chemistry) in Madrid. In 1992 he became Professor of Organic Chemistry at the UAM. He is also Professor of Research of the CSIC. In 2004 he moved to Tarragona as Group Leader at the Institute of Chemical Research of Catalonia (ICIQ). His research interests include the discovery of new catalytic methods based on the chemistry of transition metals as well as the synthesis of natural products and polyarenes. |
Introduction
For centuries, gold was considered a precious, purely decorative noble metal. It was not until 1998, in a groundbreaking report, that the hydration of alkynes catalysed by Au(I) complexes under homogeneous conditions was reported.1,2 Henceforth, numerous transformations have been developed, nourishing the field of organic synthesis.3 Gold salts and complexes emerged as powerful catalysts for the selective electrophilic activation of multiple bonds towards a variety of hetero- and carbonucleophiles under mild conditions. Cycloisomerizations and cycloadditions attracted particular attention for the construction of complex polycyclic structures present in diverse natural products.4,5In most cases, reactions catalysed by gold under homogeneous conditions proceed by multistep pathways that are rather complex. Therefore, a mechanistic understanding has been based often on analogy and speculation. Although coherent mechanistic schemes have been advanced by means of DFT calculations, as well as labelling and kinetic experiments, isolation of key intermediates has proven to be challenging.6 Here we present a critical outlook of the main mechanistic proposals in this area of homogeneous catalysis. This discussion is by no means comprehensive, but it centres on some of the best-studied gold-catalysed activation of alkynes and allenes with the aim of contributing to a more clear understanding of this intricate field of research.
Gold(I) catalysts
Although simple gold salts such as NaAuCl4 or AuCl are active enough to catalyse many transformations, precatalysts LAuCl bearing phosphine or N-heterocyclic carbene as ligands have found more widespread applications.7–9 (Fig. 1). The active species are often generated in situ by chloride abstraction using a silver salt with distinct anions. The innocence of silver in the reaction mixture has been recently questioned,10 although this aspect merits additional scrutiny. The most convenient catalysts are gold complexes [AuLL′]X or [AuLX] with weakly coordinating neutral (L′)11 or anionic ligands (X),12 which could enter catalytic cycles by associative ligand exchange with the substrate.13 The properties of the catalysts can be easily tuned sterically or electronically depending on the ligand. Thus, in general, complexes with phosphite and related ligands are more electrophilic than those bearing more donating N-heterocyclic carbenes, whereas with phosphines show intermediate electrophilicity.3d The use of chiral ligands has led to the development of efficient asymmetric processes.14,15 Several studies have shown that the basicity and coordinating ability of the counteranion also play a significant role.11b,14,16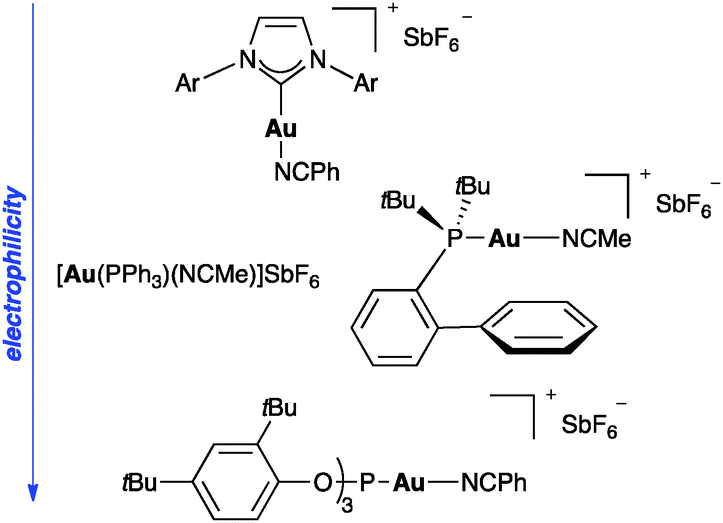 | ||
| Fig. 1 Representative gold(I) cationic catalysts. | ||
Activation of unsaturated substrates
Important structural features of gold are its aurophilicity, its linear geometry that limits the coordination potential, and the fact that it does not undergo spontaneous oxidative addition nor β-hydride elimination. Gold has the highest electronegativity among the transition metals, which is attributed to relativistic effects.17–19 Hence, the contraction of the 6s orbital in gold is much more significant than for the rest of the transition metals, which leads to an expansion of the 5d orbital, decreasing its electron–electron repulsion. Furthermore, 5d electrons are too low in energy to experience a significant backbonding to anti-bonding orbitals but not to empty non-bonding orbitals. Thus, a 3 centre-4 electron σ-bond is proposed in gold(I)-carbenes [L-Au![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR2]+ accompanied by orthogonal weak π-backbonding from the metal to both the ligand and the substrate (Fig. 2).
CR2]+ accompanied by orthogonal weak π-backbonding from the metal to both the ligand and the substrate (Fig. 2).
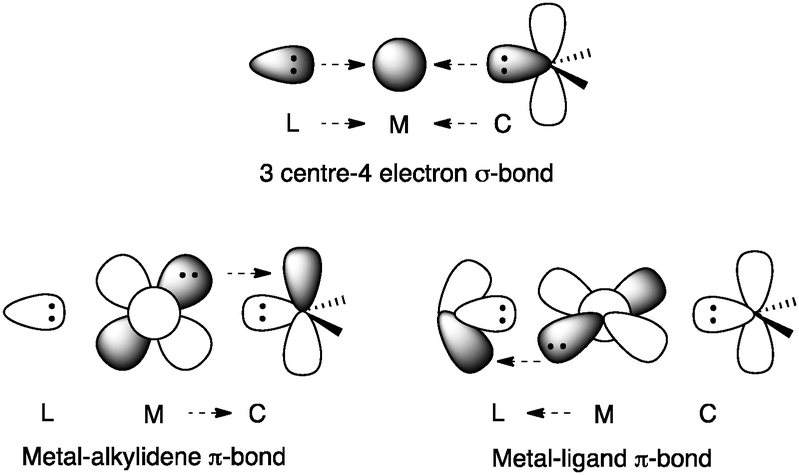 | ||
| Fig. 2 Ligand–metal-substrate orbital interactions in gold–carbenes [L-AuCR2]+. | ||
Gold(I) forms stable monomeric two coordinate π-complexes with alkenes,20–22 1,3-dienes,23 allenes,24 and substituted alkynes.19a,25,26 Variations of the bond lengths as well as the ligand–metal-substrate angle suggest that the alkene orientation is controlled largely by steric factors. Thus, terminal alkenes bind unsymmetrically with gold(I) resulting in longer bonds with the substituted carbon atom. The X-ray structure of isobutylene IPr–gold(I) complex 1a revealed a 0.086 Å difference between the metal–carbon bonds, whereas for norbornene (1b) and 2,3-dimethyl-2-butene (1c), this difference is 0.024 Å and 0.009 Å, respectively (Fig. 3).20b The angle between the metal and the centroid of the alkene also increased with the bulkiness of the alkene: 171.8° (isobutylene), 174.8° (norbornene), and 176.8° (2,3-dimethyl-2-butene).
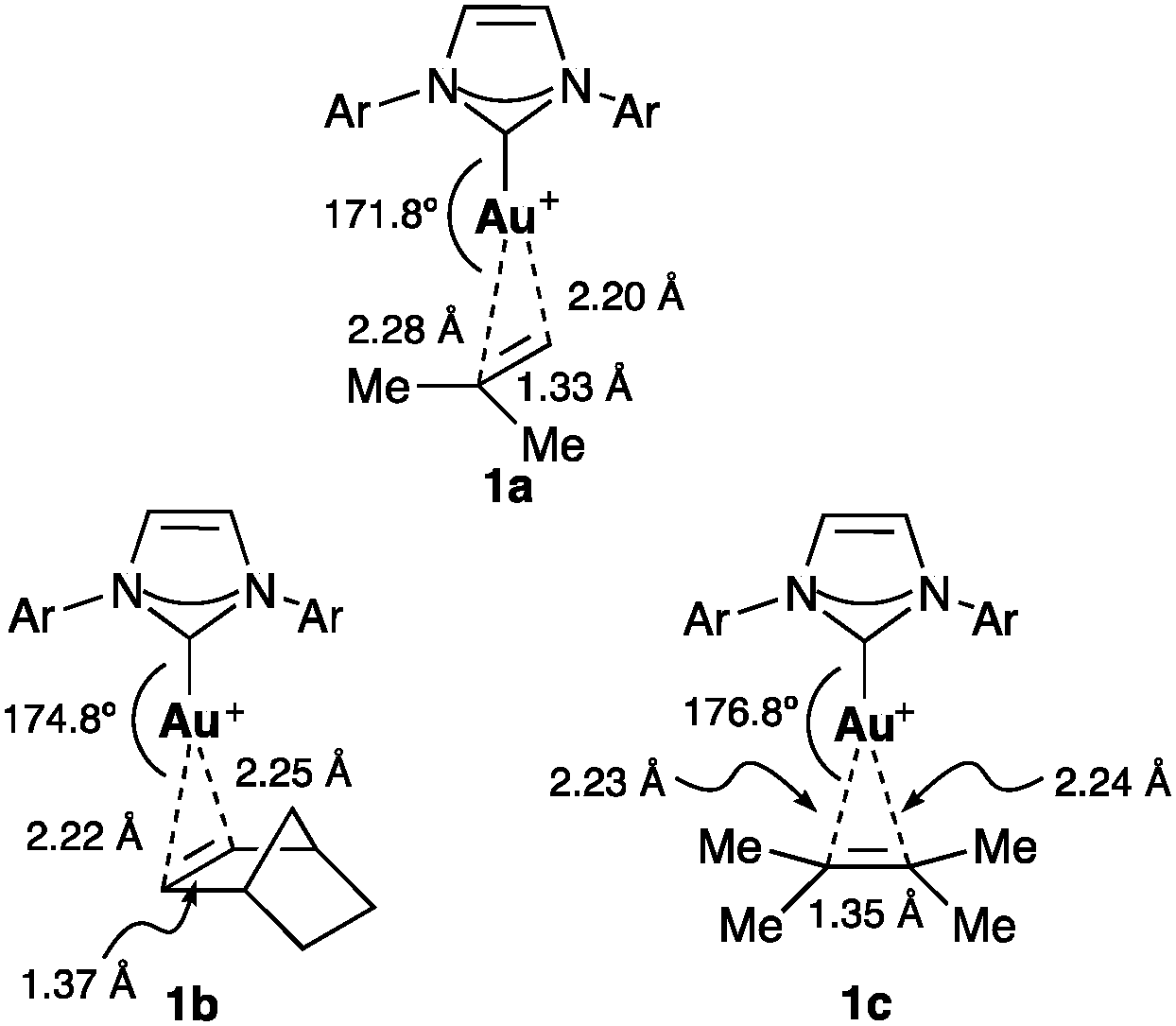 | ||
| Fig. 3 Isobutylene (1a), norbornene (1b) and 2,3-dimethyl-2-butene (1c) IPr–Au(I) complexes (Ar = 2,6-iPr2C6H3).20b | ||
In the case of allenes, structural and solution analysis demonstrate that gold(I) preferentially binds to the less substituted CC bond (Fig. 4).24
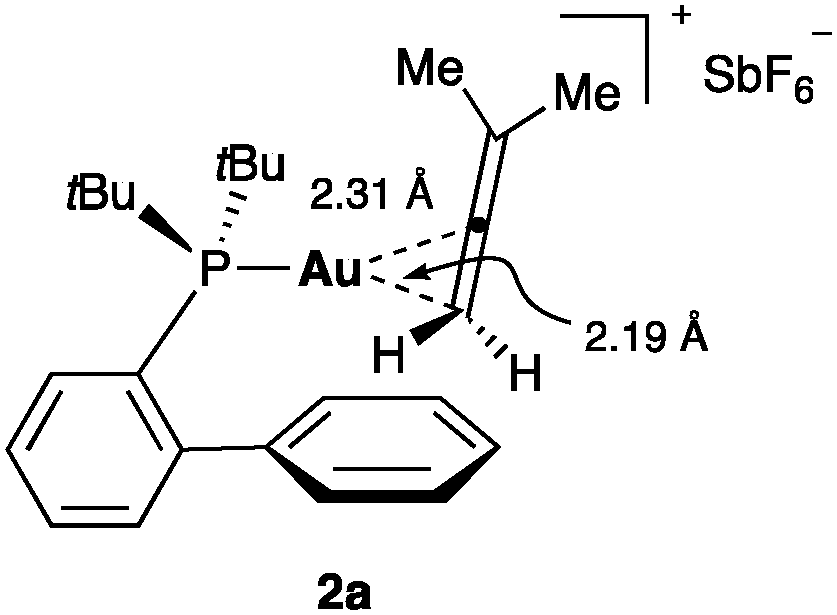 | ||
| Fig. 4 3-Methylbuta-1,2-diene gold(I) complex 2a.24 | ||
Although a theoretical study has proposed that π-coordinated gold(I) complexes 2 with model NHC or phosphite ligands could be in rapid equilibrium with η1-allyl species 2′, slightly favouring 2 (Scheme 1),27 experimental results with gold(I) complexes bearing bulky phosphine ligands rule out the involvement of 2′ in the low energy (≤10 kcal mol−1) π-face exchange processes.24
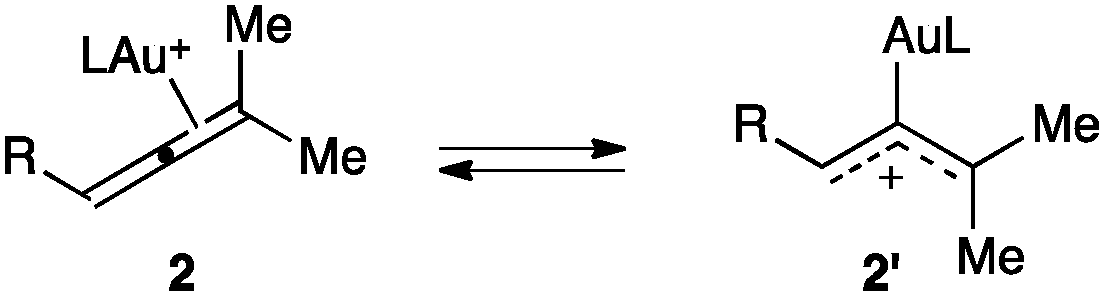 | ||
| Scheme 1 Proposed equilibrium between η2-allene (2) and η1-allyl species (2′).27 | ||
Internal alkyne gold(I) complex 3a shows the characteristic of almost symmetrical η2-coordination (Fig. 5).25e The triple bond length is identical to that of a free alkyne, although there is significant bending back of the alkyl substituents.
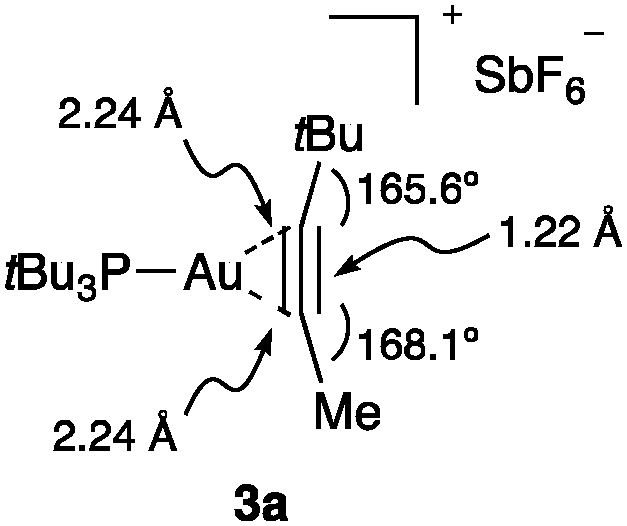 | ||
| Fig. 5 Structure of η2-alkyne Au(I) complex 3a.25e | ||
Nucleophilic attack
In general, attack of nucleophiles to η2-alkyne Au(I) complexes 3 gives trans-alkenyl species 4 (Scheme 2).3,28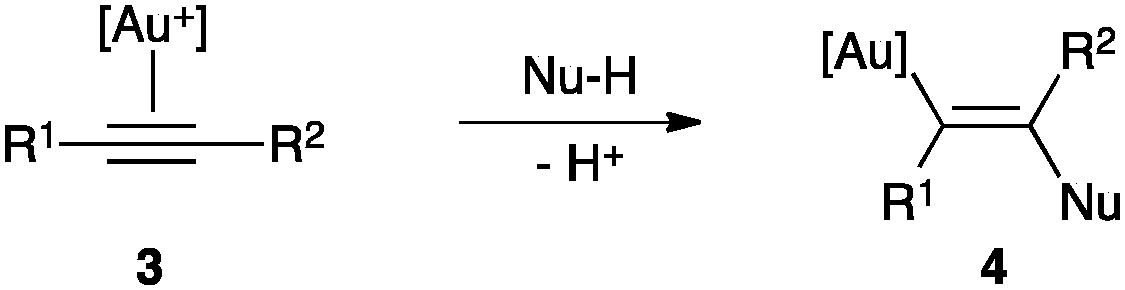 | ||
| Scheme 2 Nucleophilic attack to a Au(I) π-activated alkyne 3. | ||
Although an outer-sphere mechanism is widely accepted and it has been verified many times in the stereoselectivity of gold(I)-catalysed reactions,3 there are few exceptions. Although it is difficult to distinguish an outer-sphere attack from an insertion process, this type of mechanism was suggested in the gold(I)-catalysed hydroamination of alkynes and allenes with ammonia, since coordination to nitrogen was found to be preferred under catalytic conditions in the presence of an excess of alkyne (Scheme 3).29,30 The syn-insertion of methyl propiolate into the Au–Si bond of a gold silyl complex has been recently demonstrated.31
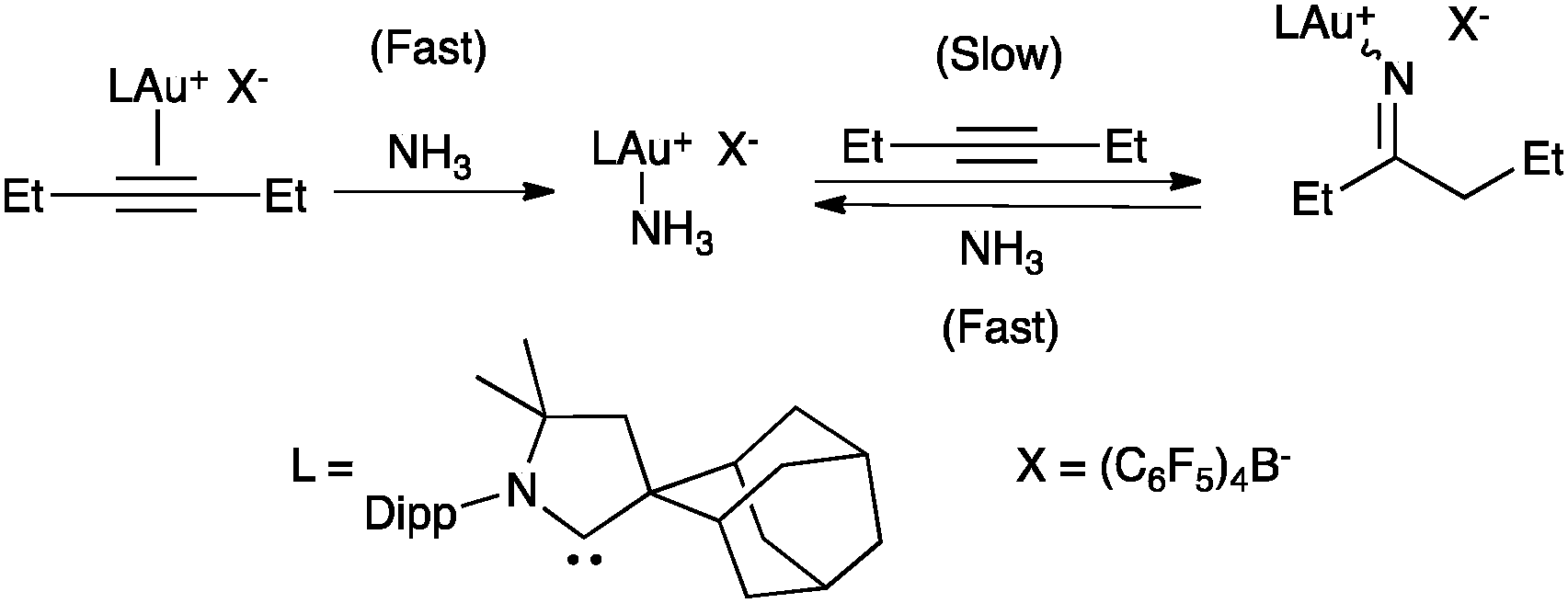 | ||
| Scheme 3 Ligand exchange prior to reaction suggesting an insertion process.29 | ||
A wide range of carbon and heteronucleophiles such as arenes,32 heteroarenes,33 alcohols,34 amines,35 imines,36 sulfoxides,37N-oxides,38 and thiols39 have been used as nucleophiles in inter- or intramolecular processes. An early example is the gold(III)-catalysed cyclisation of α-hydroxyallenes that allows the straightforward synthesis of 2,5-dihydrofurans (Scheme 4).40
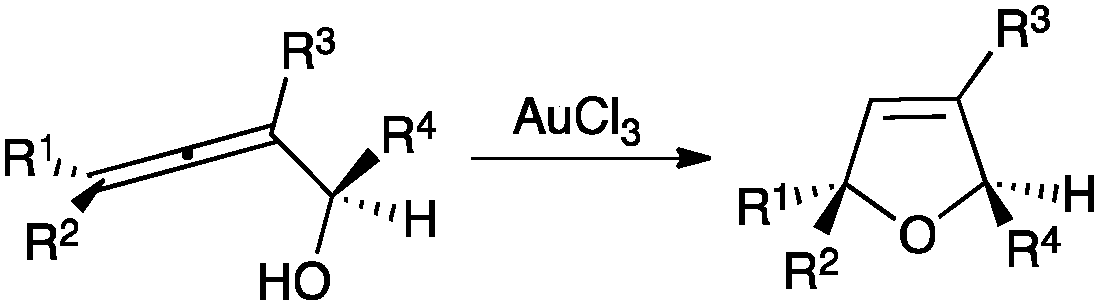 | ||
| Scheme 4 Gold(III)-catalysed cyclization of α-hydroxyallenes.40 | ||
Noteworthy, the regioselectivity in the cyclization of halogenated allenones can be controlled depending on the oxidation state of the catalyst (Scheme 5).41 Thus, gold(III) favours a mechanism in which the ketone is preferentially activated leading to cyclisation with concomitant 1,2-halogen migration through bromonium intermediate 5, whereas gold(I) coordinates to the allene leading to cyclisation without halogen migration via6.
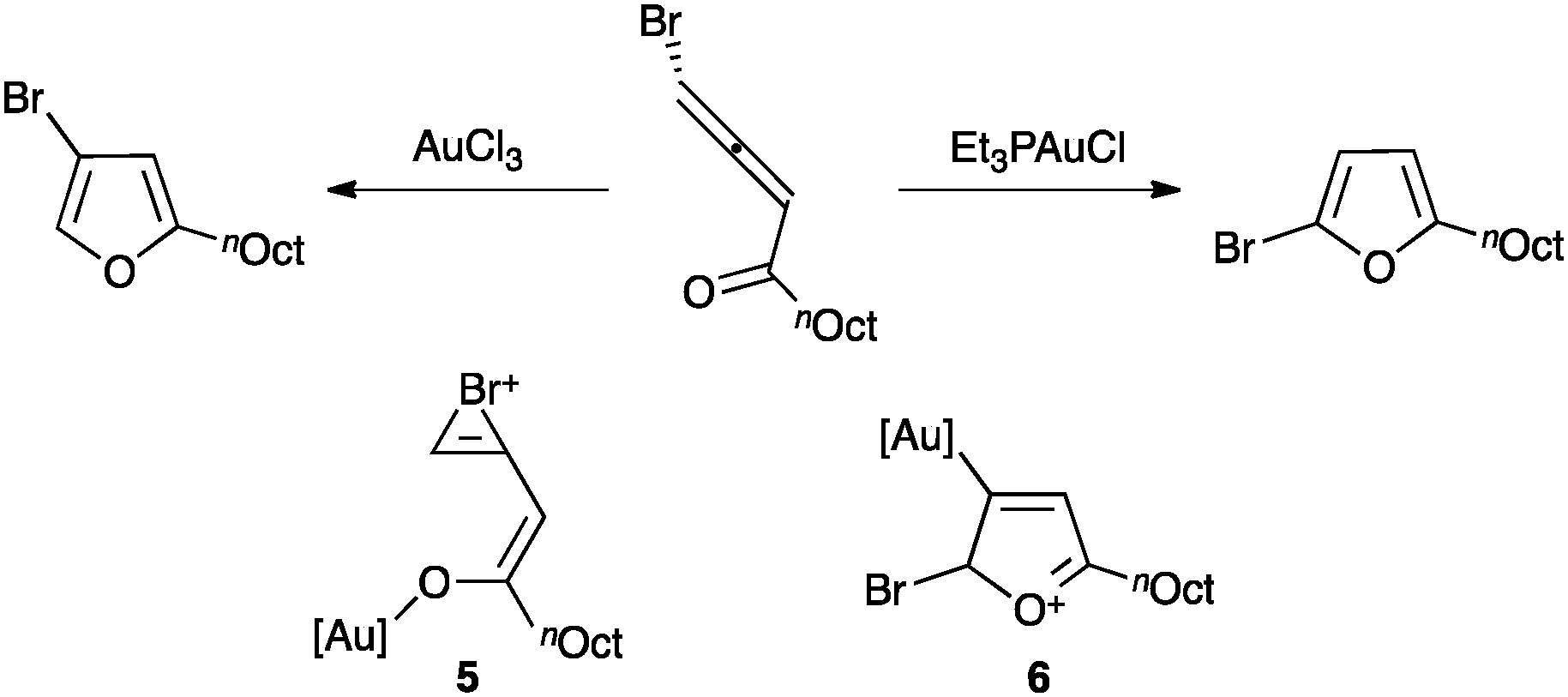 | ||
| Scheme 5 Regioselective cyclization of halogenated allenones.41 | ||
The intramolecular nucleophilic addition deserves a special mention when the nucleophile is located at the propargylic position.42 Thus, propargylic carboxylates can undergo 1,2- or 1,3-acyloxy migrations leading to the formation of vinyl gold(I) carbenoid species 7 or allene gold(I) complexes 8, which could be in rapid equilibrium (Scheme 6).43,44 A double 1,2-shift, which also leads to 8, was found to be energetically more favoured than the direct 1,3-shift, although different substitution at the substrate could significantly influence this preference.
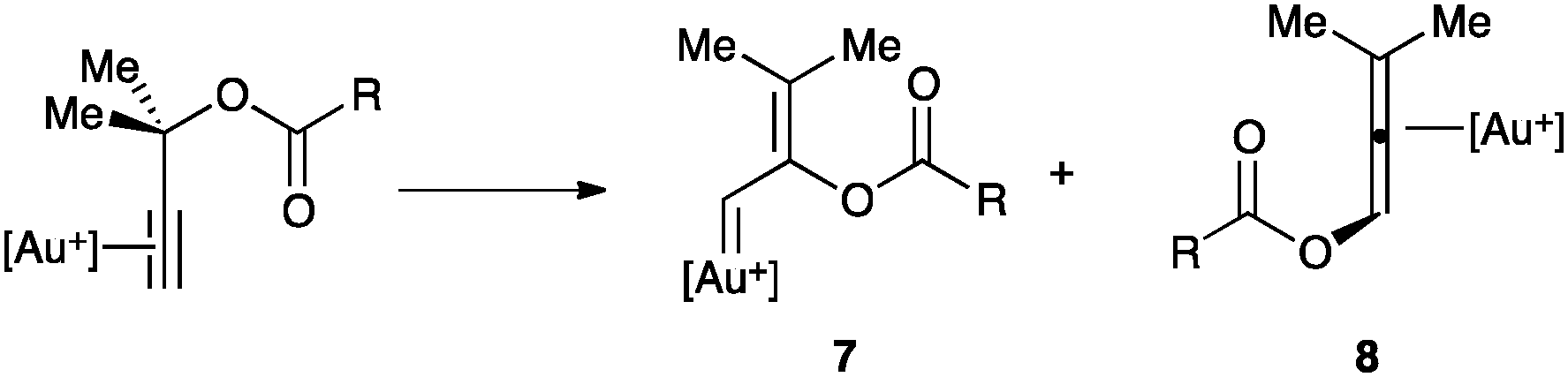 | ||
| Scheme 6 Key intermediates in the propargylic migrations. | ||
Cycloisomerisation of 1,n-enynes is a class of emblematic transformations in which an alkene acts as the nucleophile towards an alkyne activated by gold.3,45 A diverse array of reactions are possible with a significant increase in molecular complexity and in a fully atom economic manner. A representative example is the single-cleavage rearrangement of 1,6-enynes 9 to form conjugated dienes 10, which has been proposed to proceed through a cyclopropyl gold(I) carbene 11 that can also be viewed as an homoallyl carbocation (Scheme 7).46,47
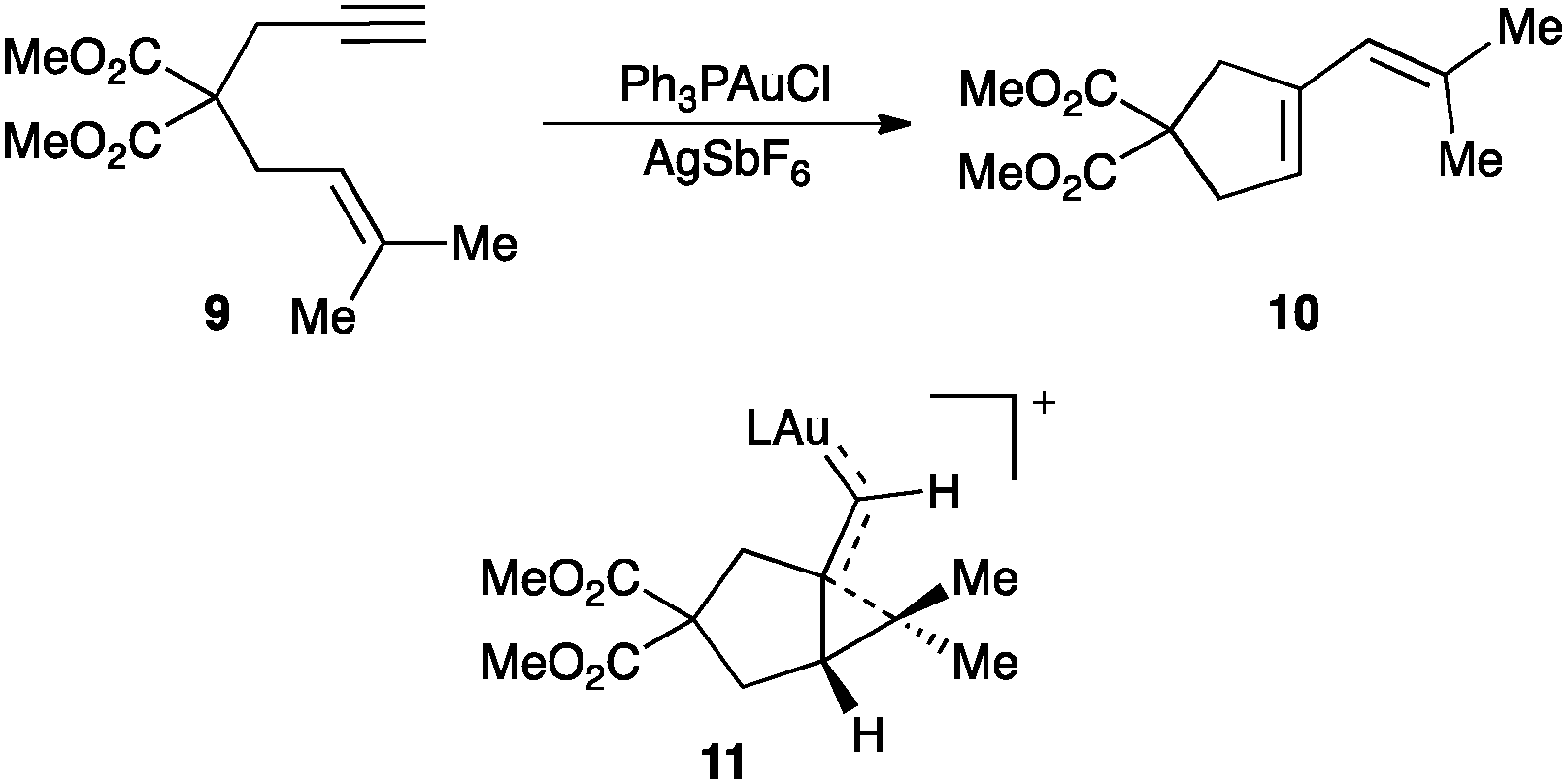 | ||
| Scheme 7 Single cleavage rearrangement of 1,6-enyne 9 to form 10 through key intermediate 11. | ||
Gold intermediates
Although many gold intermediates are highly reactive to be readily isolated, some progress has been achieved in the observation of a few key species.6In general, alkynes are selectively activated by gold(I) in the presence of alkenes. This high site-selectivity (alkynophilicity) of gold(I) is not directly related to a thermodynamic preference for the coordination to the alkynes, but to a higher reactivity of the η2-alkyne gold(I) complexes towards nucleophilic attack.48 Thus, NMR studies confirmed that both 12a and 12b coexist with free 1,7-enyne (Scheme 8), although this class of substrates exclusively reacts by intramolecular attack of the alkene to η2-alkyne gold(I) species such as 12a.49
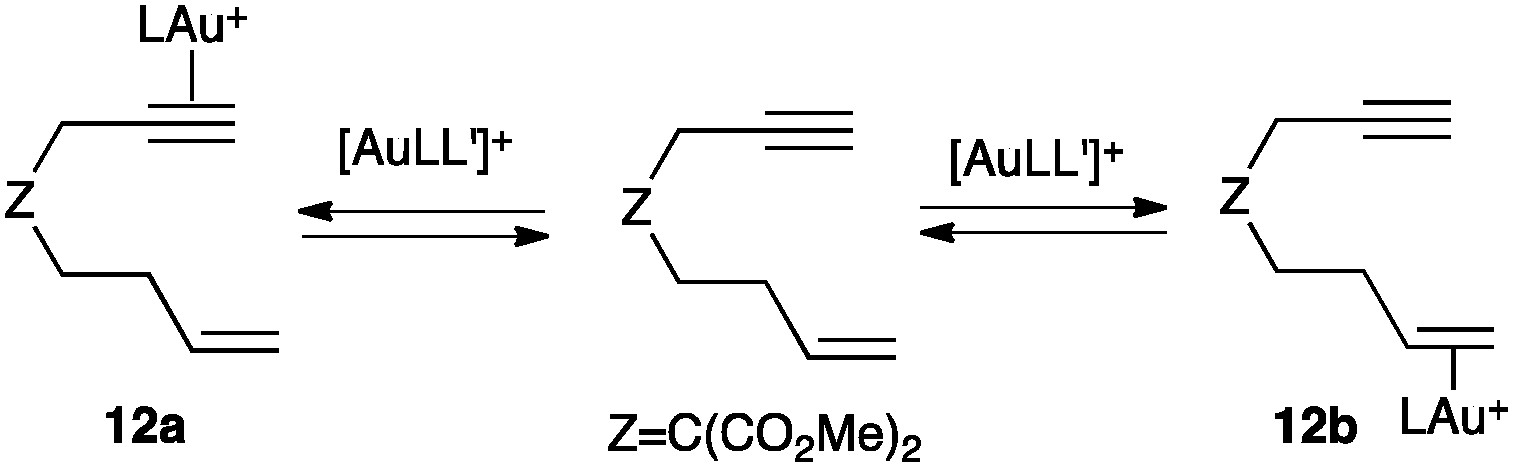 | ||
| Scheme 8 Coordination of the gold(I) complex to alkynes and alkenes (L = JohnPhos).48 | ||
The first stable organogold(I) intermediate (13) was isolated in an intramolecular reaction between an allene and an ester (Scheme 9).50,51 This result demonstrated the formation of vinyl gold(I) complexes by nucleophilic attack on allene–gold(I) complexes.
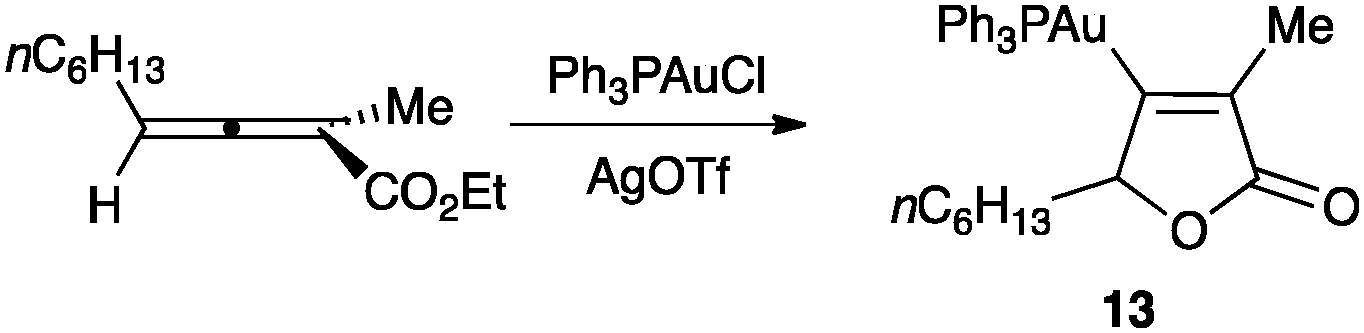 | ||
| Scheme 9 Formation of the first isolated vinyl gold(I) complex in a gold(I)-catalysed cyclization.50 | ||
An interesting debate has been centred on the nature of the gold–carbon bond in complexes of type [LAuCHR]+.52,53 For the parent reaction between alkynes and alkenes, gold(I) carbenes (14a), gold(I) stabilized carbocations (14b), homoallyl (14c) or even cyclobutyl carbocations (14d) could be conceived (Scheme 10).46 This transformation was realized intermolecularly between aryl alkynes and electron-rich alkenes using cationic gold(I) complexes bearing bulky phosphines (Scheme 10).54 This reaction has also been applied for the synthesis of large macrocycles.55 The observed regiochemistry as well as trapping experiments were consistent with a reaction proceeding through a highly distorted cyclopropyl gold(I) carbene 15, intermediate between 14a–c, which undergoes ring expansion to form cyclobutyl carbocation 16. Related distorted cyclopropyl gold(I) carbenes 15 (Scheme 7) were also proposed in the reaction of propiolic acid with alkenes56 as well as in the addition of nucleophiles to enynes.46,47,57,58
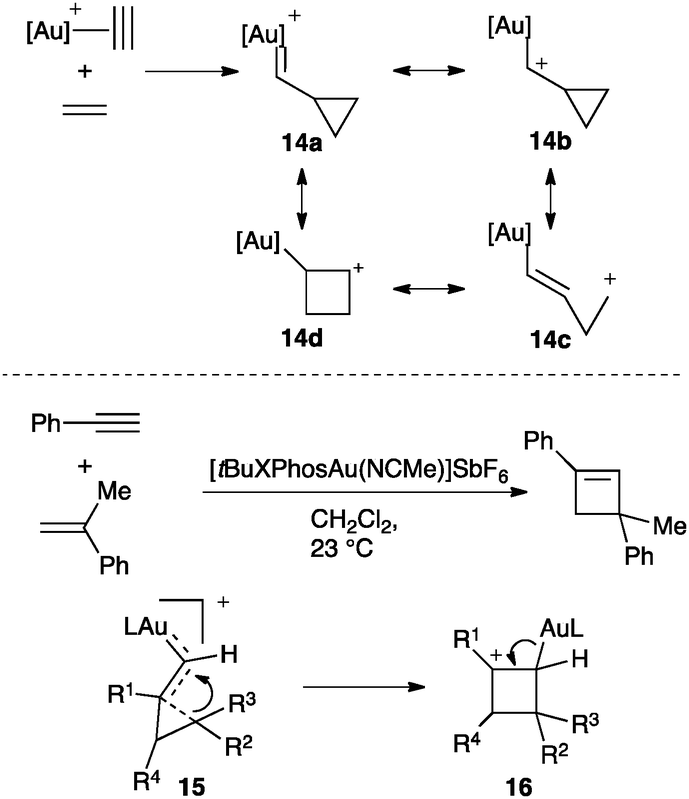 | ||
| Scheme 10 Possible intermediates involved in the gold(I)-catalysed intermolecular [2+2] cycloaddition of alkynes with alkenes. | ||
The regioselective cyclization of enynes 17 with a pending carboxylic acid forms selectively lactones 18 and/or 19 (Scheme 11).59 Following the Stork–Eschenmoser model for cyclizations of squalene and oxidosqualene,60 these types of cascade cyclizations were rationalized as proceeding through concerted transition states such as 20, not involving cyclopropyl gold(I) carbenes as discrete intermediates.61 Analogous reasoning was put forward in the asymmetric hydroarylation of 1,6-enynes62 and in other related processes.63
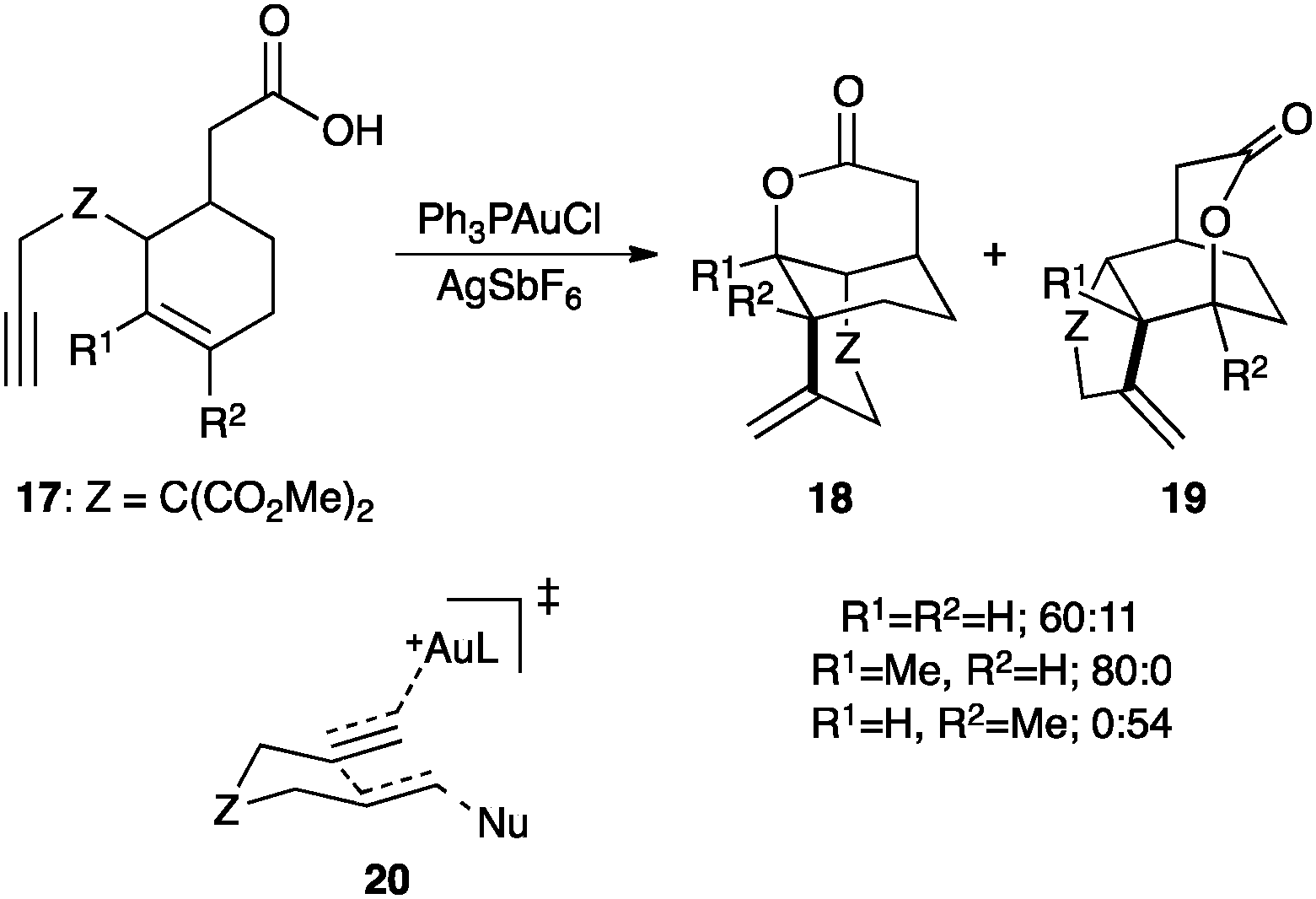 | ||
| Scheme 11 Cascade cyclization of 1,6-enynes 17.59 | ||
However, other studies strongly suggest that gold(I)-catalysed 1,n-enyne cyclizations occur step-wise involving cyclopropyl gold(I) carbenes (11) and related types of intermediates.45–47,64 Thus, for example, reaction of 1,6-enyne 21 with indole leads to adducts 22 and 23 by nucleophilic attack at the cyclopropyl (b) or carbene (a) carbons of intermediate 24 (Scheme 12).65 The use of complex [IPrAu(NCPh)]SbF6 as the catalyst with a highly donating NHC ligand enhances the carbene-like nature of the intermediate, favouring nucleophilic attack at the carbene carbon leading to 23.
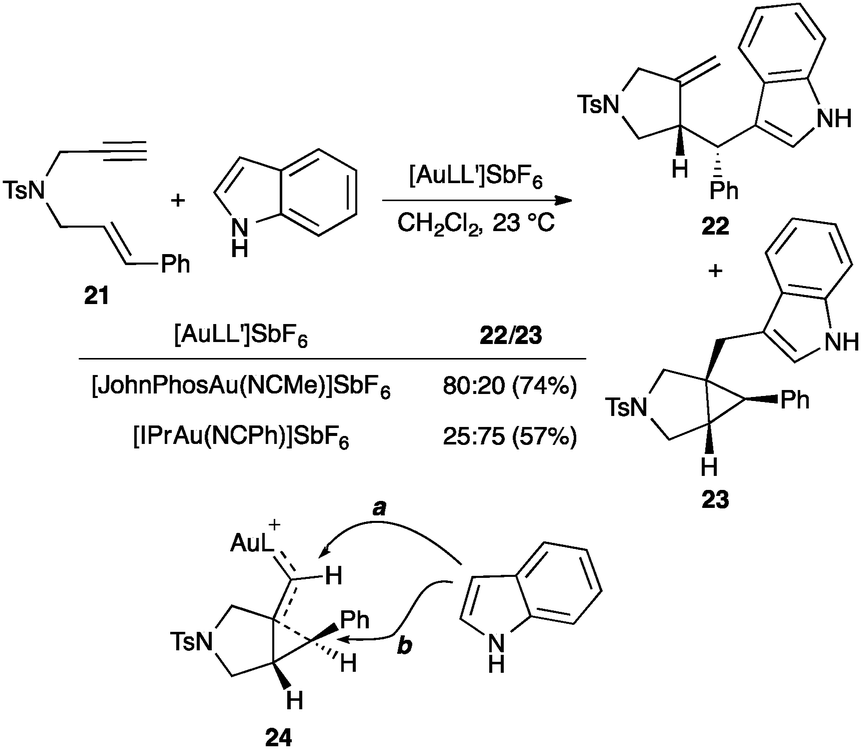 | ||
| Scheme 12 Nucleophilic addition to the gold(I) carbene position.65 | ||
Spectroscopic or structural data for carbene-like structures of relevance in homogeneous catalysis are lacking. The interesting earlier structure of gold(I) carbene 25 showed C–Au length within the range of a single bond between a sp2 carbon atom and the metal while the C–N bond is shorter than a typical imine (Fig. 6).66 However, this scaffold is far from the intermediates generated in a catalytic cycle.
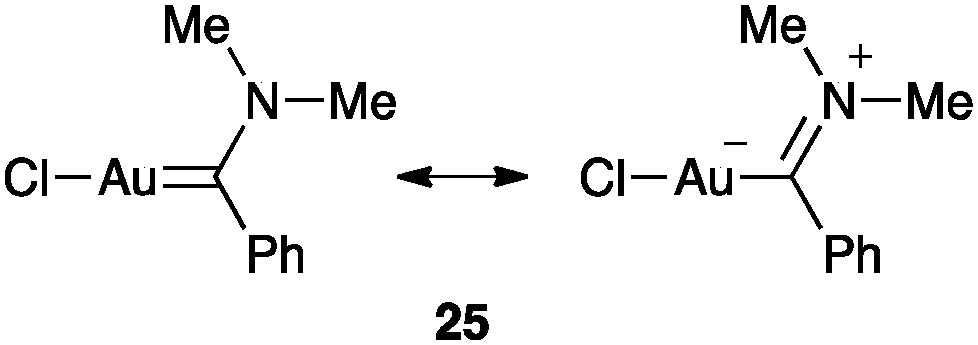 | ||
| Fig. 6 Gold(I) carbene 25.66 | ||
Opening of cyclopropenone acetal 26 with gold(I) leads to Z-27 that isomerises to E-27 presumably through gold(I) carbene 28 (Scheme 13).67 The spectroscopic data of Z-27 and E-27 revealed an oxocarbenium cationic structure.
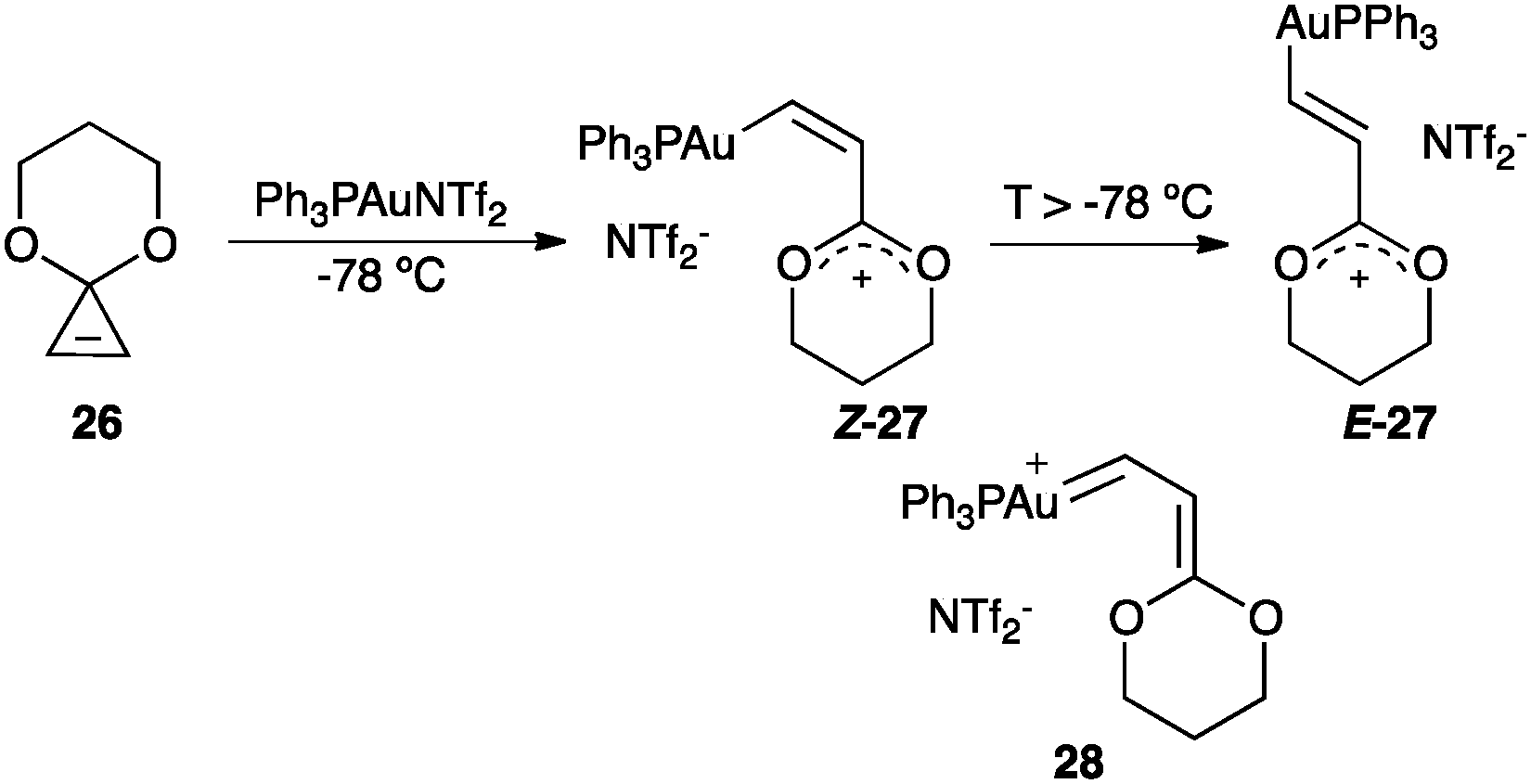 | ||
| Scheme 13 Generation of carbocationic organogold(I) complexes.67 | ||
An in-depth theoretical analysis of the bond rotation energy for different carbocations demonstrates that AuL has a similar stabilizing effect as OMe on an allyl carbocation (Fig. 7).52 Moreover, the bond length between gold and the carbene carbon decreased with strong σ-donating ligands such as chloride or N-heterocyclic carbenes, but it increases with less donating, π-acidic ligands such as phosphines or phosphites by reducing the back-donation to the substrate. This study concluded a continuum character of the organogold(I) species ranging from metal stabilized singlet carbene to metal coordinated carbocation depending on the substitution pattern and the ligand on gold.
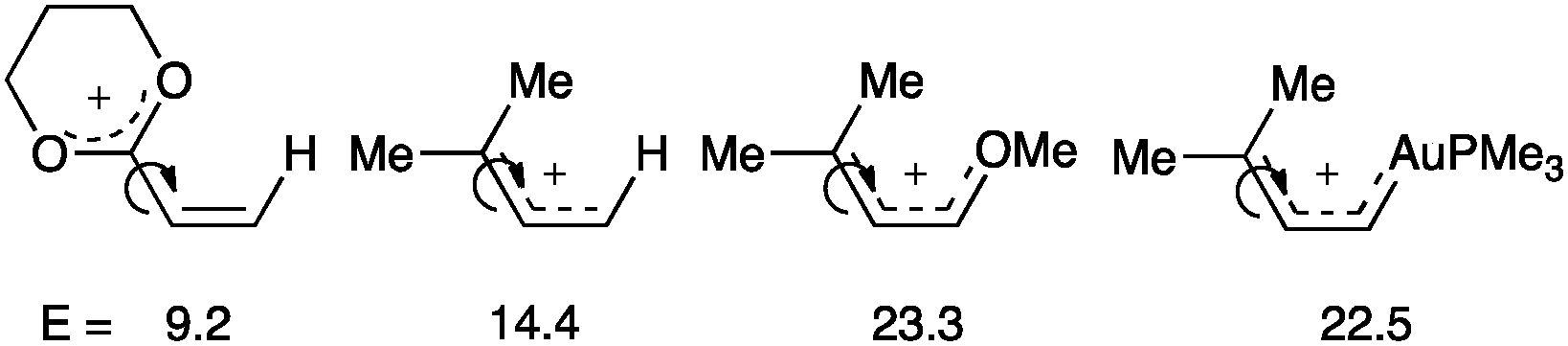 | ||
| Fig. 7 Calculated rotation energies depending on the substituents (M06, kcal mol−1).52 | ||
Gold(I) allenylidenes such as 29 have been recently structurally characterized (Fig. 8).68 Theoretical and experimental analysis suggested that the heteroatom stabilization favoured an Au-propargyl carbocation (29c-d). In contrast, without this stabilization provided by the heteroatoms, the structure depends on the ligand, ranging from gold(I)-propargyl carbocations to gold(I) allenylidenes. Gold(III) allenylidenes with similar substitution at the terminus carbon have also been structurally characterized.69
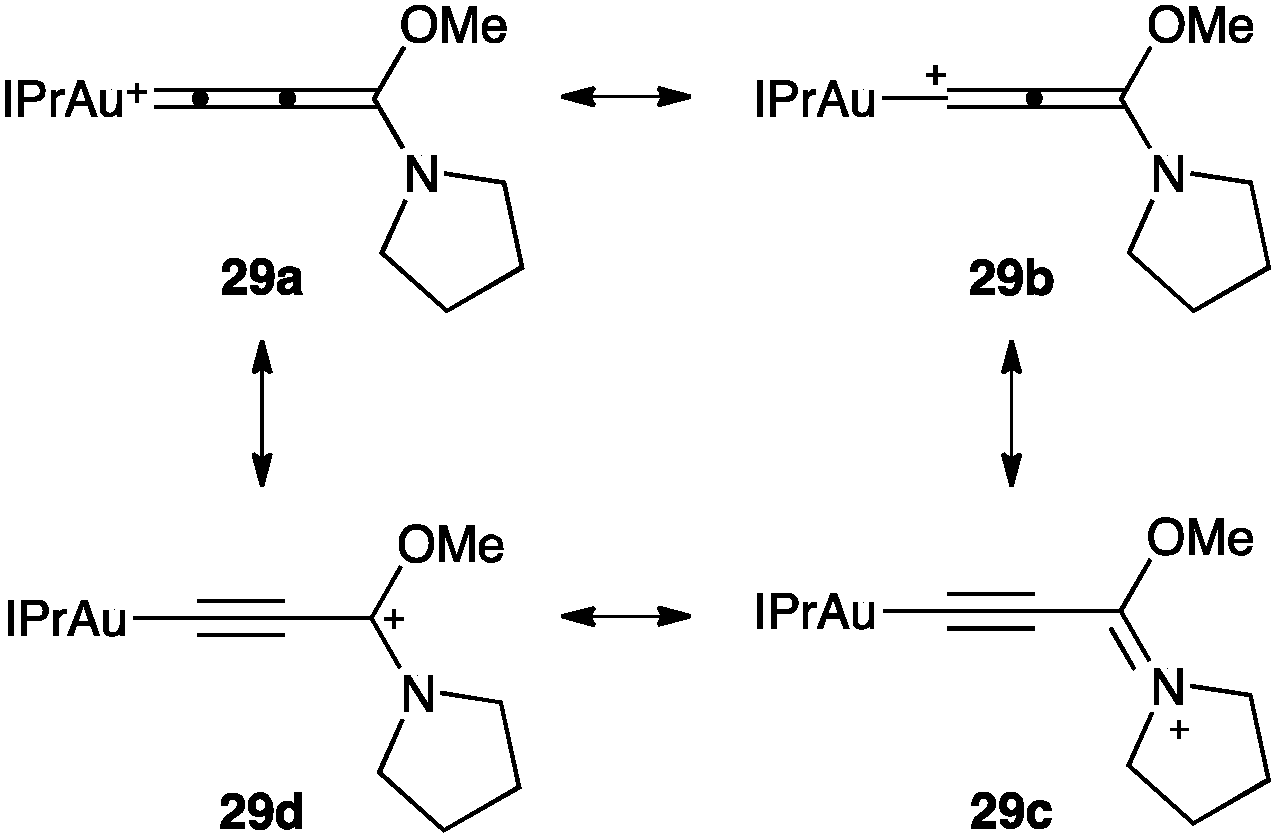 | ||
| Fig. 8 Gold(I) allenylidene 29.68 | ||
Evolution of the key gold intermediates
Subsequent to the gold activation of the unsaturated scaffold and its nucleophilic attack, the intermediates can evolve through many different pathways leading to a huge variety of complex products.3 The simplest evolution of the alkenyl gold(I) intermediates is their reaction with an electrophile, most usually by protodeauration regenerating the active catalyst (Scheme 14).6,50,51 Similarly, reaction with iodine and related electrophiles leads to the corresponding halo-derivatives.50,70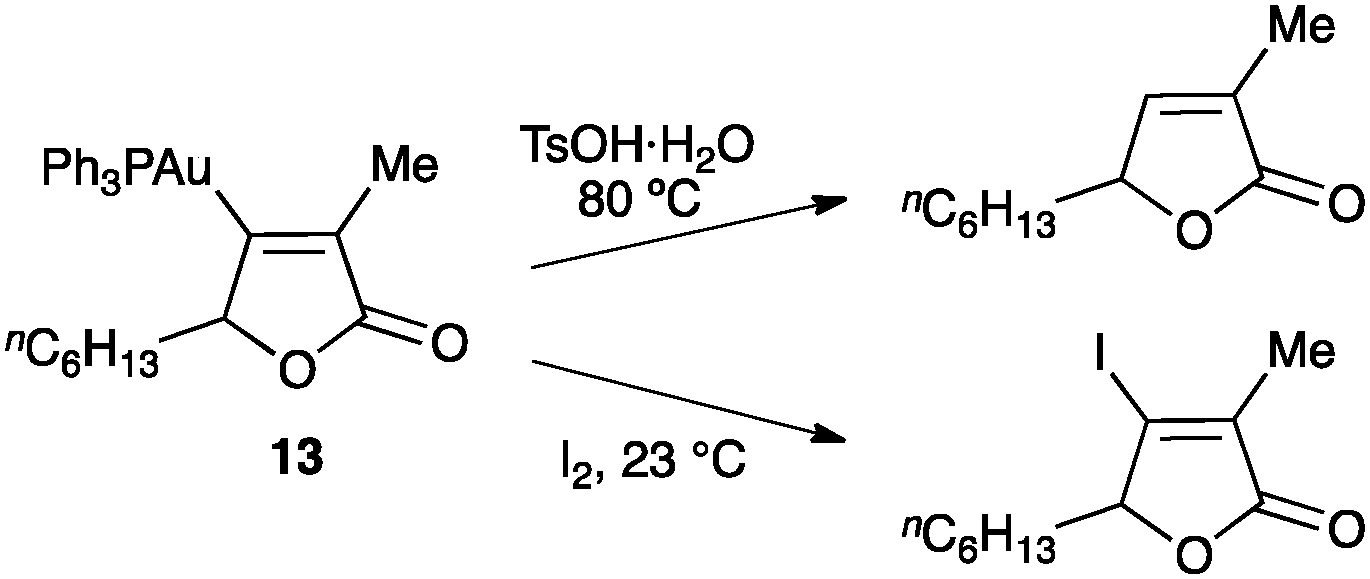 | ||
| Scheme 14 Electrophilic attack to an alkenyl gold(I) complex.50 | ||
A mechanistic study of the intermolecular hydroamination of allenes by nucleophilic attack of a carbazate followed by protodeauration showed that the catalyst resting state was the η2-allenegold(I) complex (Scheme 15).71 Kinetic analysis showed zero order for the carbazate, suggesting that this nucleophile is not involved in the rate-determining step. DFT calculations predicted that the activation of the allene was the step with the highest activation energy, 11.6 kcal mol−1. A two-step, no intermediate process followed by an irreversible protonation was proposed for this transformation.
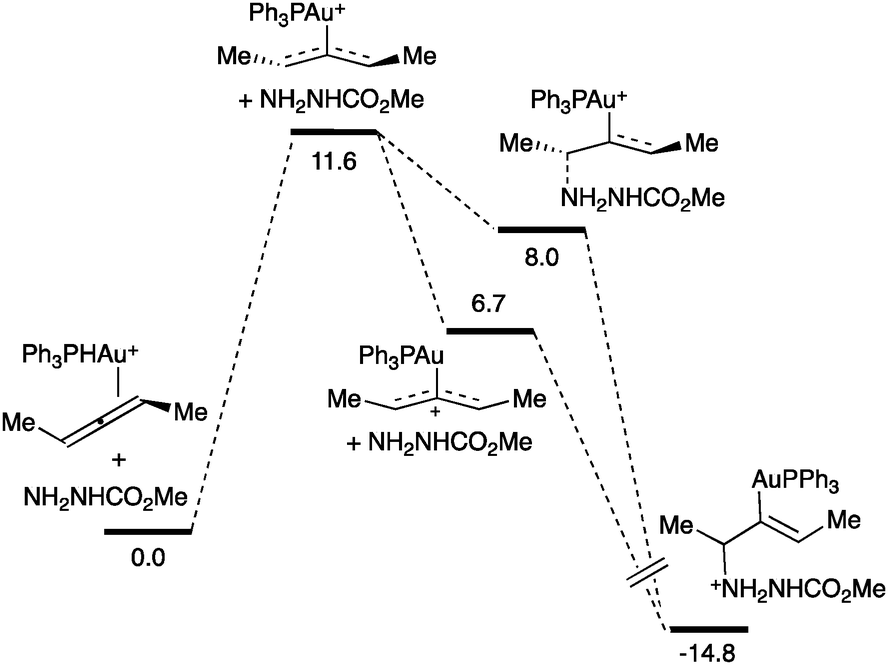 | ||
| Scheme 15 Reaction coordinate for the hydroamination of allenes (kcal mol−1).71 | ||
The alkenyl gold(I) intermediate could further react in a multistep process. As an important example, the enantioselective gold(I)-catalysed Rautenstrauch rearrangement of 1,4-enyne 30 with a propargylic pivaloyl group to form cyclopentenone 31 proceeded through 1,2-migration generating a vinyl gold(I) complex 32, which underwent cyclisation with the alkene to form five-membered ring 34 (Scheme 16).72 DFT calculations suggested that it is the helicity of the pentadienyl cation intermediate 33 that retains the chiral information.73
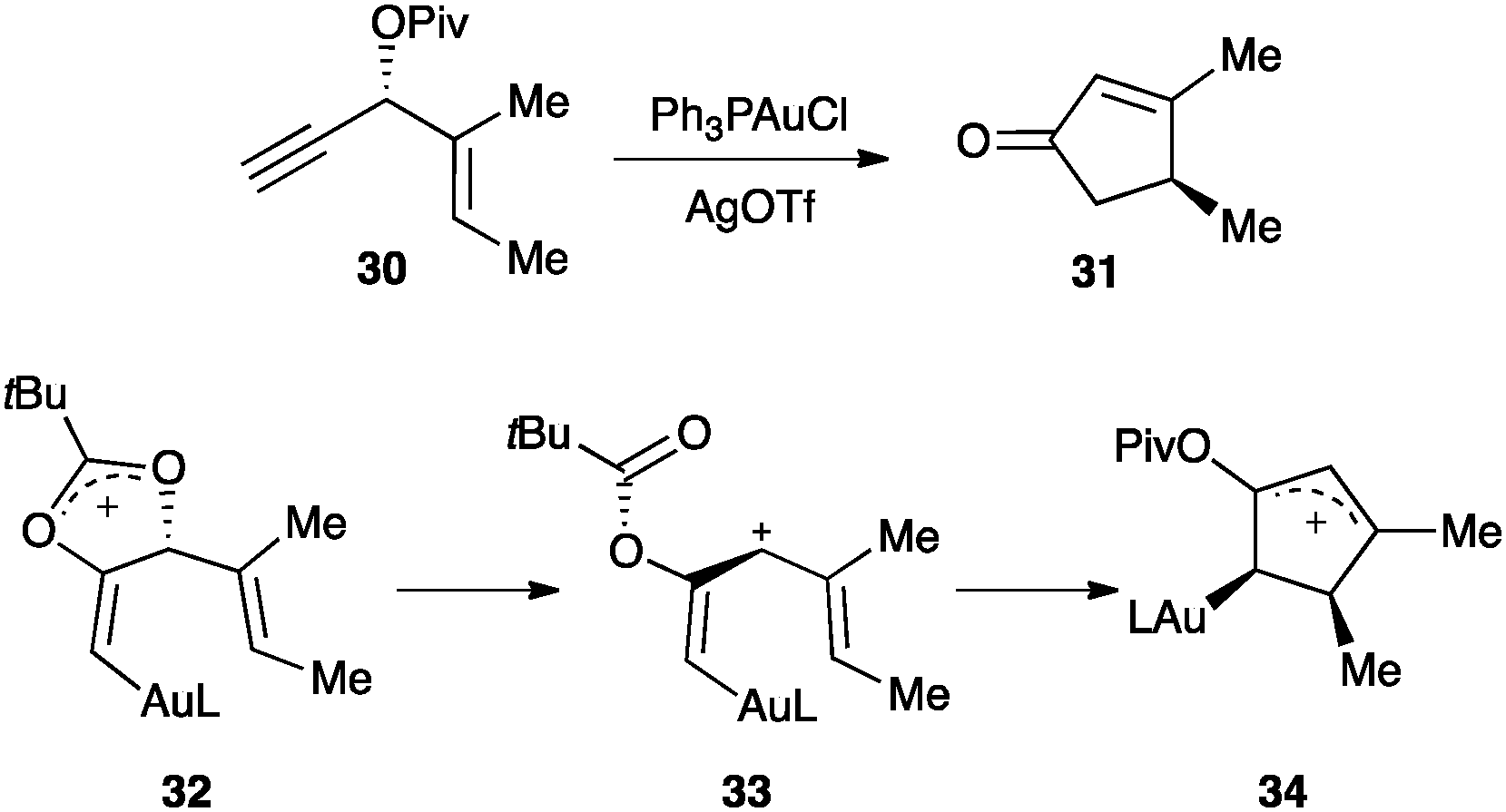 | ||
| Scheme 16 Enantioselective gold(I)-catalysed Rautenstrauch rearrangement.72,73 | ||
Similarly, a gold(I)-catalysed 1,3-propargylic acetate migration of substrates such as 35 generates intermediate 36 that undergoes Nazarov-type electrocyclisation through 37 generating gold(I) carbene 38 (Scheme 17).74 This intermediate can further react by intramolecular cyclopropanation to form tricyclic structure 39.
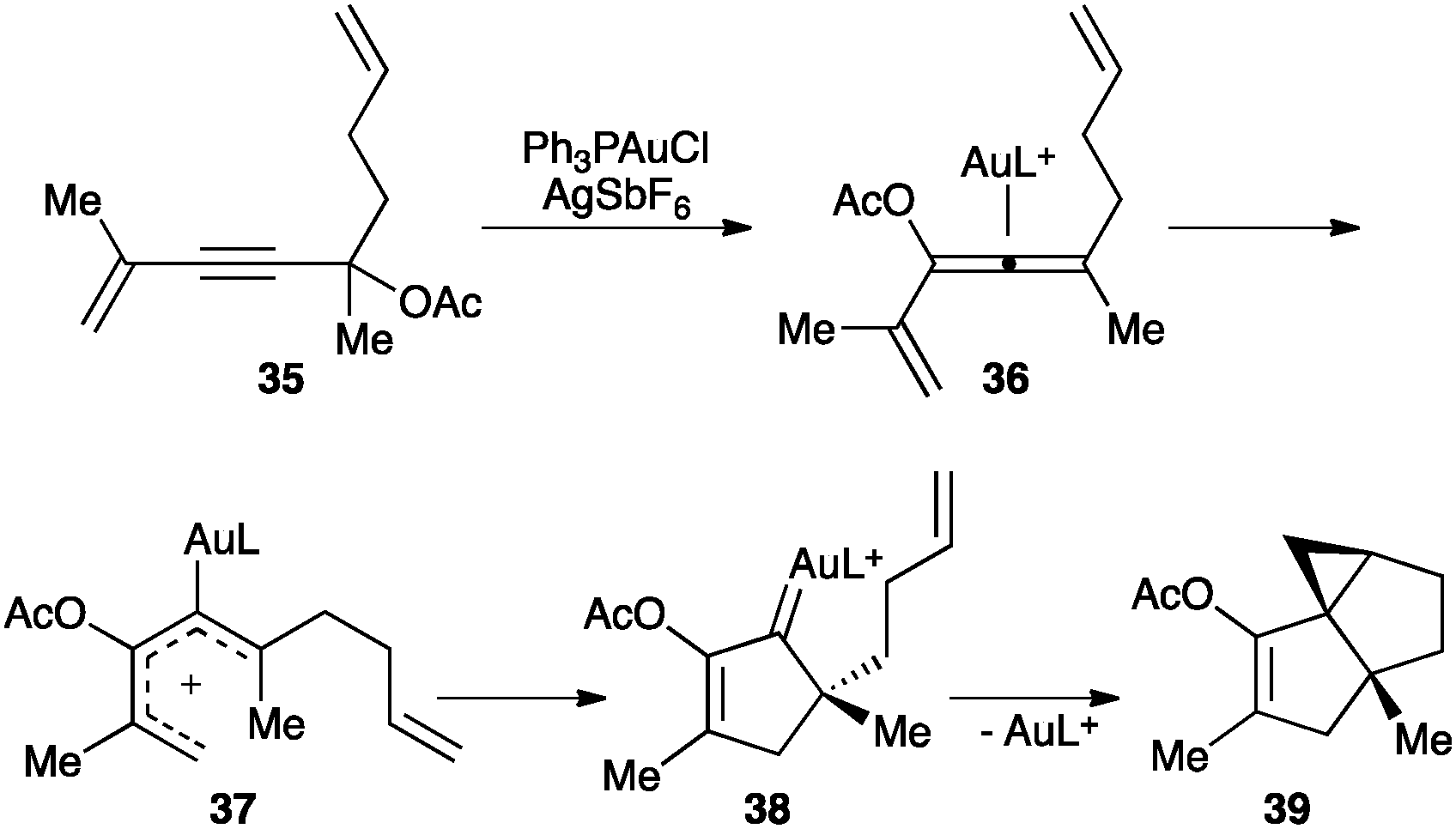 | ||
| Scheme 17 Nazarov-type cyclization of substrates 35.74 | ||
An interesting cyclization of allenes with dienes proceeds by formal [4+3] and [4+2] cycloaddition forming seven or six-membered rings (Scheme 18).27 The competitive cyclization processes are initiated by the selective activation of the allene with the gold(I) catalyst (40) to form the allyl cation 41 as the rate-determining step, which would rapidly lead to gold(I) carbene 42 in a concerted manner. This intermediate can then evolve through a 1,2-hydrogen shift leading to 43 or through a 1,2-alkyl shift resulting in a ring contraction forming 44. Gold(I) carbene 42 could be trapped by oxidation with diphenylsulfoxide forming a bicyclic ketone.27a
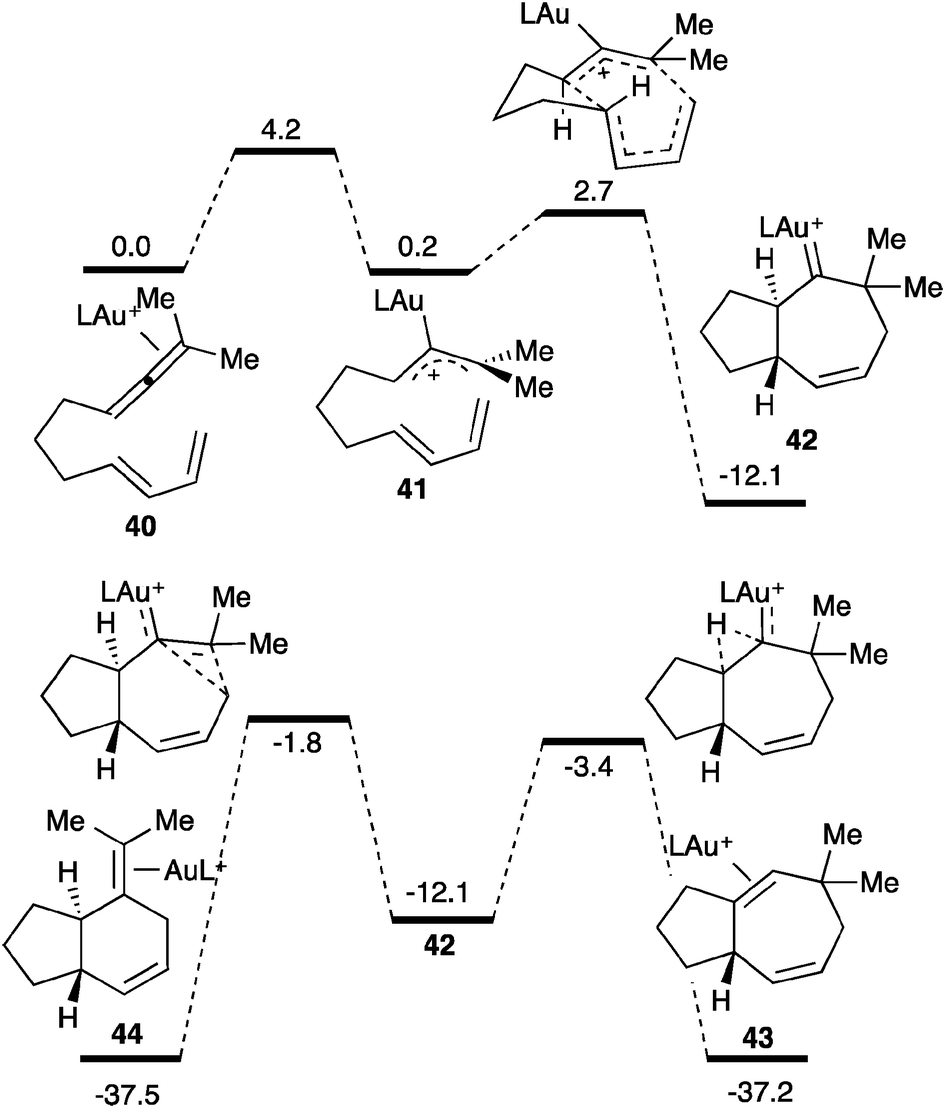 | ||
| Scheme 18 Reaction coordinate of the cycloaddition between allenes and dienes (kcal mol−1), L = P(OMe)3.27 | ||
A different type of behaviour (push–pull reactivity) was suggested for certain alkenyl gold(I) complexes.37a,75–77 An early example is the rearrangement of alkynyl sulfoxides.78 In this case, the alkyne activation followed by endo or exo nucleophilic attack of the sulfoxide depending on R and formation of an alkenyl gold(I) intermediate (45 or 46, respectively) was initially proposed (Scheme 19). These species were proposed to evolve by retro-donating electron density from the metal and pushing the sulphide moiety forming a α-carbonyl gold(I) carbene 47 that underwent an intramolecular Friedel–Crafts reaction to give 48 and then 49. However, recent studies supported theoretically and experimentally a completely different progression after the initial cyclization to 46.78c Since the prepared α-carbonyl gold(I) carbene did not undergo the intramolecular Friedel–Crafts reaction, further DFT calculations were performed. Thus, a subtle change in the conformation of 46 allows a [3,3]-sigmatropic rearrangement to form 48, which explains the observed results. Formation of α-carbonyl gold(I) carbenes has also been considered to be unlikely in other reactions of alkynes with sulfoxides.37c
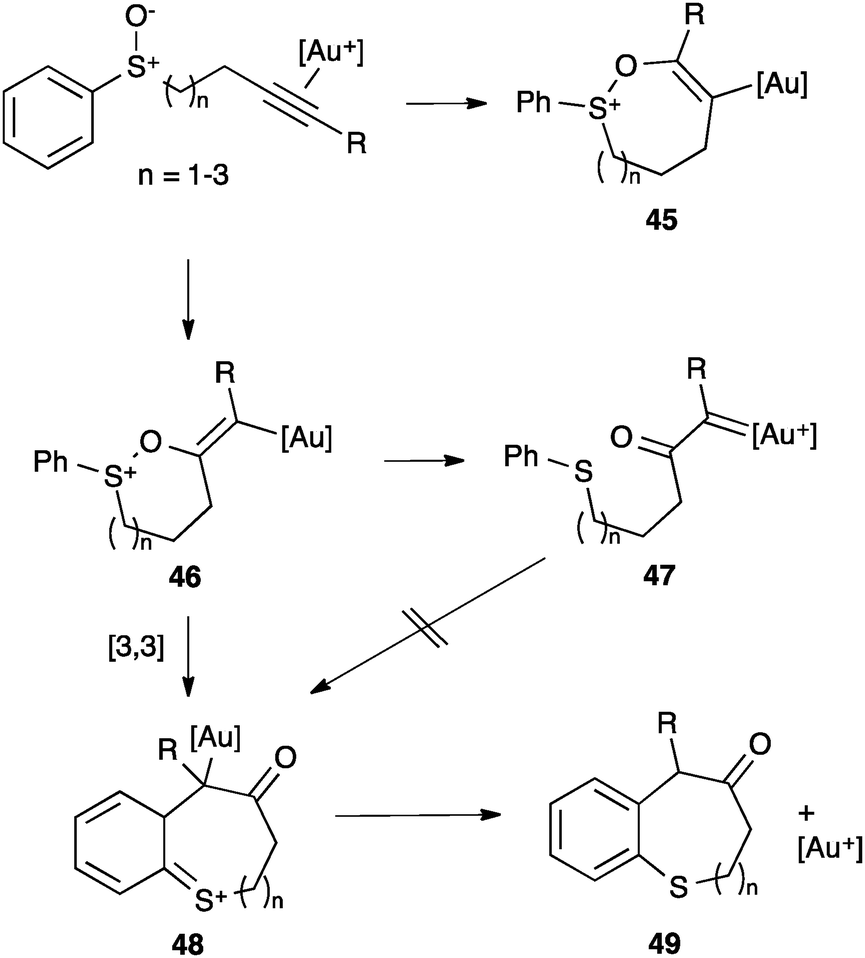 | ||
| Scheme 19 Cyclization of alkynyl sulfoxides.78c | ||
The involvement of α-oxo gold(I) carbenes38,79,80 was also questioned in the related reaction of alkynyl amine N-oxides.76 This time DFT calculations revealed another completely different pathway (Scheme 20).81 Hence, the gold-coordinated amine N-oxide 50 attacks the alkyne to form 51, which evolves by 1,5-hydrogen shift to form iminium cation 52, which undergoes an intramolecular Mannich reaction to form the final piperidinone.
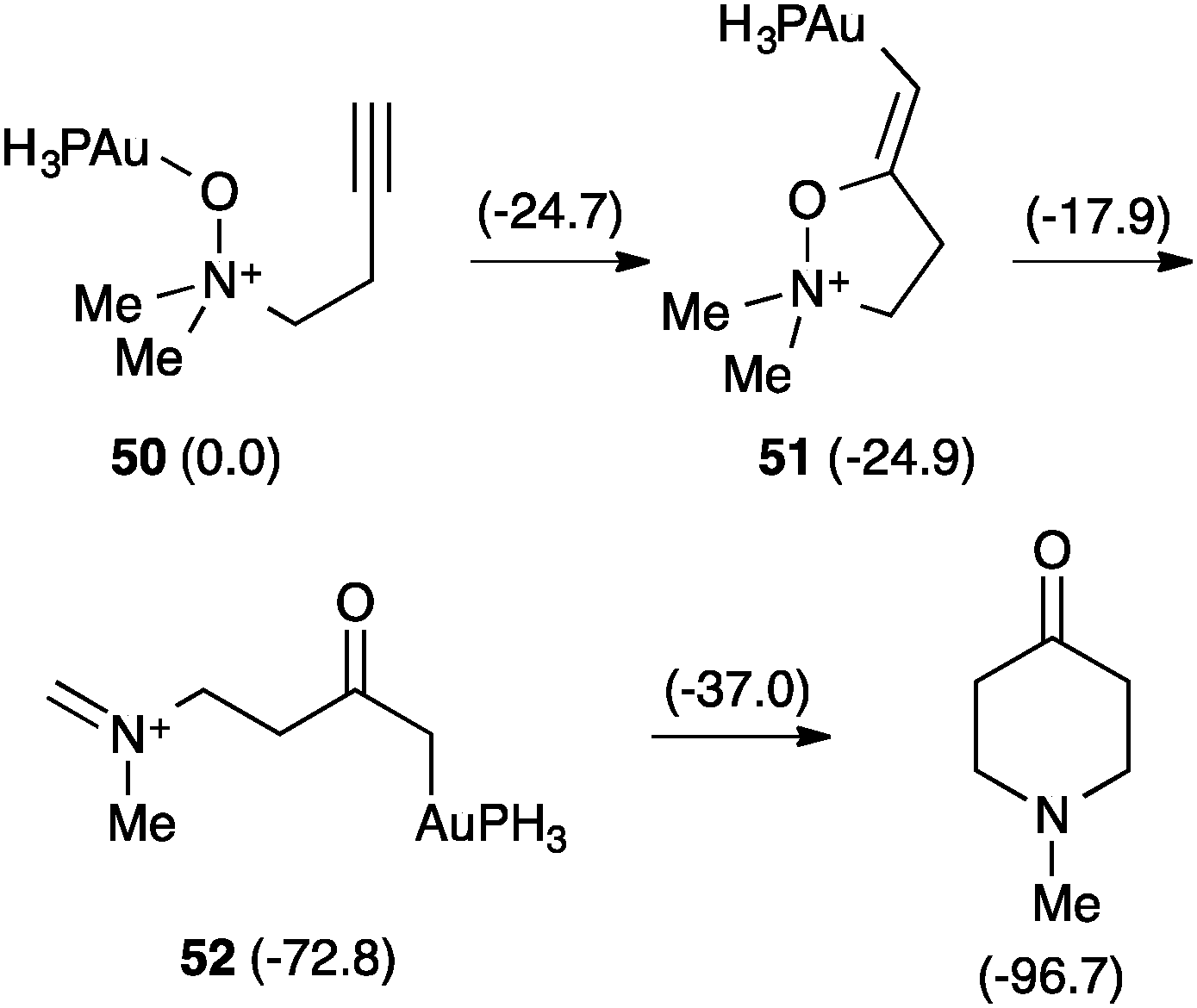 | ||
| Scheme 20 Rearrangement of alkynyl amine N-oxides (kcal mol−1).81 | ||
Nevertheless, a α-carbonyl gold(I) carbene was indeed trapped by oxidation with diphenylsulfoxide during the nucleophilic attack of an epoxide to an alkyne (53) in the cyclization to the α,β-unsaturated ketone 54 (Scheme 21).75d
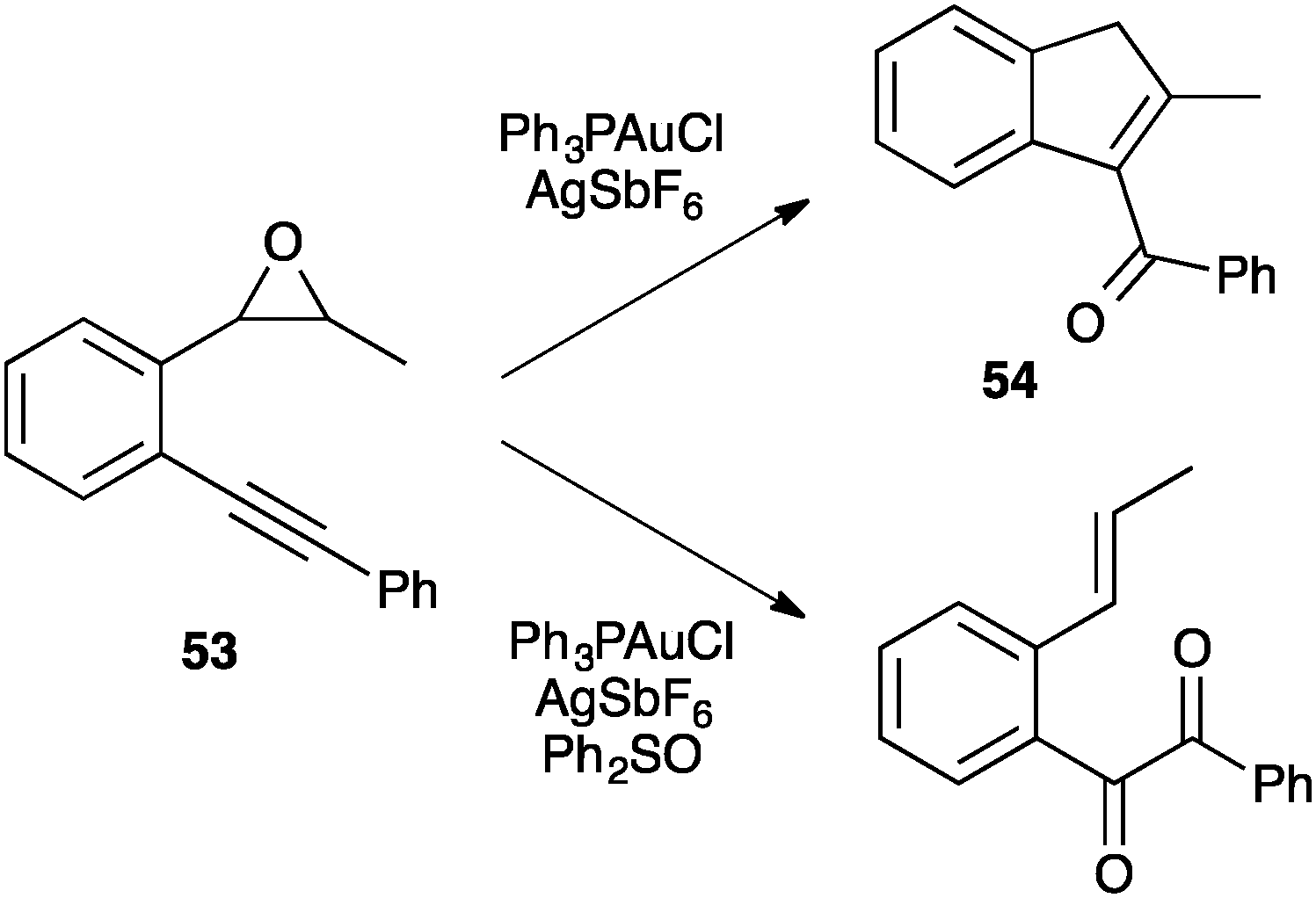 | ||
| Scheme 21 Gold(I) carbene trapping in the cycloisomerization of epoxide-alkyne functionalities. | ||
1,n-Enynes react stereospecifically with a variety of nucleophiles.46,47,57,58,65 As an illustrative example, 1,6-enyne 55 reacts with a gold(I) catalyst to form 56 as a result of the intermolecular anti attack of methanol on the cyclopropane-type ring of intermediate 57 (Scheme 22). Oxygen transfer to the carbene-like carbon of similar intermediates from diphenylsulfoxide, as a nucleophilic oxidant, leads to the corresponding aldehydes.82
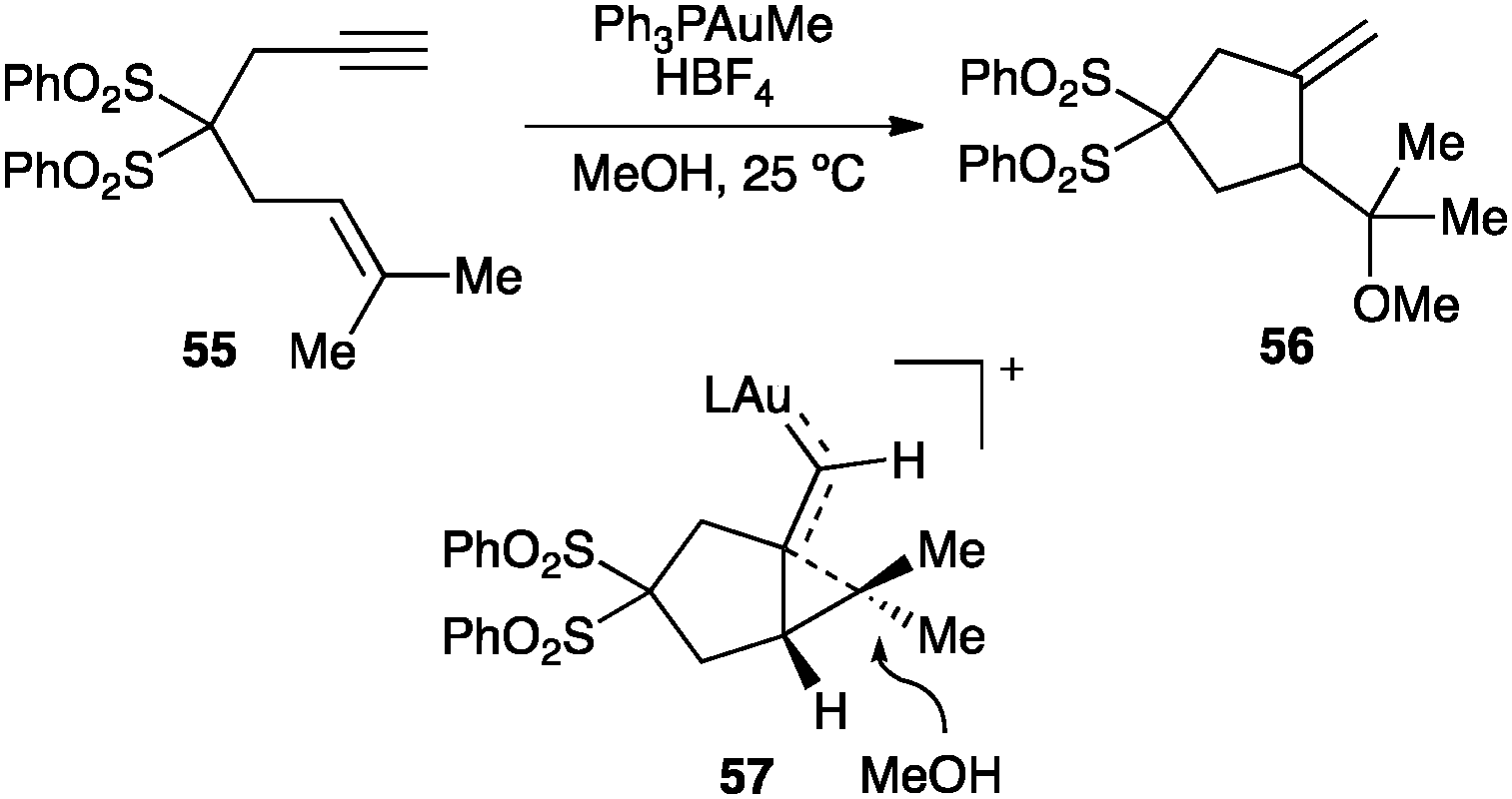 | ||
| Scheme 22 Cyclopropane ring opening by nucleophilic attack (57).46 | ||
Carbon nucleophiles react by similar mechanistic pathways.65 A particular case is illustrated by the cycloaddition of aryl substituted enynes, such as 58, which react readily with cationic gold(I) catalysts to form tricyclic derivatives 59 through intermediates of type 60 (Scheme 23).83 This formal [4+2] cycloaddition is stereospecific and, according to DFT calculations, proceeds in a stepwise fashion in which formation of 60 is rate-determining.83b
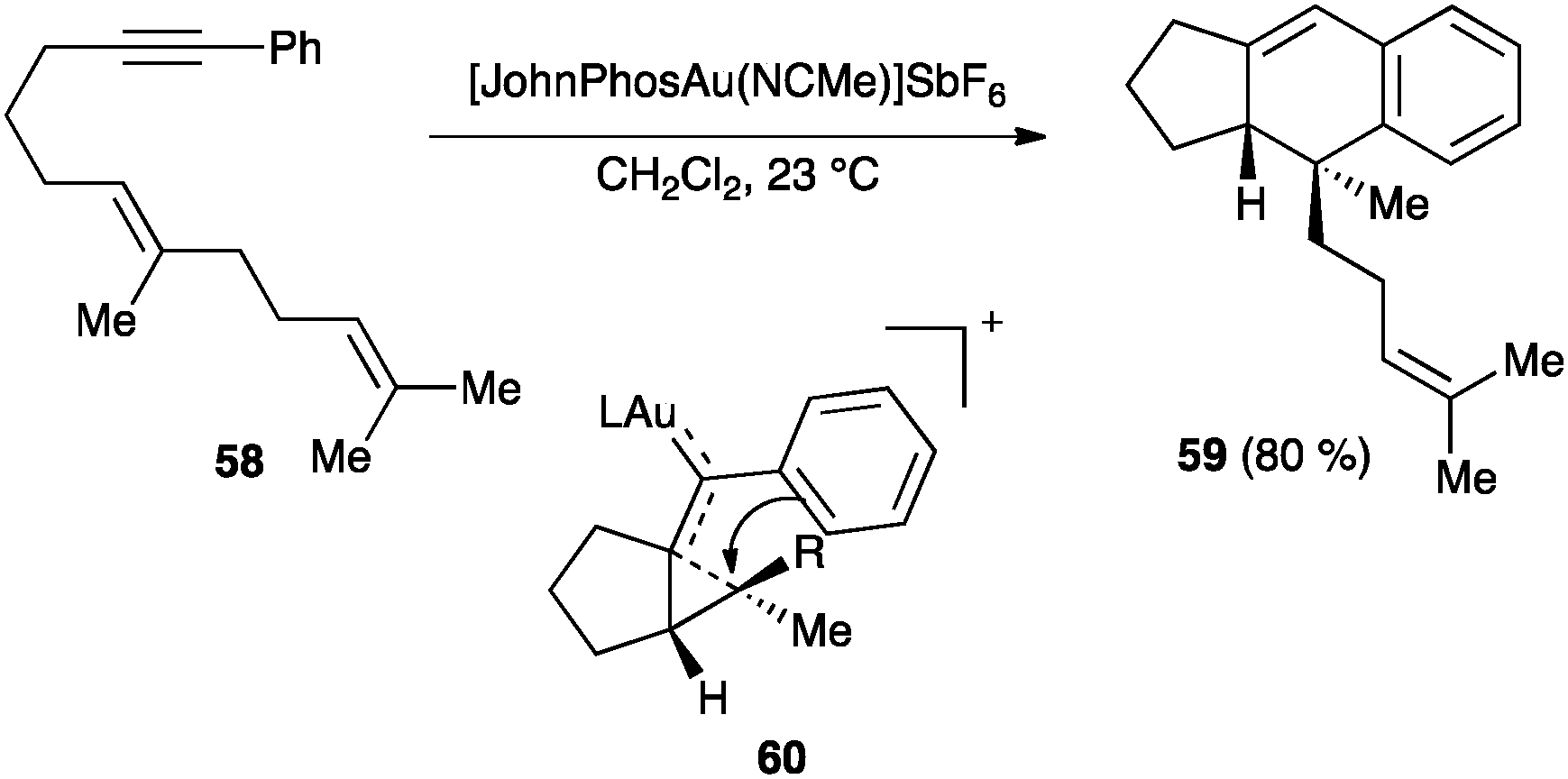 | ||
| Scheme 23 [4+2] Cycloaddition of aryl substituted enynes.83 | ||
In the absence of external or internal nucleophiles, enynes react with gold(I) by a series of fascinating rearrangements and related processes3,46,84 These types of cycloisomerisations have also been observed with other metal catalysts,85–90 although cationic gold(I) complexes are by far the most reactive and selective catalysts.3,46 In the case of 1,6-enynes that bear an aryl substituted alkyne and a terminally unsubstituted alkene, which react sluggishly with electrophilic metal catalysts, the initial gold(I)-activated substrate 61 could evolve by three pathways: anti-5-exo-dig (via62), 6-endo-dig (via63), and syn-5-exo-dig (via64) (Scheme 24).91 In general agreement with previous mechanistic work,58,92 recent DFT calculations show that the syn-5-exo-dig cyclization does not compete with the other two pathways, whose relative preference depends on the substitution pattern of the enyne.91 Products of double-cleavage rearrangement 66 could be formed through intermediate 65, in which the methylene is formally inserted into the alkyne carbons. On the other hand, dienes 70 of single-cleavage rearrangement can be formed by expansion of the cyclopropane of 63 to form cyclobutene–Au(I) complex 67, followed by cleavage to give diene–Au(I) complex 68. Intermediates 63 can also evolve to form bicyclo[4.1.0]hept-2-ene derivatives.46,93
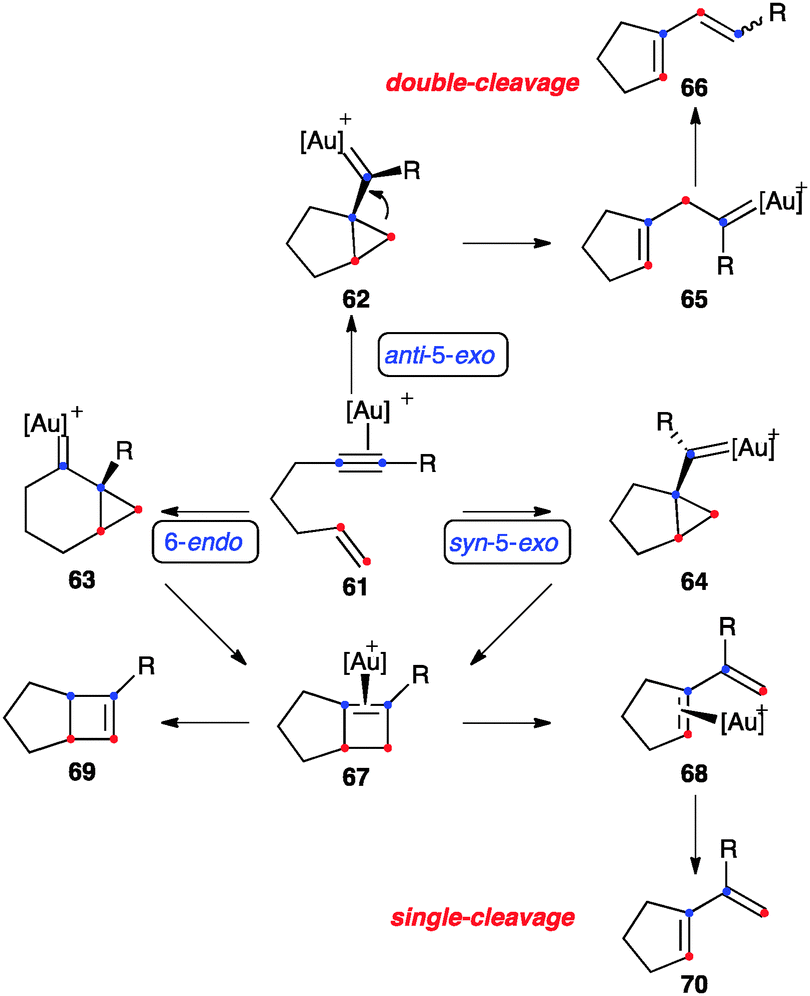 | ||
| Scheme 24 General pathways for the skeletal rearrangement of [2+2] cycloaddition of 1,6-enynes.91 | ||
Only a few examples of highly strained bicyclo[3.2.0]hept-5-enes have been reported in gold(I) catalysed isomerisations,91,94,95 whereas products of formal [2+2] cycloaddition have been observed in the parent intermolecular reactions of alkynes with alkenes,54 as well as in the cyclisation of 1,7-, 1,8-46,96 and higher enynes.55 Alternatively, 67 could give the product of alkene isomerisation 69.83 This type of isomerisation has been observed very recently by NMR at low temperature.95
1,6-Enynes substituted at the terminal carbon of the alkene (71) can directly give 1,3-dienes 74 of single-cleavage rearrangement by direct evolution of intermediates 72 formed in the anti-5-exo-dig pathway to form cations 73 (Scheme 25).58 An endocyclic version of this rearrangement has also been documented.46b,97 In general, this is a stereospecific process, although (E)-1,6-enynes bearing electron-donating R substituents at the alkene lead selectively to 1,3-dienes with the Z configuration.98
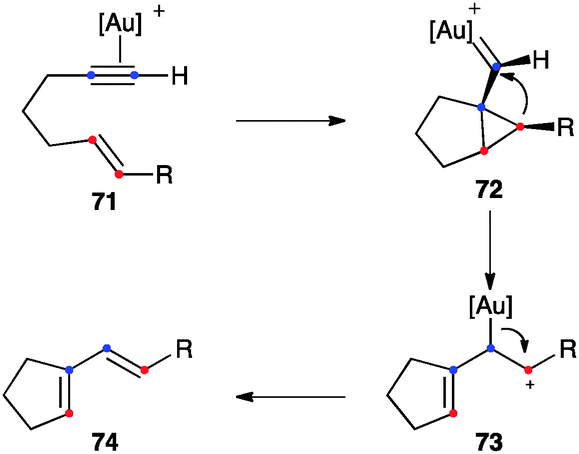 | ||
| Scheme 25 Alternative mechanism for the single-cleavage skeletal rearrangement.58 | ||
Cyclopropyl gold(I) carbene intermediates can also be trapped by carbonyl groups inter-99,100 or intramolecularly.101,102 Thus, oxo-1,6-enynes 75 undergo a [2+2+2] cycloaddition to form oxatricyclic compounds 76 (Scheme 26). As a side-reaction, diene 77 is also obtained. The reaction presumably proceeds through intermediate 78, which evolves to form 79 that undergoes a Prins-type cyclization leading to tricyclic scaffold 80. Cation 80 then furnishes oxatricyclic derivatives 76 by metal elimination or dienes 77 by fragmentation.
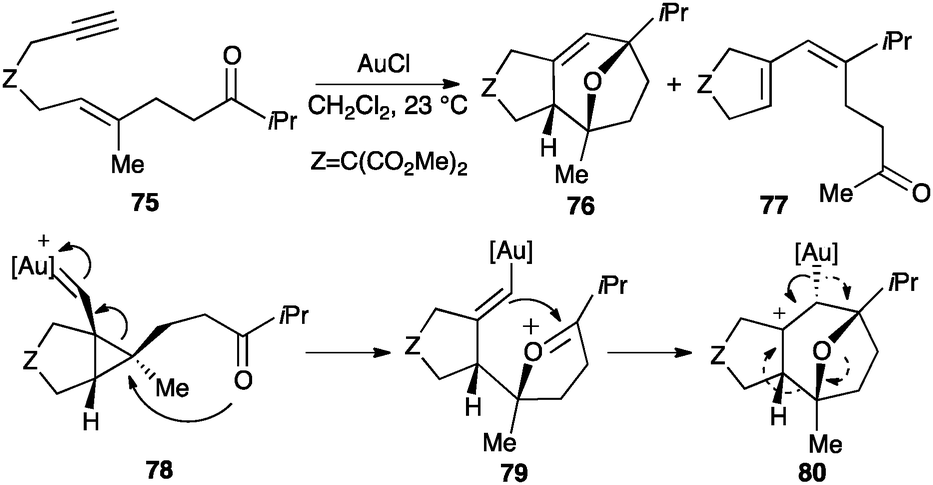 | ||
| Scheme 26 Intramolecular [2+2+2] cycloaddition of oxoeynes.101 | ||
The carbene-like character of the intermediates generated by reaction of 1,n-enynes with gold(I) is more clearly revealed by their trapping with alkenes. Thus, for example, reaction of dienyne 81 leads stereoselectively to tetracyclic compound 82 (Scheme 27).46 DFT calculations are consistent with a concerted, asynchronous cyclopropanation through intermediate 83.46 A similar model 84 was proposed for the intermolecular cyclopropanation of 1,6-enynes by alkenes.103 The cyclopropanation was found to be concerted for symmetrical or less polarized alkenes, whereas styrenes react in a stepwise manner. Nevertheless, even in this case, the overall process is stereospecific since formation of the second carbon–carbon occurred with a very small energy. Similarly, double 1,5-enyne 85 furnished 86 by intramolecular cyclopropanation of the intermediate endo-carbene.104 Gold(I) carbenes generated through 1,2-migration of propargylic carboxylates were also trapped stereospecifically with alkenes.105,106
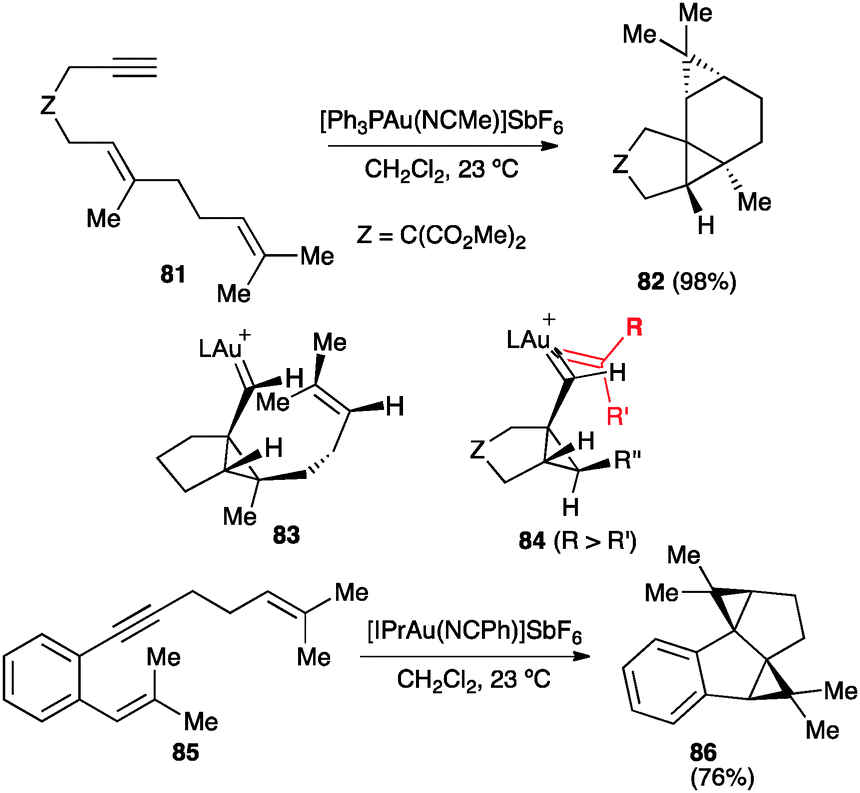 | ||
| Scheme 27 Intramolecular cyclopropanation of 1,6- and 1,5-enynes.46,103,104 | ||
Dienynes such as 87 bearing an alkoxy group at the propargylic position react differently leading to tricyclic products 88 as a result of a cyclization cascade process that involves a formal 1,5-migration of the OR group (Scheme 28).107 The reaction proceeds through intermediate 89, which evolves by intramolecular attack of the OR at the electrophilic site of the cyclopropane to form 90. α,β-Unsaturated gold(I) carbene 91, generated by cleavage of the oxonium bridge, then undergoes cyclopropanation with the pending alkene. Intermediate 89 can also be trapped intermolecularly with an external nucleophile to generate the corresponding epimeric derivative.
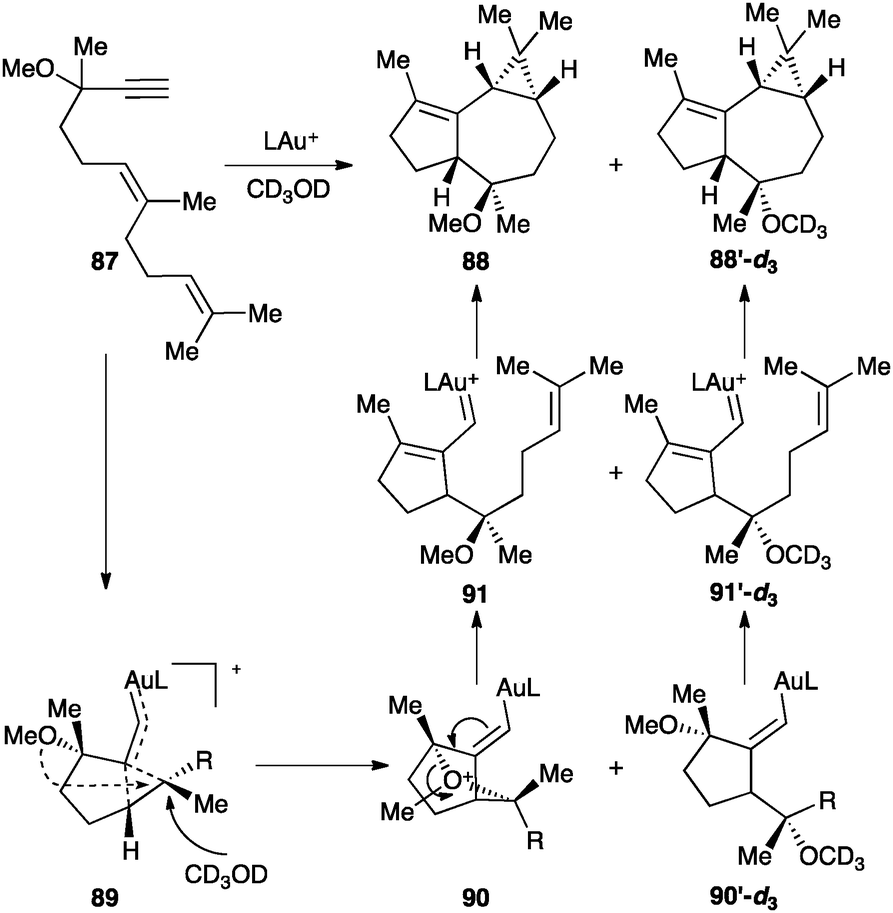 | ||
| Scheme 28 1,5-Propargyl ether migration of a 1,6-enyne followed by intramolecular cyclopropanation (L = JohnPhos).107 | ||
Gold(I) carbene intermediates can also be generated by reaction of diazoacetates with gold(I) complexes.108 The retro-Buchner reaction of 7-substituted cycloheptatrienes 92 also generates gold(I) carbenes (Scheme 29).109 Cycloheptatrienes 92 react with gold(I) through norcaradienes 93 to give gold(I) carbenes 94, which are trapped by alkenes leading to the corresponding cyclopropanes. A similar process was observed in the gas phase under electrospray ionization conditions.110
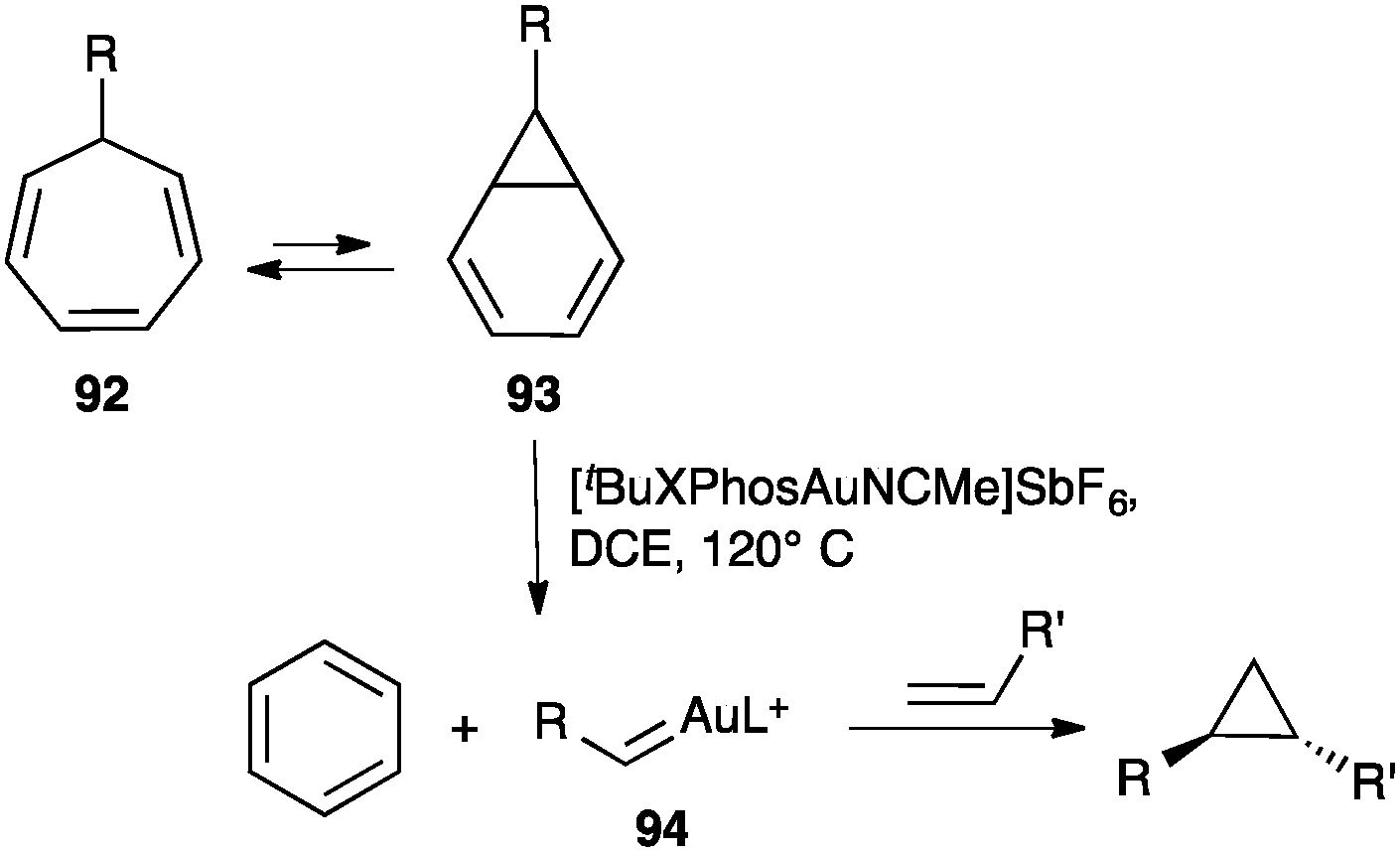 | ||
| Scheme 29 Cyclopropanation of alkenes by retro-Buchner reaction of cycloheptatrienes.109 | ||
More recently, gold(I) catalysis has also been combined with strong oxidants,111 organocatalysts,112 palladium, nickel or rhodium,113 and photoredox reactions114 leading to a completely new set of interesting transformations.
Digold complexes
Digold(I) species were proposed to be involved as key intermediates in the cyclization of 1,5-allenynes such as 95, which proceed by a stereospecific intramolecular hydrogen transfer from the allene to the alkyne (Scheme 30).115 Accordingly, gold(I) coordinates with the alkyne making the proton more acidic, leading to an alkynyl gold(I) complex that reacts with a second equivalent of the catalyst to form 97. Nucleophilic attack of the allene generates allyl stabilized carbocation 98 in the rate-determining step, which is followed by an intramolecular 1,5-hydrogen shift leading to 96 through diaurated species 99.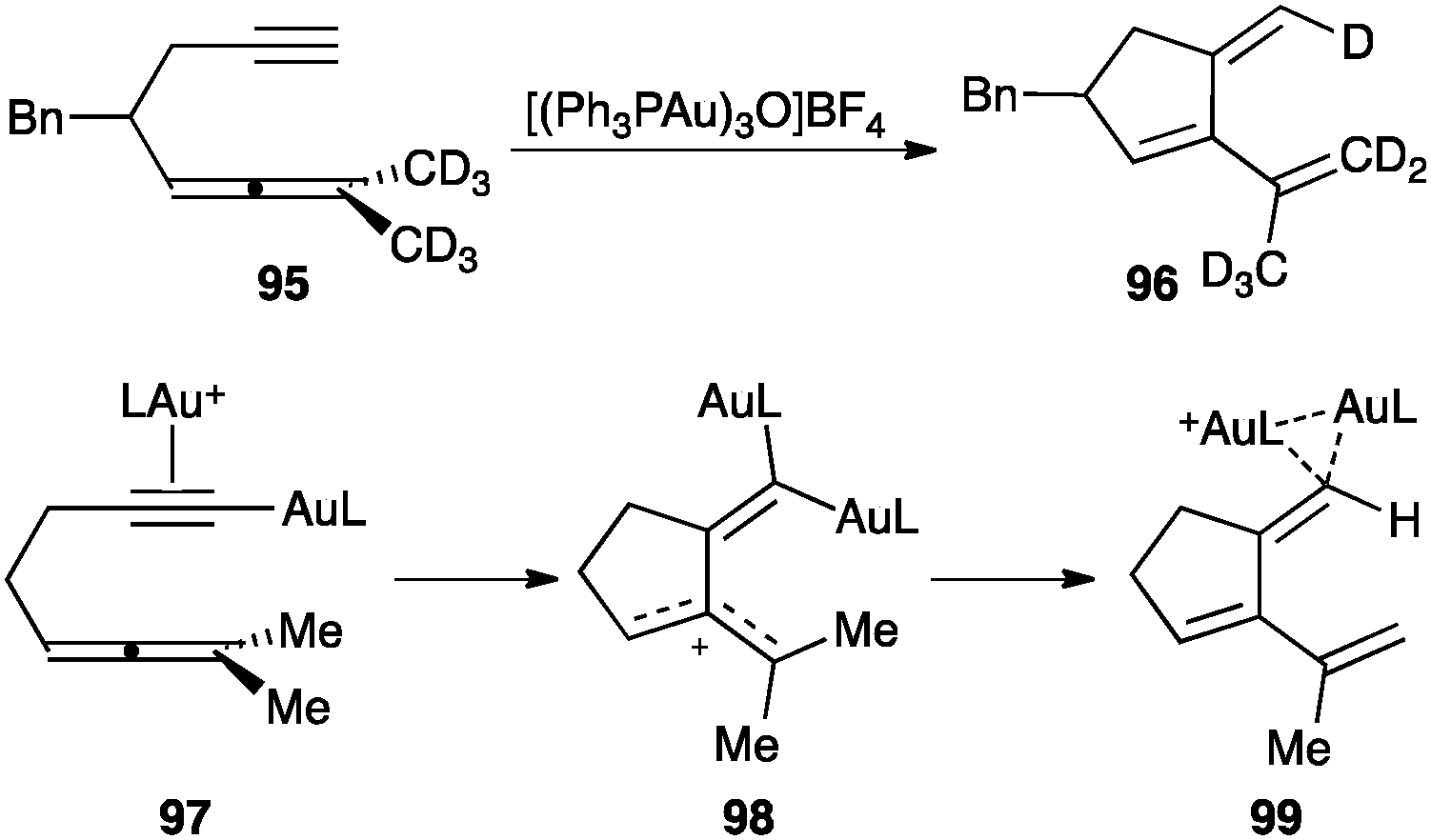 | ||
| Scheme 30 Cycloisomerization of 1,5-allenynes.115 | ||
Digold(I) complexes with the bridging 3-centre 2-electron bond related to 99 had been observed before in other contexts,116 and were later found to play relevant roles in catalysis. Thus, for example, during the catalytic intramolecular hydroarylation of allenes 100 to give 101, complex 102 was isolated as a catalyst resting state (Scheme 31).117 Similar species were observed in the intramolecular allene hydroalkoxylation.118
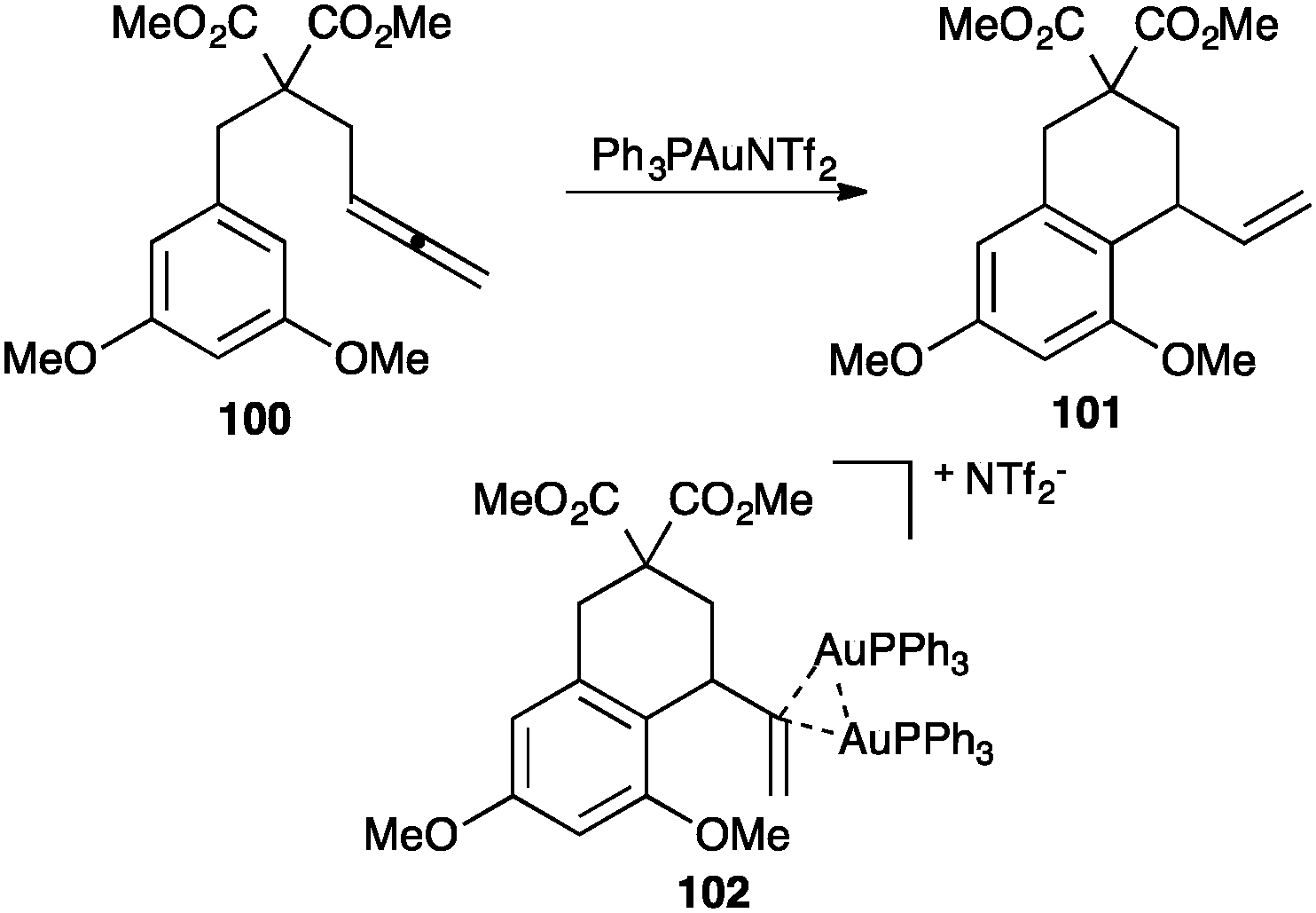 | ||
| Scheme 31 Intramolecular hydroarylation of allenes.117 | ||
A species of this type was generated and fully characterized by gold–boron transmetalation from 103 (Scheme 32).119 In this case, the monogold(I) complex 104 could not be observed. The analysis of the X-ray structure of 105 revealed an important stabilization from the oxygen atom and two almost regular carbon–gold σ-bonds.
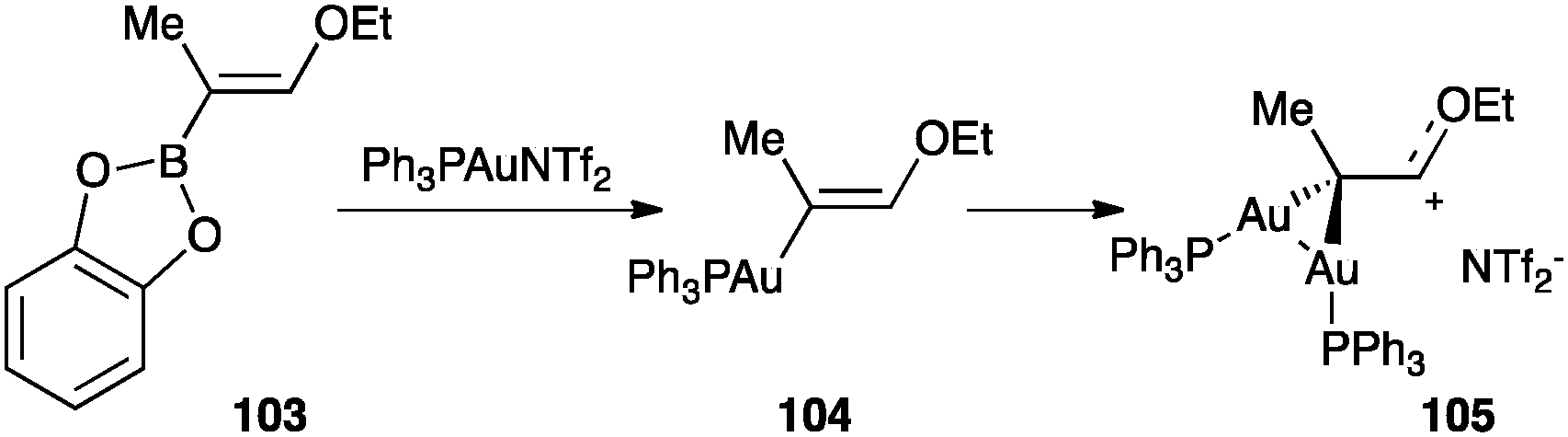 | ||
| Scheme 32 Formation of digold(I) species by gold–boron transmetalation.119 | ||
The nature of the 3-centre 2-electron interaction was also investigated by studying the equilibrium between aryl–gold 106 and digold(I) complexes 107 as a function of the electronic effects of the R substituents and the counteranions (Scheme 33).120 Thus, it was found that formation of 107 was favoured with less coordinating counteranions as well as with more electron-rich substrates supporting the proposal of an electrodeficient Au2C bond.
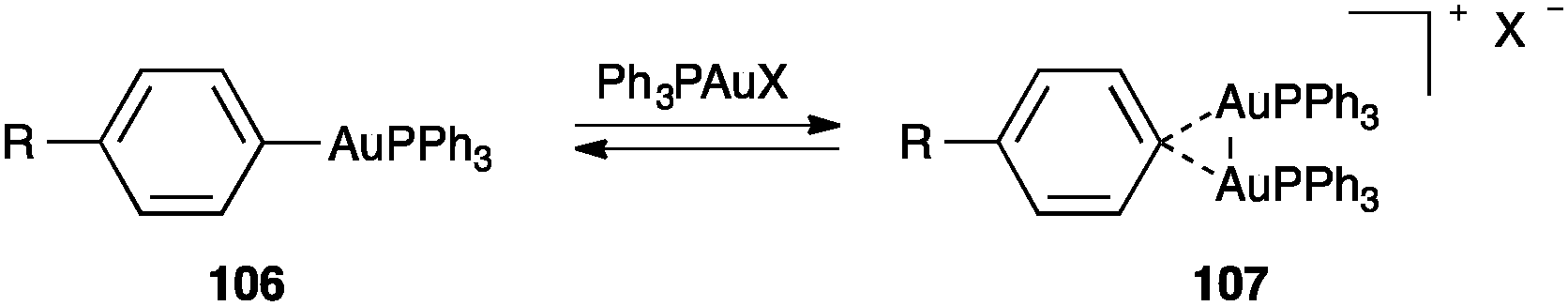 | ||
| Scheme 33 Equilibrium between mono- and digold(I) species.120 | ||
Alkynyl digold(I) complexes have been formed from terminal alkynes.25e,121 At −78 °C complex 108 was formed, which then formed digold(I) complex 109 at higher temperatures (Scheme 34).121
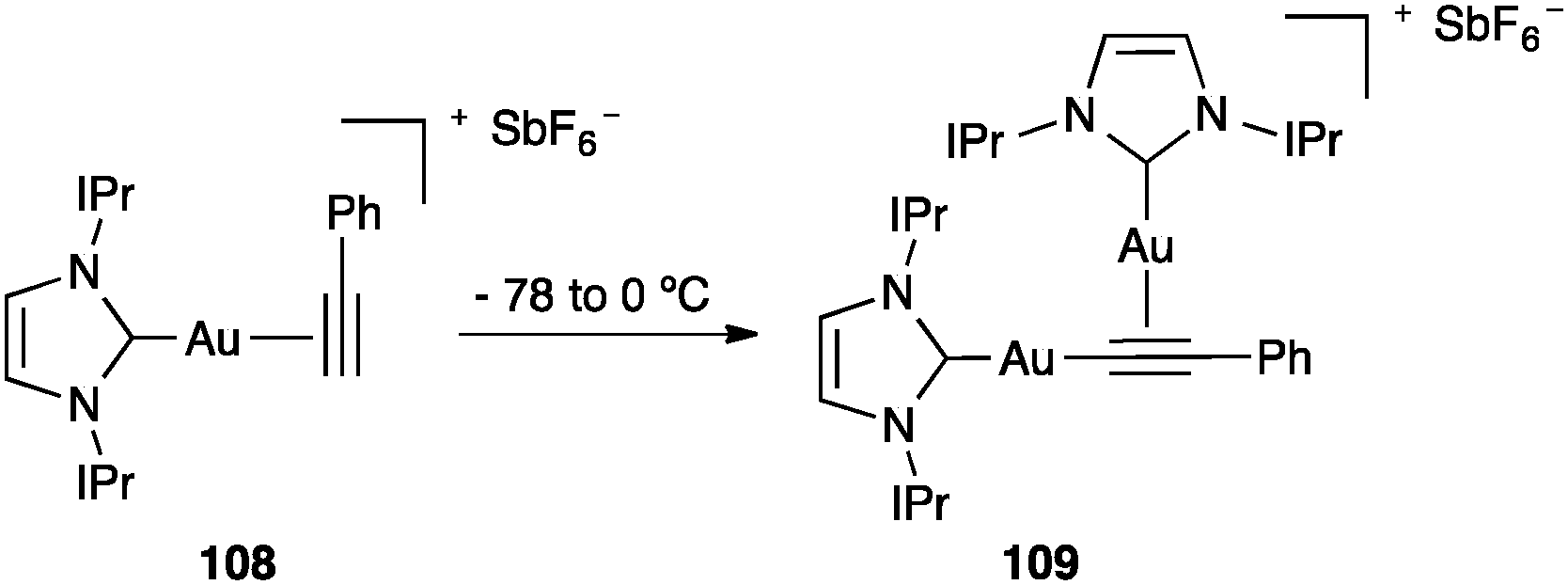 | ||
| Scheme 34 Formation of alkynyl digold(I) complex 109.121 | ||
Similar digold(I) complexes with phosphine ligands were detected in the intermolecular [2+2] cycloaddition of alkynes with alkenes.122 Complex 110 is also formed in the intermolecular [2+2+2] cycloaddition of alkynes with oxoalkenes, leading to oxatricyclic products 111 (Scheme 35),123 a process mechanistically related to the intramolecular reaction of oxo-1,6-enynes (see Scheme 26). Although the digold(I) complex 110 was identified as the resting state of the catalytic cycle, only traces of product 111 were obtained when this complex was used as the catalyst. The catalytic activity was recovered in the presence of an acid, which cleaved the gold–acetylide bond. The results demonstrated that the reactive species is a π-coordinated monogold(I) complex, which was confirmed by DFT calculations.
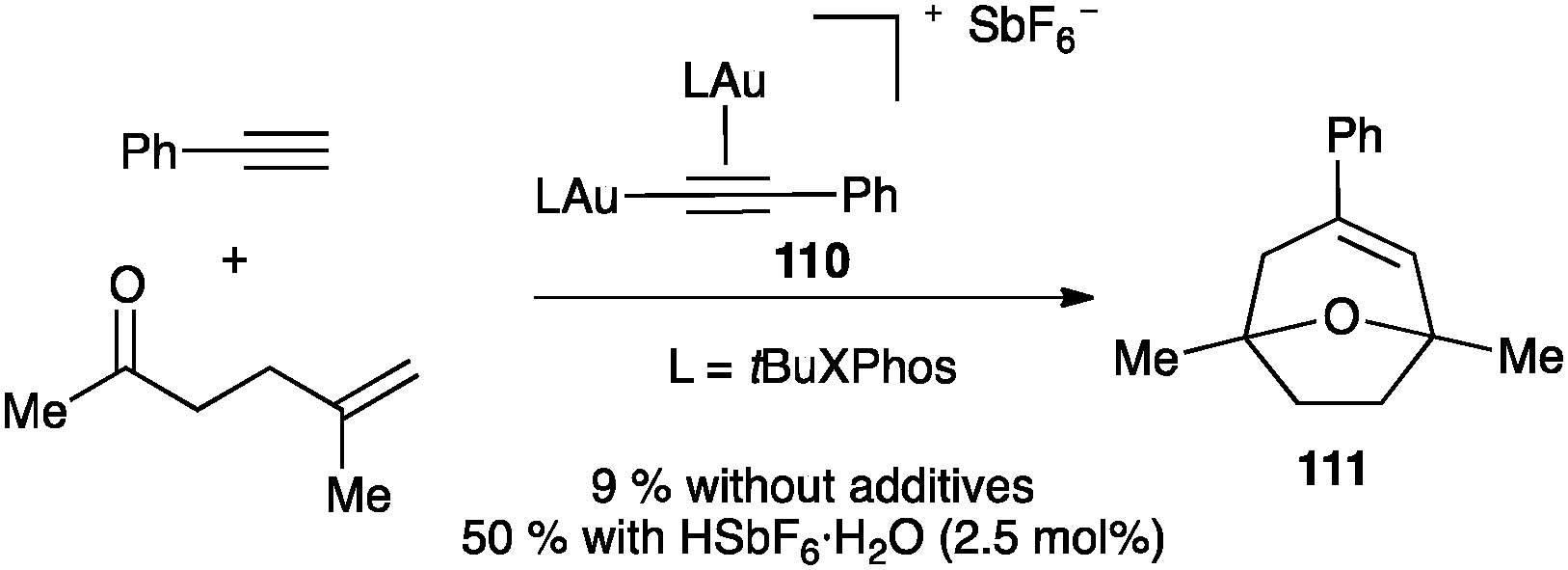 | ||
| Scheme 35 Digold(I) species 110 as a precatalyst.123 | ||
The possible role of digold(I) complexes in cycloisomerization reactions of 1,6-enynes has also been examined.124 Experimental and computational work suggested that these types of σ,π-digold(I) complexes are unreactive in the cycloisomerization process.
However, σ-alkynyl gold complexes react with alkynes in intramolecular transformations leading to a variety of interesting cyclic systems. Thus, diyne 112 was cyclised to form 1,2-dihydrocyclopenta[a]indene 113 (Scheme 36).125 The reaction was proposed to proceed by formation of alkynyl gold(I) species 114, which evolved by attack of the σ-alkynyl gold to the π-activated non-terminal alkyne in a 5-endo-dig cyclization. The resulting gold(I) vinylidene 115 could undergo a C–H insertion followed by protodeauration to form 113. Theoretical work suggested that formation of 116 by 6-endo-dig cyclization could also be possible. A similar transformation has been applied for the synthesis of cyclopentapyridinones.126
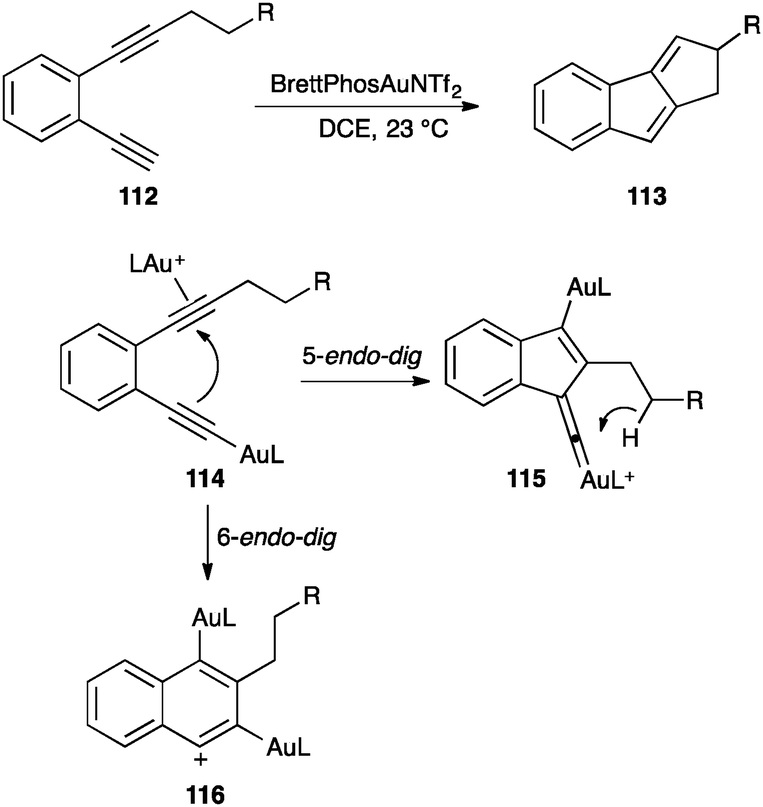 | ||
| Scheme 36 Dual activation followed by C–H insertion.125 | ||
In a parallel study, the reaction of 117 led to digold(I) complex 118 (Scheme 37).127 Additionally, the intermolecular trapping of the vinylidene gold(I) complexes of type 115 with alkenes was found to give cyclobutane through a cyclopropanation reaction, followed by ring expansion.128 Inter- and intramolecular reactions of vinylidene gold(I) complexes with arene rings have also been reported.129 It is interesting that, in contrast to that found in the reactions between alkynes and alkenes, these types of diyne cyclizations are smoothly catalysed with σ,π-digold(I) alkyne complexes.129–131
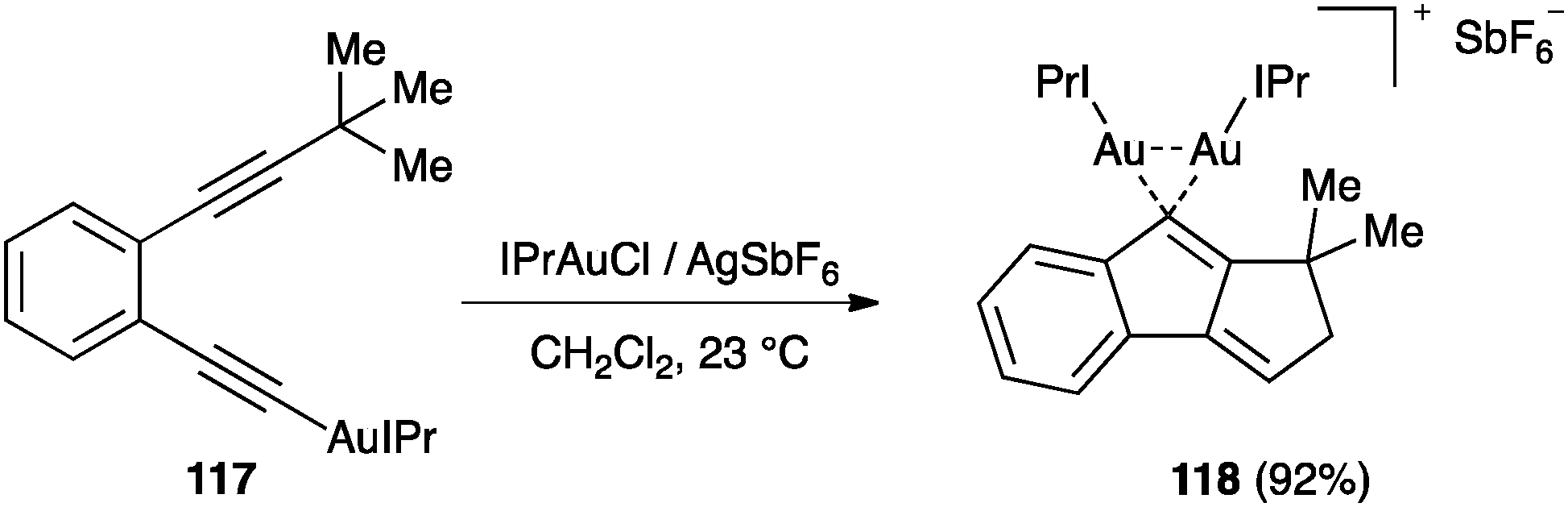 | ||
| Scheme 37 Formation of digold(I) species 118 during the dual activation of diynes.128 | ||
Conclusions
Many reactions catalysed by gold, or by other electrophilic catalysts, closely resemble proton-initiated carbocationic processes. However, gold(I) very often provides exquisite control on these transformations, channeling the process towards the desired products by stabilising the key reactive intermediates. The basic mechanisms involved in the cycloisomerization of substrates containing alkynes and allenes are now better understood and have guided the discovery of synthetically useful cascade processes for the build up of complex molecular architectures. However, there are still some elusive aspects in this area, particularly in intermolecular reactions. Closer scrutiny of some early mechanistic proposals will certainly lead to the discovery of new types of reactivity. The recent outburst in digold catalysis, which suggests that more complex species are involved in certain cases, augurs well for the future discovery of yet more complex transformations.We thank the MEC (CTQ2010-16088/BQU), the MECD (PFU fellowship to C.O.), the European Research Council (Advanced Grant No. 321066), the AGAUR (2009SGR47), and the ICIQ Foundation for financial support.
Notes and references
- For other precedents see: (a) R. O. C. Norman, W. J. E. Parr and C. B. Thomas, J. Chem. Soc., Perkin Trans. 1, 1976, 1983–1987 RSC; (b) Y. Fukuda and K. Utimoto, J. Org. Chem., 1991, 56, 3729–3731 CrossRef CAS.
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 CrossRef CAS.
- (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed; (b) E. Jiménez-Núñez and A. M. Echavarren, Chem. Commun., 2007, 333–346 RSC; (c) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed; (d) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed; (e) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (f) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC; (g) N. D. Shapiro and F. D. Toste, Synlett, 2010, 675–691 CAS; (h) H. G. Raubenheimer and H. Schmidbaur, S. Afr. J. Sci., 2011, 107, 31–34 CrossRef CAS PubMed; (i) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS PubMed.
- (a) N. Krause, V. Belting, C. Deutsch, J. Edrsack, H. T. Fan, B. Gockel, A. Hoffmann-Röder, N. Morita and F. Volz, Pure Appl. Chem., 2008, 80, 1063–1069 CrossRef CAS; (b) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448–2462 RSC; (c) W. E. Brenzovich Jr., Angew. Chem., Int. Ed., 2012, 51, 8933–8935 CrossRef PubMed.
- (a) E. Jiménez-Núñez, K. Molawi and A. M. Echavarren, Chem. Commun., 2009, 7327–7329 RSC; (b) K. Molawi, N. Delpont and A. M. Echavarren, Angew. Chem., Int. Ed., 2010, 49, 3517–3519 CrossRef CAS PubMed; (c) M. Gaydou, R. E. Miller, N. Delpont, J. Ceccon and A. M. Echavarren, Angew. Chem., Int. Ed., 2013, 52, 6396–6399 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232–5241 CrossRef CAS PubMed; (b) L. P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129–3139 RSC; (c) I. Braun, A. M. Asiri and A. S. K. Hashmi, ACS Catal., 2013, 3, 1902–1907 CrossRef CAS.
- P. Pérez-Galán, N. Delpont, E. Herrero-Gómez, F. Maseras and A. M. Echavarren, Chem.–Eur. J., 2010, 16, 5324–5332 CrossRef PubMed.
- (a) D. V. Partyka, T. J. Robilotto, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2008, 27, 28–32 CrossRef CAS; (b) A. S. K. Hashmi, T. Hengst, C. Lothschütz and F. Rominger, Adv. Synth. Catal., 2010, 352, 1315–1337 CrossRef CAS; (c) G. C. Fortman and S. P. Nolan, Organometallics, 2010, 29, 4579–4583 CrossRef CAS.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- (a) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed; (b) Y. Zhu, C. S. Day, L. Zhang, K. J. Hauser and A. C. Jones, Chem.–Eur. J., 2013 DOI:10.1002/chem.201302152; (c) A. Homs, I. Escofet and A. M. Echavarren, 2013, submitted.
- (a) M. Raducan, C. Rodríguez-Escrich, X. C. Cambeiro, E. C. Escudero-Adán, M. A. Pericàs and A. M. Echavarren, Chem. Commun., 2011, 47, 4893–4895 RSC; (b) M. Raducan, M. Moreno, C. Bour and A. M. Echavarren, Chem. Commun., 2012, 48, 52–54 RSC.
- (a) N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef PubMed; (b) L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704–4707 CrossRef CAS.
- P. N. Dickson, A. Wehrli and G. Geier, Inorg. Chem., 1988, 27, 2921–2925 CrossRef CAS.
- (a) M. P. Muñoz, J. Adrio, J. C. Carretero and A. M. Echavarren, Organometallics, 2005, 24, 1293–1300 CrossRef; (b) G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496–499 CrossRef CAS PubMed; (c) F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS PubMed; (d) S. G. Sethofer, T. Mayer and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8276–8277 CrossRef CAS PubMed; (e) C. Wang, Z. Y. Han, H. W. Luo and L. Z. Gong, Org. Lett., 2010, 12, 2266–2269 CrossRef CAS PubMed; (f) K. Aikawa, M. Kojima and K. Mikami, Adv. Synth. Catal., 2010, 352, 3131–3135 CrossRef CAS; (g) H. Teller, S. Flügge, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 1949–1953 CrossRef CAS PubMed; (h) M. J. Campbell and F. D. Toste, Chem. Sci., 2011, 2, 1369–1378 RSC; (i) G. Zhou, F. Liu and J. Zhang, Chem.–Eur. J., 2011, 17, 3101–3104 CrossRef CAS PubMed; (j) J. F. Brazeau, S. Zhang, I. Colomer, B. K. Corkey and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 2742–2749 CrossRef CAS PubMed; (k) R. J. Felix, D. Weber, O. Gutierrez, D. J. Tantillo and M. R. Gagné, Nat. Chem., 2012, 4, 405–409 CrossRef CAS PubMed; (l) J. Francos, F. Grande-Carmona, H. Faustino, J. Iglesias-Sigüenza, E. Díez, I. Alonso, R. Fernández, J. M. Lassaletta, F. López and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322–14325 CrossRef CAS PubMed; (m) S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912–2915 CrossRef CAS PubMed; (n) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916–11919 CrossRef CAS PubMed.
- (a) R. A. Widenhoefer, Chem.–Eur. J., 2008, 14, 5382–5391 CrossRef CAS PubMed; (b) N. T. Patil, Chem.–Asian J., 2012, 7, 2186–2194 CrossRef CAS PubMed; (c) N. Huguet and A. M. Echavarren, Asymmetric Synthesis II, ed. M. Christman and S. Bräse, Wiley-VCH, Ch. 26, 2013, pp. 205–2013 Search PubMed; (d) P. de Mendoza and A. M. Echavarren, Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and D. F. Toste, Wiley-VCH Verlag GmbH & Co., Ch. 5, 2012, pp. 135–152 Search PubMed.
- (a) A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhemsen, M. Rudolph and F. Rominger, Chem.–Eur. J., 2013, 19, 1058–1065 CrossRef CAS PubMed; (b) P. W. Davies and N. Martin, Org. Lett., 2009, 11, 2293–2296 CrossRef CAS PubMed; (c) J. H. Kim, S. W. Park, S. R. Park, S. Lee and E. J. Kang, Chem.–Asian J., 2011, 6, 1982–1986 CrossRef CAS PubMed; (d) D. Zuccaccia, L. Belpassi, F. Tarantelli and A. Macchioni, J. Am. Chem. Soc., 2009, 131, 3170–3171 CrossRef CAS PubMed; (e) A. Homs, C. Obradors, D. Leboeuf, A. M. Echavarren, 2013, submitted.
- (a) P. Pyykkö, Angew. Chem., Int. Ed., 2002, 41, 3573–3578 CrossRef; (b) P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed; (c) H. Schwarz, Angew. Chem., Int. Ed., 2003, 42, 4442–4454 CrossRef CAS PubMed.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed.
- (a) N. D. Shapiro and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2779–2782 CrossRef CAS; (b) M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295–301 CrossRef CAS PubMed; (c) Y. Yamamoto, J. Org. Chem., 2007, 72, 7818–7831 CrossRef PubMed.
- (a) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, Chem. Commun., 2009, 6451–6453 RSC; (b) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 6350–6351 CrossRef CAS PubMed; (c) R. E. M. Broomer and R. A. Widenhoefer, Organometallics, 2012, 31, 768–771 CrossRef; (d) R. E. M. Brooner, T. J. Brown and R. A. Widenhoefer, Chem.–Eur. J., 2013, 19, 8276–8284 CrossRef CAS PubMed.
- (a) D. Zuccaccia, L. Belpassi, F. Tarantelli and A. Macchioni, J. Am. Chem. Soc., 2009, 131, 3170–3171 CrossRef CAS PubMed; (b) N. Salvi, L. Belpassi, D. Zuccaccia, F. Tarantelli and A. Macchioni, J. Organomet. Chem., 2010, 695, 2679–2686 CrossRef CAS PubMed.
- P. Nava, D. Hagebaum-Reignier and S. Humbel, ChemPhysChem, 2012, 13, 2090–2096 CrossRef CAS PubMed.
- (a) R. A. Sanguramath, T. N. Hooper, C. P. Butts, M. Green, J. E. McGrady and C. A. Russell, Angew. Chem., Int. Ed., 2011, 50, 7592–7595 CrossRef CAS PubMed; (b) R. E. M. Brooner and R. A. Widenhoefer, Organometallics, 2011, 30, 3182–3193 CrossRef CAS; (c) I. Krossing, Angew. Chem., Int. Ed., 2011, 50, 11576–11578 CrossRef CAS PubMed.
- T. J. Brown, A. Sugie, M. G. D. Leed and R. A. Widenhoefer, Chem.–Eur. J., 2012, 18, 6959–6971 CrossRef CAS PubMed.
- (a) G. Wittig and S. Fischer, Chem. Ber., 1972, 105, 3542–3552 CrossRef CAS; (b) V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569–13573 CrossRef CAS PubMed; (c) S. Flügge, A. Anoop, R. Goddard, W. Thiel and A. Fürstner, Chem.–Eur. J., 2009, 15, 8558–8565 CrossRef PubMed; (d) T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313–2315 RSC; (e) T. J. Brown and R. A. Widenhoefer, J. Organomet. Chem., 2011, 696, 1216–1220 CrossRef CAS PubMed; (f) T. J. Brown and R. A. Widenhoefer, Organometallics, 2011, 30, 6003–6009 CrossRef CAS; (g) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940–3943 CrossRef CAS PubMed; (h) A. Das, C. Dash, M. A. Celik, M. Yousufuddin, G. Frenking and H. V. R. Dias, Organometallics, 2013, 32, 3135–3144 CrossRef CAS; (i) M. Fianchini, C. F. Campana, B. Chilukuri, T. R. Cundari, V. Petricek and H. V. R. Dias, Organometallics, 2013, 32, 3034–3041 CrossRef CAS.
- Study of π-alkyne–gold(I) complexes in solution: (a) R. Huettel and H. Forkl, Chem. Ber., 1972, 105, 1664–1673 CrossRef CAS; (b) P. Schulte and U. Behrens, Chem. Commun., 1998, 1633–1634 RSC; (c) D. Zuccaccia, L. Belpassi, L. Rocchigiani, F. Tarantelli and A. Macchioni, Inorg. Chem., 2010, 49, 3080–3082 CrossRef CAS PubMed; (d) T. J. Brown and R. A. Widenhoefer, J. Organomet. Chem., 2011, 696, 1216–1220 CrossRef CAS PubMed; (e) G. Ciancaleoni, L. Belpassi, F. Tarantelli, D. Zuccaccia and A. Macchioni, Dalton Trans., 2013, 42, 4122–4131 RSC.
- (a) B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, Chem.–Eur. J., 2009, 15, 3336–3339 CrossRef CAS PubMed; (b) I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020–13030 CrossRef CAS PubMed.
- J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736–7737 CrossRef CAS PubMed.
- V. Lavallo, G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5224–5228 CrossRef CAS PubMed.
- For other examples see: (a) E. Mizushima, T. Hayashi and M. Tanaka, Org. Lett., 2003, 5, 3349–3352 CrossRef CAS PubMed; (b) N. Nishina and Y. Yamamoto, Angew. Chem., Int. Ed., 2006, 45, 3314–3317 CrossRef CAS PubMed; (c) N. Nishina and Y. Yamamoto, Tetrahedron, 2009, 65, 1799–1808 CrossRef CAS PubMed; (d) E. L. Noey, Y. Luo, L. Zhang and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 1078–1084 CrossRef CAS PubMed; (e) M. J. López-Gómez, D. Martin and G. Bertrand, Chem. Commun., 2013, 49, 4483–4485 RSC.
- M. Joost, P. Gualco, S. Mallet-Ladeira, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2013, 52, 7160–7163 CrossRef CAS PubMed.
- (a) M. T. Reetz and K. Sommer, Eur. J. Org. Chem., 2003, 3485–3496 CrossRef CAS; (b) C. Nevado and A. M. Echavarren, Synthesis, 2005, 167–182 CAS.
- (a) A. S. K. Hashmi, P. Haufe, C. Schmid, A. Rivas Nass and W. Frey, Chem.–Eur. J., 2006, 12, 5376–5382 CrossRef CAS PubMed; (b) C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105–1109 CrossRef CAS PubMed.
- (a) E. Mizushima, K. Sato, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2002, 41, 4563–4565 CrossRef CAS; (b) C. M. Krauter, A. S. K. Hashmi and M. Pernpointner, ChemCatChem, 2010, 2, 1226–1230 CrossRef CAS.
- (a) F. M. Istrate and F. Gagosz, Org. Lett., 2007, 9, 3181–3184 CrossRef CAS PubMed; (b) J. Qian, Y. Liu, J. Cui and Z. Xu, J. Org. Chem., 2012, 77, 4484–4490 CrossRef CAS PubMed.
- (a) H. Kusama, Y. Miyashita, J. Takay and and N. Iwasawa, Org. Lett., 2006, 8, 289–292 CrossRef CAS PubMed; (b) E. Benedetti, G. Lemière, L. L. Chapellet, A. Penoni, G. Palmisano, M. Malacria, J. P. Goddard and L. Fensterbank, Org. Lett., 2010, 12, 4396–4399 CrossRef CAS PubMed.
- (a) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160–4161 CrossRef CAS PubMed; (b) P. W. Davies and S. J. C. Albrecht, Angew. Chem., Int. Ed., 2009, 48, 8372–8375 CrossRef CAS PubMed; (c) S. Shi, T. Wang, W. Yang, M. Rudolph and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 6576–6580 CrossRef CAS PubMed.
- L. Ye, L. Cui, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 3258–3259 CrossRef CAS PubMed.
- (a) I. Nakamura, T. Sato and Y. Yamamoto, Angew. Chem., Int. Ed., 2006, 45, 4473–4475 CrossRef CAS PubMed; (b) I. Nakamura, T. Sato, M. Terada and Y. Yamamoto, Org. Lett., 2007, 9, 4081–4083 CrossRef CAS PubMed.
- A. Hoffmann-Röder and N. Krause, Org. Lett., 2001, 3, 2537–2538 CrossRef PubMed.
- A. W. Sromek, M. Rubina and V. Gevorgyan, J. Am. Chem. Soc., 2005, 127, 10500–10501 CrossRef CAS PubMed.
- (a) N. Marion and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2750–2752 CrossRef CAS PubMed; (b) S. Wang, G. Zhang and L. Zhang, Synlett, 2010, 692–706 CAS; (c) R. K. Shiroodi and V. Gevorgyan, Chem. Soc. Rev., 2013, 42, 4991–5001 RSC.
- A. Correa, N. Marion, L. Fensterbank, M. Malacria, S. P. Nolan and L. Cavallo, Angew. Chem., Int. Ed., 2008, 47, 718–721 CrossRef CAS PubMed.
- T. de Haro, E. Gómez-Bengoa, R. Cribiú, X. Huang and C. Nevado, Chem.–Eur. J., 2012, 18, 6811–6824 CrossRef CAS PubMed.
- A. M. Echavarren and E. Jiménez-Núñez, Top. Catal., 2010, 53, 924–930 CrossRef CAS and references cited therein.
- (a) C. Nieto-Oberhuber, M. P. Muñoz, E. Buñuel, C. Nevado, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 2402–2406 CrossRef CAS PubMed; (b) C. Nieto-Oberhuber, M. P. Muñoz, S. López, E. Jiménez-Núñez, C. Nevado, E. Herrero-Gómez, M. Raducan and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 1677–1693 CrossRef CAS PubMed.
- C. Nieto-Oberhuber, S. López, E. Jiménez-Núñez and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 5916–5923 CrossRef CAS PubMed.
- M. García-Mota, N. Cabello, F. Maseras, A. M. Echavarren, J. Pérez-Ramírez and N. Lopez, ChemPhysChem, 2008, 9, 1624–1629 CrossRef PubMed.
- N. Cabello, C. Rodríguez and A. M. Echavarren, Synlett, 2007, 1753–1758 CAS.
- L. P. Liu, B. Xu, M. S. Mashuta and G. B. Hammond, J. Am. Chem. Soc., 2008, 130, 17642–17643 CrossRef CAS PubMed.
- W. Wang, G. B Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697–5705 CrossRef CAS PubMed.
- D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard III and F. D. Toste, Nat. Chem., 2009, 1, 482–486 CrossRef CAS PubMed.
- A. M. Echavarren, Nat. Chem., 2009, 1, 431–433 CrossRef CAS PubMed.
- V. López-Carrillo and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 9292–9294 CrossRef PubMed.
- C. Obradors, D. Leboeuf, J. Aydin and A. M. Echavarren, Org. Lett., 2013, 15, 1576–1579 CrossRef CAS PubMed.
- (a) H.-S. Yeom, J. Koo, H.-S. Park, Y. Wang, Y. Liang, Z.-X. Yu and S. Shin, J. Am. Chem. Soc., 2012, 134, 208–211 CrossRef CAS PubMed; (b) S. R. Park, C. Kim, D. Kim, D. Thrimurtulu, H.-S. Yeom, J. Jun, S. Shin and Y. H. Rhee, Org. Lett., 2013, 15, 1166–1169 CrossRef CAS PubMed.
- C. Nieto-Oberhuber, M. P. Muñoz, S. López, E. Jiménez-Núñez, C. Nevado, E. Herrero-Gómez, M. Raducan and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 1677–1693 CrossRef CAS PubMed.
- C. Nieto-Oberhuber, S. López, M. P. Muñoz, D. J. Cárdenas, E. Buñuel, C. Nevado and A. M. Echavarren, Angew. Chem., Int. Ed., 2005, 44, 6146–6148 CrossRef CAS PubMed.
- A. Fürstner and L. Morency, Angew. Chem., Int. Ed., 2008, 47, 5030–5033 CrossRef PubMed.
- (a) A. Eschenmoser, L. Ruzicka, O. Jeger and D. Arigoni, Helv. Chim. Acta, 1955, 38, 1890–1904 CrossRef CAS; (b) G. Stork and A. W. Burgstahler, J. Am. Chem. Soc., 1955, 77, 5068–5077 CrossRef CAS; (c) A. Eschenmoser and D. Arigoni, Helv. Chim. Acta, 2005, 88, 3011–3050 CrossRef CAS.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2008, 47, 6754–6756 CrossRef CAS PubMed.
- C. M. Chao, M. R. Vitale, P. Y. Toullec, J. P. Genêt and V. Michelet, Chem.–Eur. J., 2009, 15, 1319–1323 CrossRef CAS PubMed.
- S. G. Sethofer, T. Mayer and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8276–8277 CrossRef CAS PubMed.
- P. Pérez-Galán, N. J. A. Martin, A. G. Campaña, D. J. Cárdenas and A. M. Echavarren, Chem.–Asian J., 2001, 6, 482–486 CrossRef PubMed.
- (a) C. H. M. Amijs, C. Ferrer and A. M. Echavarren, Chem. Commun., 2007, 698–700 RSC; (b) C. H. M. Amijs, V. López-Carrillo, M. Raducan, P. Pérez-Galán, C. Ferrer and A. M. Echavarren, J. Org. Chem., 2008, 73, 7721–7730 CrossRef CAS PubMed.
- U. Schubert, K. Ackermann and R. Aumann, Cryst. Struct. Commun., 1982, 11, 591–594 CAS.
- G. Seidel, R. Mynott and A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 2510–2513 CrossRef CAS PubMed.
- M. M. Hansmann, F. Rominger and A. S. K. Hashmi, Chem. Sci., 2013, 4, 1552–1559 RSC.
- X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem.–Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed.
- L. Ye and L. Zhang, Org. Lett., 2009, 11, 3646–3649 CrossRef CAS PubMed.
- Z. J. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard III and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 13064–13071 CrossRef CAS PubMed.
- X. Shi, D. J. Gorin and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 5802–5803 CrossRef CAS PubMed.
- O. Nieto Faza, C. Silva López, R. álvarez and A. R. de Lera, J. Am. Chem. Soc., 2006, 128, 2434–2437 CrossRef CAS PubMed.
- G. Lemière, V. Gandon, K. Cariou, A. Hours, T. Fukuyama, A. L. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2009, 131, 2993–3006 CrossRef PubMed.
- (a) G. Li and L. Zhang, Angew. Chem., Int. Ed., 2007, 46, 5156–5159 CrossRef CAS PubMed. For nitrones see: (b) H. S. Yeom, J. E. Lee and S. Shin, Angew. Chem., Int. Ed., 2008, 47, 7040–7043 CrossRef CAS PubMed; (c) H. S. Yeom, Y. Lee, J. Jeong, E. So, S. Hwang, J. E. Lee, S. S. Lee and S. Shin, Angew. Chem., Int. Ed., 2010, 49, 1611–1614 CrossRef CAS PubMed. For epoxides see: (d) G. Y. Lin, C. W. Li, S. H. Hung and R. S. Liu, Org. Lett., 2008, 10, 5059–5062 CrossRef CAS PubMed; (e) A. S. K. Hashmi, M. Bührle, R. Salathé and J. Bats, Adv. Synth. Catal., 2008, 350, 2059–2064 CrossRef CAS; (f) A. M. Jadhav, S. Bhunia, H. Y. Liao and R. S. Liu, J. Am. Chem. Soc., 2011, 133, 1769–1771 CrossRef CAS PubMed.
- (a) L. Cui, Y. Peng and L. Zhang, J. Am. Chem. Soc., 2009, 131, 8394–8395 CrossRef CAS PubMed; (b) L. Cui, L. Ye and L. Zhang, Chem. Commun., 2010, 46, 3351–3353 RSC.
- D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462–1479 CrossRef CAS.
- (a) L. Cui, Y. Peng and L. Zhang, J. Am. Chem. Soc., 2009, 131, 8394–8395 CrossRef CAS PubMed; (b) L. Cui, L. Ye and L. Zhang, Chem. Commun., 2010, 46, 3351–3353 RSC; (c) B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo and L. Zhang, J. Am. Chem. Soc., 2013, 135, 8512–8524 CrossRef CAS PubMed.
- (a) W. He, L. Xie, Y. Xu, J. Xiang and L. Zhang, Org. Biomol. Chem., 2012, 10, 3168–3171 RSC; (b) K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508–6512 CrossRef CAS PubMed.
- Recent lead references: (a) S. Ghorpade, M.-D. Su and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4229–4234 CrossRef CAS PubMed; (b) G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277–6282 CrossRef CAS PubMed.
- E. L. Noey, Y. Luo, L. Zhang and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 1078–1084 CrossRef CAS PubMed.
- C. A. Witham, P. Mauleón, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838–5839 CrossRef CAS PubMed.
- (a) C. Nieto-Oberhuber, S. López and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178–6179 CrossRef CAS PubMed; (b) C. Nieto-Oberhuber, P. Pérez-Galán, E. Herrero-Gómez, T. Lauterbach, C. Rodríguez, S. López, C. Bour, A. Rosellón, D. J. Cárdenas and A. M. Echavarren, J. Am. Chem. Soc., 2008, 130, 269–279 CrossRef CAS PubMed.
- C. Ferrer, M. Raducan, C. Nevado, C. K. Claverie and A. M. Echavarren, Tetrahedron, 2007, 63, 6306–6316 CrossRef CAS PubMed.
- L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS.
- (a) B. M. Trost and G. J. Tanoury, J. Am. Chem. Soc., 1988, 110, 1636–1638 CrossRef CAS; (b) B. M. Trost and M. K. Trost, Tetrahedron Lett., 1991, 32, 3647–3650 CrossRef CAS; (c) B. M. Trost and G. A. Doherty, J. Am. Chem. Soc., 2000, 122, 3801–3810 CrossRef CAS; (d) B. M. Trost, M. Yanai and K. Hoogsteed, J. Am. Chem. Soc., 1993, 115, 5294–5295 CrossRef CAS; (e) B. M. Trost and A. S. K. Hashmi, Angew. Chem., Int. Ed. Engl., 1993, 32, 1085–1087 CrossRef; (f) B. M. Trost and A. S. K. Hashmi, J. Am. Chem. Soc., 1994, 116, 2183–2184 CrossRef CAS; (g) B. M. Trost, A. S. K. Hashmi and R. G. Ball, Adv. Synth. Catal., 2001, 343, 490–494 CrossRef CAS.
- S. I. Lee and N. Chatani, Chem. Commun., 2009, 371–384 RSC.
- (a) A. Fürstner, H. Szillat and F. Stelzer, J. Am. Chem. Soc., 2000, 122, 6785–6786 CrossRef; (b) A. Fürstner, F. Stelzer and H. Szillat, J. Am. Chem. Soc., 2001, 123, 11863–11869 CrossRef PubMed; (c) A. Fürstner, H. Szillat, B. Gabor and R. Mynott, J. Am. Chem. Soc., 1998, 120, 8305–8314 CrossRef.
- (a) E. Mainetti, V. Mouriès, L. Fensterbank, M. Malacria and J. Marco-Contelles, Angew. Chem., Int. Ed., 2002, 41, 2132–2135 CrossRef CAS; (b) Y. Harrak, C. Blaszykowski, M. Bernard, K. Cariou, E. Mainetti, V. Mouriès, A.-L. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2004, 126, 8656–8657 CrossRef CAS PubMed; (c) C. Blaszykowski, Y. Harrak, M.-H. Gonçalves, J.-M. Cloarec, A.-L. Dhimane, L. Fensterbank and M. Malacria, Org. Lett., 2004, 6, 3771–3774 CrossRef CAS PubMed; (d) J. Marco-Contelles, N. Arroyo, S. Anjum, E. Mainetti, N. Marion, K. Cariou, G. Lemière, V. Mouriès, L. Fensterbank and M. Malacria, Eur. J. Org. Chem., 2006, 4618–4633 CrossRef CAS; (e) C. Blaszykowski, Y. Harrak, C. Brancour, K. Nakama, A.-L. Dhimane, L. Fensterbank and M. Malacria, Synthesis, 2007, 2037–2049 CAS.
- J. W. Faller and P. P. Fontaine, J. Organomet. Chem., 2006, 691, 1912–1915 CrossRef CAS PubMed.
- A. Escribano-Cuesta, P. Pérez-Galán, E. Herrero-Gómez, M. Sekine, A. A. C. Braga, F. Maseras and A. M. Echavarren, Org. Biomol. Chem., 2012, 12, 6105–6111 Search PubMed.
- S. Oi, I. Tsukamoto, S. Miyano and Y. Inoue, Organometallics, 2001, 20, 3704–3709 CrossRef CAS.
- R. B. Dateer, B. S. Shaibu and R. S. Liu, Angew. Chem., Int. Ed., 2012, 51, 113–117 CrossRef CAS PubMed.
- S. I. Lee, S. M. Kim, M. R. Choi, S. Y. Kim and Y. K. Chung, J. Org. Chem., 2006, 71, 9366–9372 CrossRef CAS PubMed.
- R. E. M. Brooner, T. J. Brown and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 6259–6261 CrossRef CAS PubMed.
- Y. Odabachian and F. Gagosz, Adv. Synth. Catal., 2009, 351, 379–386 CrossRef CAS.
- N. Cabello, E. Jiménez-Núñez, E. Buñuel, D. J. Cárdenas and A. M. Echavarren, Eur. J. Org. Chem., 2007, 4217–4223 CrossRef CAS.
- E. Jiménez-Núñez, C. K. Claverie, C. Bour, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2008, 47, 7892–7895 CrossRef PubMed.
- A. Escribano-Cuesta, V. López-Carrillo, D. Janssen and A. M. Echavarren, Chem.–Eur. J., 2009, 11, 5646–5650 CrossRef PubMed.
- (a) M. Schelwies, A. L. Dempwolff, F. Rominger and G. Helmchen, Angew. Chem., Int. Ed., 2007, 46, 5598–5601 CrossRef PubMed; (b) M. Schelwies, R. Moser, A. L. Dempwolff, F. Rominger and G. Helmchen, Chem.–Eur. J., 2009, 15, 10888–10900 CrossRef CAS PubMed.
- E. Jiménez-Núñez, C. K. Claverie, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5452–5455 CrossRef PubMed.
- N. Huguet, and A. M. Echavarren, Synlett, 2012, 49–53 CAS.
- (a) S. López, E. Herrero-Gómez, P. Pérez-Galán, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 6029–6032 CrossRef PubMed; (b) P. Pérez-Galán, H. Herrero-Gómez, D. T. Hog, N. J. A. Martin, F. Maseras and A. M. Echavarren, Chem. Sci., 2011, 2, 141–149 RSC.
- V. López-Carrillo, N. Huguet, A. Mosquera and A. M. Echavarren, Chem.–Eur. J., 2011, 17, 10972–10978 CrossRef.
- M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 18002–18003 CrossRef CAS PubMed.
- (a) Y. Zou, D. Garayalde, Q. Wang, C. Nevado and A. Goeke, Angew. Chem., Int. Ed., 2008, 47, 10110–10113 CrossRef CAS PubMed; (b) D. Garayalde, K. Kruger and C. Nevado, Angew. Chem., Int. Ed., 2010, 50, 911–915 CrossRef PubMed.
- E. Jiménez-Núñez, M. Raducan, T. Lauterbach, K. Molawi, C. R. Solorio and A. M. Echavarren, Angew. Chem., Int. Ed., 2009, 48, 6152–6155 CrossRef PubMed.
- (a) M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS PubMed; (b) M. R. Fructos, M. M. Díaz-Requejo and P. J. Pérez, Chem. Commun., 2009, 5153–5155 RSC; (c) A. Prieto, M. R. Fructos, M. M. Díaz-Requejo, P. J. Pérez, P. Pérez-Galán, N. Delpont and A. M. Echavarren, Tetrahedron, 2009, 65, 1790–1793 CrossRef CAS PubMed.
- C. R. Solorio, Y. Wang and A. M. Echavarren, J. Am. Chem. Soc., 2011, 133, 11952–11955 CrossRef PubMed.
- (a) A. Fedorov, M. E. Moret and P. Chen, J. Am. Chem. Soc., 2008, 130, 8880–8881 CrossRef CAS PubMed; (b) A. Fedorov and P. Chen, Organometallics, 2010, 29, 2994–3000 CrossRef CAS; (c) A. Fedorov, L. Batiste, A. Bach, D. M. Birney and P. Chen, J. Am. Chem. Soc., 2011, 133, 12162–12171 CrossRef CAS PubMed; (d) D. H. Ringger and P. Chen, Angew. Chem., Int. Ed., 2013, 52, 4686–4689 CrossRef CAS PubMed.
- M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248–8262 CrossRef CAS PubMed and references cited therein.
- For some illustrative examples see: (a) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9533–9537 CrossRef CAS PubMed; (b) M. Bandini, A. Bottoni, M. Chiarucci, G. Cera and G. P. Miscione, J. Am. Chem. Soc., 2012, 134, 20690–20700 CrossRef CAS PubMed; (c) C. Praveen, B. Montaignac, M. R. Vitale, V. Ratovelomanana-Vida and V. Michelet, ChemCatChem, 2013, 5, 2395–2404 CrossRef CAS.
- J. J. Hirner, Y. Shi and S. A. Blum, Acc. Chem. Res., 2011, 44, 603–613 CrossRef CAS PubMed and references cited therein.
- B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS PubMed.
- P. H. Y. Cheong, P. Morganelli, M. R. Luzung, K. N. Houk and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 4517–4526 CrossRef CAS PubMed.
- (a) K. A. Porter, A. Schier and H. Schmidbaur, Organometallics, 2003, 22, 4922–4927 CrossRef CAS; (b) K. I. Grandberg and V. P. Dyadchenko, J. Organomet. Chem., 1994, 474, 1–21 CrossRef CAS; (c) K. I. Grandberg, Russ. Chem. Rev., 1982, 51, 249–262 CrossRef PubMed; (d) A. N. Nesmeyanov, E. G. Perevalova, K. I. Grandberg, D. A. Lemenovskii, T. V. Baukova and O. B. Afanassova, J. Organomet. Chem., 1974, 65, 131–144 CrossRef CAS.
- (a) D. Weber, M. A. Tarselli and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 5733–5736 CrossRef CAS PubMed; (b) D. Weber and M. R. Gagné, Chem. Sci., 2012, 4, 335–338 RSC.
- T. J. Brown, D. Weber, M. R. Gagné and R. A. Widenhoefer, J. Am. Chem. Soc., 2012, 134, 9134–9137 CrossRef CAS PubMed.
- G. Seidel, C. W. Lehmann and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 8466–8470 CrossRef CAS PubMed.
- D. Weber, T. D. Jones, L. L. Adduci and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 2452–2456 CrossRef CAS PubMed.
- T. J. Brown and R. A. Widenhoefer, Organometallics, 2011, 30, 6003–6009 CrossRef CAS.
- A. Grirrane, H. Garcia, A. Corma and E. álvarez, ACS Catal., 2011, 1, 1647–1653 CrossRef CAS.
- C. Obradors and A. M. Echavarren, Chem.–Eur. J., 2013, 19, 3547–3551 CrossRef CAS PubMed.
- A. Simonneau, F. Jaroschik, D. Lesage, M. Karanik, R. Guillot, M. Malacria, J. C. Tabet, J. P. Goddard, L. Fensterbank, V. Gandon and Y. Gimbert, Chem. Sci., 2011, 2, 2417–2422 RSC.
- (a) L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef CAS PubMed; (b) Y. Wang, A. Yepremyan, S. Ghorai, R. Todd, D. H. Aue and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7795–7799 CrossRef CAS PubMed.
- D. D. Vachhani, M. Galli, J. Jacobs, L. Van Meervelt and E. V. Van der Eycken, Chem. Commun., 2013, 49, 7171–7173 RSC.
- A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456–4460 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Wieteck, I. Braun, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 10633–10637 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644–661 CrossRef CAS; (b) A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhelmsen, M. Rudolph and F. Rominger, Chem.–Eur. J., 2012, 19, 1058–1065 CrossRef PubMed; (c) A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nösel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555–562 CrossRef CAS.
- M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 2593–2598 CrossRef CAS PubMed.
- P. Nösel, T. Lauterbach, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 8634–8641 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |