DOI:
10.1039/C4BM00065J
(Paper)
Biomater. Sci., 2014,
2, 1367-1376
A facile strategy to prepare redox-responsive amphiphilic PEGylated prodrug with high drug loading content and low critical micelle concentration†
Received
27th February 2014
, Accepted 7th May 2014
First published on 24th June 2014
Abstract
A redox-responsive amphiphilic polymeric prodrug was synthesized in a facile way by polycondensation of oligo(ethylene glycol) with dicarboxylic acids including malic acid and 3,3′-dithiodipropionic acid , followed by esterification with ibuprofen, which was used as a model drug. Because of its amphiphilic nature and relatively high molecular weight, this polymeric prodrug can form stable micelles in aqueous media with a low critical micellar concentration (CMC). Free ibuprofen molecules can be steadily incorporated into the core of these micelles with a surprisingly high loading content (38.9 wt%), owing to hydrophobic interaction and π–π stacking with the ibuprofen moieties in the copolymer. The in vitro release results indicate that there was a relatively slow and sustained release of the conjugated ibuprofen moieties, while encapsulated ibuprofen molecules showed a rapid release. Furthermore, for both the conjugated ibuprofen and the encapsulated ibuprofen there was an accelerated release in the presence of 10 mM DL-dithiothreitol due to cleavage of the disulfide bonds, which lead to disassociation of the micelles. Notably, this prodrug was revealed to have excellent cell compatibilities via a cell counting kit-8 (CCK-8) assay. Confocal laser scanning microscope observations indicated that the micelles based on the polymeric prodrug can be taken up quickly by cells and present a redox-responsive drug release in cytoplasm. This kind of polymeric nanocarrier with a high drug loading content, low CMC, excellent biocompatibility and rapid response to a reductive environment may have tremendous scope in the area of controlled drug delivery.
Introduction
The clinical use of most drugs is limited because of their poor water solubility, low selectivity, rapid blood clearance and the severe side effects to healthy tissues with few drugs reaching the desired site in vivo.1,2 The development of smart vehicles which selectively deliver drug molecules to target cells is urgent and has attracted rapidly increasing attention.3–5 Various polymeric drug delivery systems, such as polymeric micelles,6–8 polymer–drug conjugates (polymeric prodrugs)9–12 and polymeric gels13,14 have been developed to address these problems.
Polymeric micelles with their unique core–shell architecture, good stability under physiological conditions and the ability to target cancerous tissues by passive accumulation via the tumors’ enhanced permeability and retention (EPR) effect has received tremendous interest in the biomedical area.15–18 Hydrophobic drugs can be encapsulated into the hydrophobic core of micelle carriers and protected by a hydrophilic shell. However, many studies have shown that the contents of encapsulated drugs are generally not greater than 10% to suppress premature burst release.19–22
Recent attention has been focused on the conjugation of drugs with polymers, termed polymeric prodrugs, which can improve solubility, bioavailability, stability of the drug, and can prolong the circulation time of a drug to make it less likely to be prematurely released.23–26 Notably, using a drug as part of a nanocarrier can also minimize the use of other inert materials in the drug carrier with unexpected side effects, and can increase the drug loading content.27 Among these, PEGylated prodrugs have been widely investigated as biocompatible drug carriers for the reason that PEG can reduce nonspecific interactions with proteins and cells, as well as having been approved by the U.S. Food and Drug Administration (FDA).28–32 Recently, Shen and coworkers reported a PEGylated prodrug, synthesized through the conjugation of camptothecin (CPT) to an oligo(ethylene glycol) (OEG) terminal via an ester bond, resulting in a high drug loading content.33 But for this kind of method, a short OEG chain is required in order to achieve a high drug loading capacity and for micelle formation. The obtained low molecular weight prodrugs, however, exhibit poor stability because of the high critical micellar concentration (CMC). To dissipate this defect, many papers have reported on the development o PEGylated prodrugs with a high molecular weight, based on amphiphilic block copolymers with pendant functional groups for drug attachment,34–36 synthesized by the ring-opening polymerization of a functional cyclic monomer with PEG as the macroinitiator. Nevertheless, the preparation is difficult and complicated with protection/deprotection, and comonomers without functional groups are required to reduce the steric hindrance, resulting in a low drug loading content.37
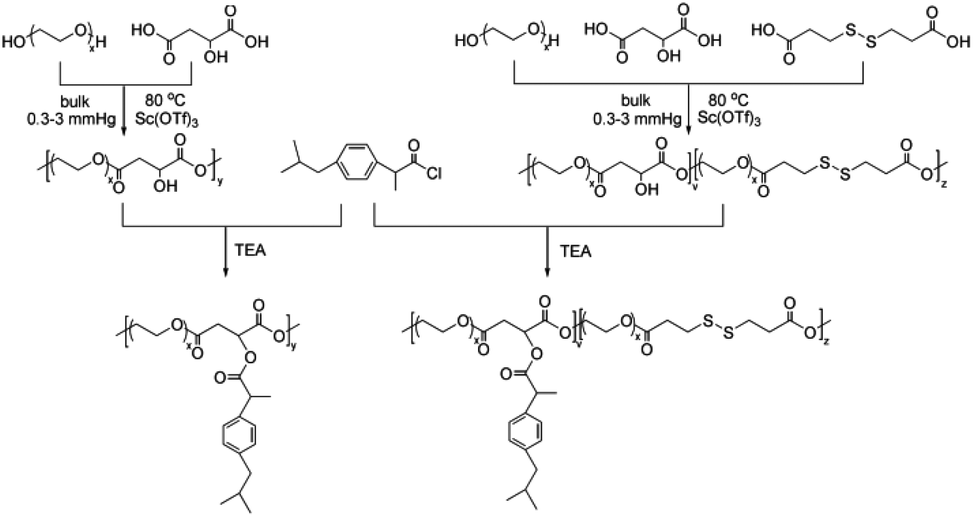
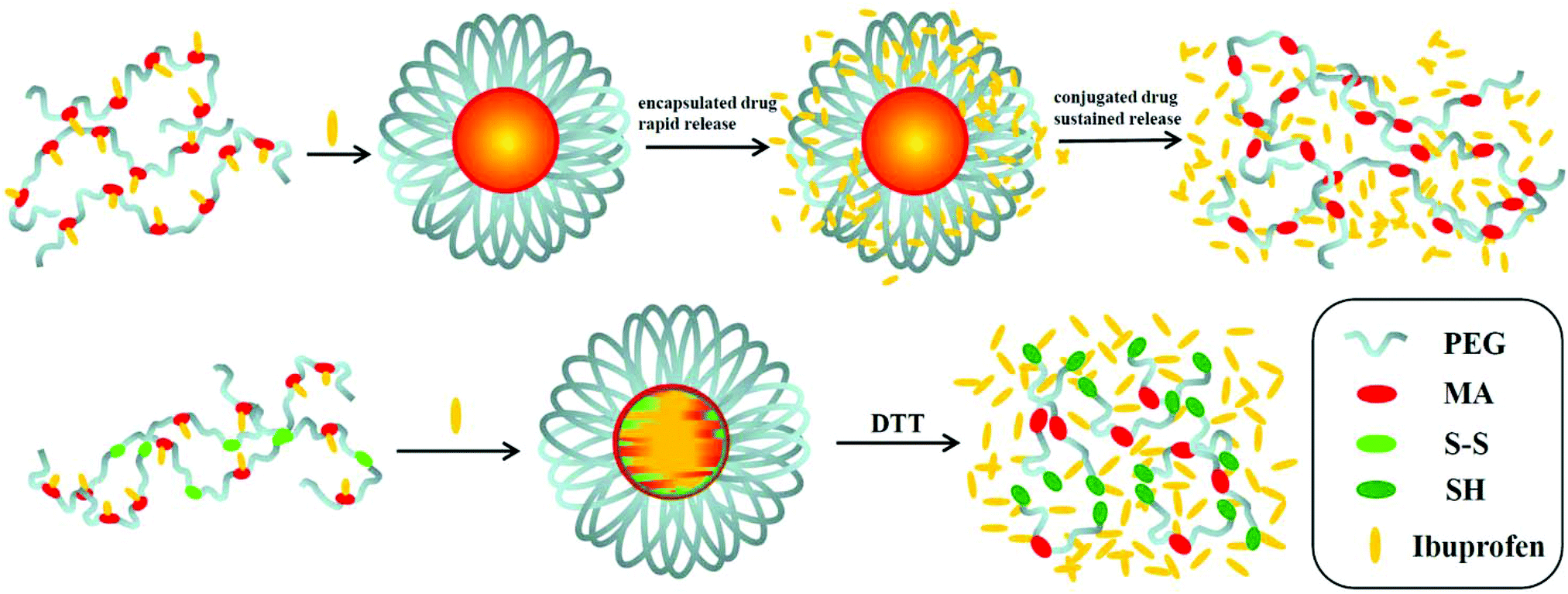
Here, in order to overcome the above mentioned shortcomings of PEGylated prodrugs, we develop a facile strategy to fabricate redox-responsive PEGylated prodrugs with a high drug loading content and a low CMC based on a multifunctional PEG derivative, poly[oligo(ethylene glycol) dithiodipropionicate-co-oligo(ethylene glycol) malicate] (POEGSSM), which was synthesized through the direct chemoselective polycondensation of OEG diol with corresponding dicarboxylic acids under mild conditions using Sc(OTf)3 as a chemoselective catalyst.38–41 Ibuprofen, a potent non-steroidal anti-inflammatory drug used in the treatment of rheumatism, arthritis, fever and other conditions, was selected as a model of a poorly water-soluble drug and was conjugated onto the POEGSSM backbone through ester bonds. Owing to the much reduced steric hindrance from the neighboring pendent hydroxyl groups in POEGSSM, a high drug loading PEG-ibuprofen polymeric prodrug was thus prepared, as shown in Scheme 1. As a control, a non-reducing cleavable polymeric prodrug based on poly[oligo(ethylene glycol) malicate] (POEGM) was also prepared in a similar manner. The prodrugs can form stable micelles in aqueous media with low CMC. Additionally, free ibuprofen molecules can be steadily encapsulated into the micelle. The in vitro release of the conjugated ibuprofen from POEGSSIBu micelles was relatively slow and sustained, while physically encapsulated ibuprofen showed a rapid release. Both of them showed a DL-dithiothreitol (DTT)-triggered release as shown in Scheme 2. The biocompatibility and cell uptake of the polymeric prodrug micelles were also studied, and the results showed that this kind of biodegradable and biocompatible polymeric nanocarrier with a high drug loading content, low CMC and reductive response has great potential in biomedical applications.
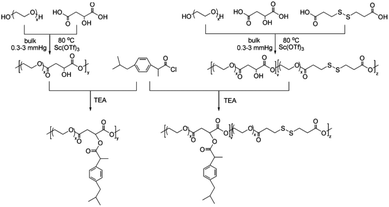 |
| | Scheme 1 Synthetic routes for PEG-ibuprofen polymeric prodrugs. | |
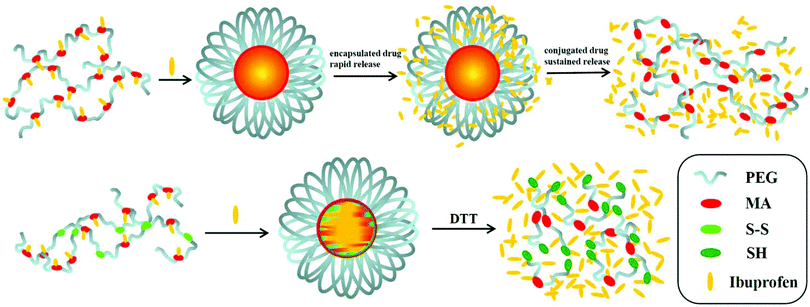 |
| | Scheme 2 Illustrative preparation of ibuprofen-loaded prodrug micelles and their redox-responsive ibuprofen release. | |
Experimental
Materials
Oligo(ethylene glycol) diols (OEG400, Mn = 400 and OEG600, Mn = 600, Aladdin, China) were dried by azeotropic distillation in the presence of dry toluene. Ibuprofen (IBu, Juhua Group Corporation; China), DL-dithiothreitol (DTT, Aladdin, China), DL-malic acid (MA, Aladdin, China), 3,3′-dithiodipropionic acid (DTPA, Aladdin, China), Nile red (NR, Sigma) and other reagents were used as received. Sc(OTf)3 was synthesized according to our previous report.42
Preparation of POEGSSM and POEGM
28.0 g of OEG400 (70 mmol), 4.7 g of MA (35 mmol), 7.36 g of DPTA (35 mmol) and 0.35 g of Sc(OTf)3 (0.7 mmol) were stirred in a 250 mL round-bottomed reactor at 80 °C under an argon atmosphere for 4 h. Then the pressure was gradually decreased to 0.3–3 mmHg with stirring for 24 h. The crude product was purified by neutral alumina column chromatography with methylene chloride as the eluent. After concentrating the eluate, the solution was poured into ethyl ether to precipitate POEGSSM, which was dried under vacuum overnight to a constant weight. Yield: 35.7 g (95%). POEGM was synthesized from 27.0 g of OEG600 (45 mmol), 6.03 g of MA (45 mmol) and 0.22 g of Sc(OTf)3 (0.45 mmol) in the same manner as described above. Yield: 30.2 g (96%). The 1H NMR spectra of POEGSSM and POEGM were recorded using a Bruker Avance DMX500 spectrometer in CDCl3 with tetramethylsilane as the internal standard. The molecular weight and molecular weight distribution of the obtained copolymers POEGSSM and POEGM were determined by gel permeation chromatography (GPC), with a Waters degasser, a Waters 1515 Isocratic HPLC pump, a Wyatt Optilab DSP interferometric refractometer, and Styragel, HT 3 and HT 4 columns. Tetrahydrofuran (THF) was used as the mobile phase at a flow rate of 1.0 mL min−1 at 25 °C and polystyrenes with molecular weights ranging from 580 to 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 were used as the calibration standards.
000 were used as the calibration standards.
Preparation of POEGSSMIBu and POEGMIBu via acylation reaction
IBu (10 g, 49 mmol) was dissolved into excess thionyl chloride and a drop of DMF was added as the catalyst. The mixture was stirred at 80 °C for 10 h, and then excess thionyl chloride was removed under vacuum to give IBu-acyl chloride (IBu-COCl) (9.8 g, 90%). POEGSSM (2.0 g, 1.8 mmol) and TEA (0.77 mL, 5.4 mmol) were dissolved in 25 mL of anhydrous CH2Cl2, after stirring at 0 °C for 30 min, IBu-COCl (1.23 g, 5.4 mmol) was added dropwise within 25 min. The solution was stirred overnight at room temperature and then washed successively with aqueous solutions of NaHCO3 and NaCl. The organic phase was dried over anhydrous MgSO4, filtered, evaporated to dryness and recrystallized from CH2Cl2–ethyl ether. The resulting solid POEGSSMIBu was dried under vacuum overnight to a constant weight. Yield: 2.16 g (92%). POEGMIBu was prepared from POEGM (1.7 g, 2.4 mmol), TEA (1.02 mL, 7.2 mmol) and IBu-COCl (1.64 g, 7.2 mmol) in the same manner. Yield: 2.1 g (93%).
Preparation of POEGSSMIBu and POEGMIBu micelles
POEGSSMIBu (50 mg) was dissolved in 10 mL of THF, and then 10 mL of phosphate buffer solution (PBS, 0.01 M, pH 7.4) was added dropwise to the solution under stirring. The resulting solution was stirred for 2 h and dialyzed (MWCO 3500, Fisher Scientific) against PBS (0.01 M, pH 7.4) over 24 h to completely remove the THF, forming a micelle structure. The final concentration of the micellar solution was adjusted to 1.0 mg mL−1. Meanwhile, micelles of POEGMIBu were prepared in the same manner. The size and size distribution of the micelles were determined by dynamic light scattering (DLS) at 25 °C using a Zetasizer Nano-ZS instrument from Malvern Instruments with a He–Ne laser (633 nm). All of the micellar solutions had a final polymer concentration of 1.0 mg mL−1 and were filtered through a 0.45 μm filter. The morphology of the micelles was observed by using a JEM-1230 transmission electron microscope (TEM) operated at an acceleration voltage of 60 kV. A drop of 1.0 mg mL−1 micellar solution was put onto the surface of Formvar-carbon film-coated copper grids. Excess solvent was quickly removed with a filter paper and then stained with 2 wt% phosphotungstic acid aqueous solution.
Reductive degradation of POEGSSMIBu micelles
It is well known that disulfide linkages are stable under normal physiological conditions but respond to glutathione (GSH) or reductases via reversible cleavage into free thiols.43,44 In order to reduce the disulfide bonds into free thiols entirely, 75 mg of DTT (0.5 mmol, 10 mM) was dissolved in POEGSSMIBu micellar solution (50 mL, 1.0 mg mL−1) and stirred at 37 °C overnight (10 h). Then the solution was added into a dialysis membrane bag (MWCO 3500, Fisher Scientific) and dialyzed against double distilled water for another 24 h. The degraded product POEGSHMIBu was finally retrieved by lyophilization.
CMC of POEGSSMIBu, POEGMIBu and POEGSHMIBu
The CMC of POEGSSMIBu, POEGMIBu and POEGSHMIBu was estimated by fluorescence measurement using pyrene as a fluorescent probe. The pyrene concentration in the solution was fixed at 6.0 × 10−7 M and the concentration of the block copolymer POEGSSMIBu, POEGMIBu and POEGSHMIBu was varied from 1.0 × 10−6 to 1.0 mg mL−1. When the polymeric micelles were formed, pyrene was preferentially incorporated into the hydrophobic micelle core instead of the polar environment (aqueous solution). And the sharp rise in the intensity ratio (I338/I333) of pyrene in the excitation spectra indicates the onset of micellization (CMC) for the amphiphilic copolymer. Fluorescence excitation spectra (300–360 nm) of the solutions were recorded using an Hitachi F-4500 fluorescence spectrometer at an emission wavelength of 390 nm, with the excitation and emission bandwidths set at 5 nm slit widths.
Encapsulation of free IBu into POEGSSMIBu and POEGMIBu micelles
POEGSSMIBu (50 mg) and IBu (15 mg) were dissolved in 10 mL of THF, and then 5 mL of PBS (0.01 M, pH 7.4) was added dropwise to the solution under stirring. The resulting solution was stirred for 2 h and dialyzed (MWCO 3500, Fisher Scientific) against PBS (0.01 M, pH 7.4) over 24 h to remove THF, forming IBu-loaded micelles. The solution was filtered to remove unincorporated IBu. The final concentration of the micellar solution was adjusted to 1.0 mg mL−1. The encapsulation of IBu to POEGMIBu micelle was carried out in a similar way. The solid state of drug-loaded micelles was retrieved by lyophilization and the amount of IBu trapped in the micelles was determined by 1H NMR (Fig. S1 and S2†).45 The drug loading content (DLC) and drug loading efficiency (DLE) were calculated according to the following equations:
| DLC (wt%) = WDM/WP × 100% |
| DLE (wt%) = (WDM − WDC)/WDt × 100% |
WDM is the weight of IBu measured by 1H NMR spectroscopy, WDC is the weight of conjugated IBu, WP is the weight of POEGSSMIBu or POEGMIBu copolymer, WDt is the total weight of free IBu used in the drug-encapsulation experiment.
In vitro IBu release from POEGSSMIBu and POEGMIBu micelles before and after loading with free IBu
5 mL of POEGSSMIBu and IBu-loaded POEGSSMIBu micellar solutions (1.0 mg mL−1) in PBS (0.01 M, pH 7.4) were transferred into a dialysis membrane bag (MWCO 3500, Fisher Scientific) respectively, and then immersed in 50 mL of PBS (0.01 M, pH 7.4) with or without DTT (10 mM), and held at a constant temperature of 37 °C in a water bath with horizontal shaking. At a predetermined time interval, 5 mL of the incubated solution was taken out and replenished with an equal volume of corresponding PBS. In vitro release of IBu from POEGMIBu and IBu-loaded POEGMIBu micelles were determined in pure PBS (0.01 M, pH 7.4) with the same operation as described above. IBu release profiles were determined by measuring the absorbance at a wavelength of 222 nm using a Shimadzu UV2550 UV-vis spectrophotometer. Calibration curves were established from known concentrations of IBu in PBS (0.01 M, pH = 7.4) and PBS (0.01 M, 10 mM DTT, pH = 7.4), respectively (Fig. S3†). After the IBu moieties were completely released from the polymeric prodrug POEGMIBu, the remaining polymer, named Re-POEGM, was retrieved by lyophilization and determined by GPC.
In vitro cytotoxicity of POEGSSMIBu and POEGMIBu
The cytotoxicity of the polymeric prodrugs POEGMSSIBu and POEGMIBu were evaluated by the CCK-8 (Dojindo, Japan) assay. All the tests were run five times. POEGSSMIBu and POEGMIBu were prepared in PBS (0.01 M, pH 7.4) and then sterilized by filtration (0.22 μm). MC3T3-E1 cells (Cell bank of the Chinese Academy of Science, China) were pre-incubated in a 96-well plate (5 × 103 cells per well) with a culture medium of 10% FBS/α-MEM (Invitrogen Co., Carlsbad, CA) in a humidified 5% CO2-containing atmosphere at 37 °C for 24 h. Then the cells were further incubated with POEGSSMIBu and POEGMIBu at four concentrations, 0.1 mg L−1, 1 mg L−1, 10 mg L−1, and 100 mg L−1 for 24 h. Subsequently, the media was aspirated and replenished with 100 μL of fresh culture medium. 10 μL of CCK-8 reagents were added into each well, and the cells were incubated in the dark for another 1 h. The absorbance at a wavelength of 450 nm of each well was measured using a microplate reader (Sunrise™ Basic; TECAN, Zurich, Switzerland). Non-treated cells were used as a negative control, wells without cells but culture medium were used as the blank. The relative cell viability was calculated as follows:
| Cell viability (%) = [(OD 450sample − OD 450blank)/(OD 450control − OD 450blank)] × 100% |
Data are presented as the average ± SD (n = 5).
Cellular uptake and intracellular release of POEGSSMIBu and POEGMIBu micelles
The cellular uptake experiments were performed for POEGSSMIBu and POEGMIBu micelles by loading NR (red fluorescence) as a hydrophobic fluorescence probe into these micelles. NR (0.1 mg) with POEGSSMIBu (20 mg) or POEGMIBu (20 mg) were dissolved in 5 mL of tetrahydrofuran (THF) and stirred for 30 min at room temperature. Then 5 mL of PBS (0.01 M, pH 7.4) was added dropwise to the solution under stirring. The resulting solution was dialyzed against PBS (0.01 M, pH 7.4) for 24 h to remove THF and form NR-loaded POEGSSMIBu and POEGMIBu micelles. After that, the solution was filtered to remove unincorporated NR and the concentration of the micelles was adjusted to 1.0 mg mL−1.
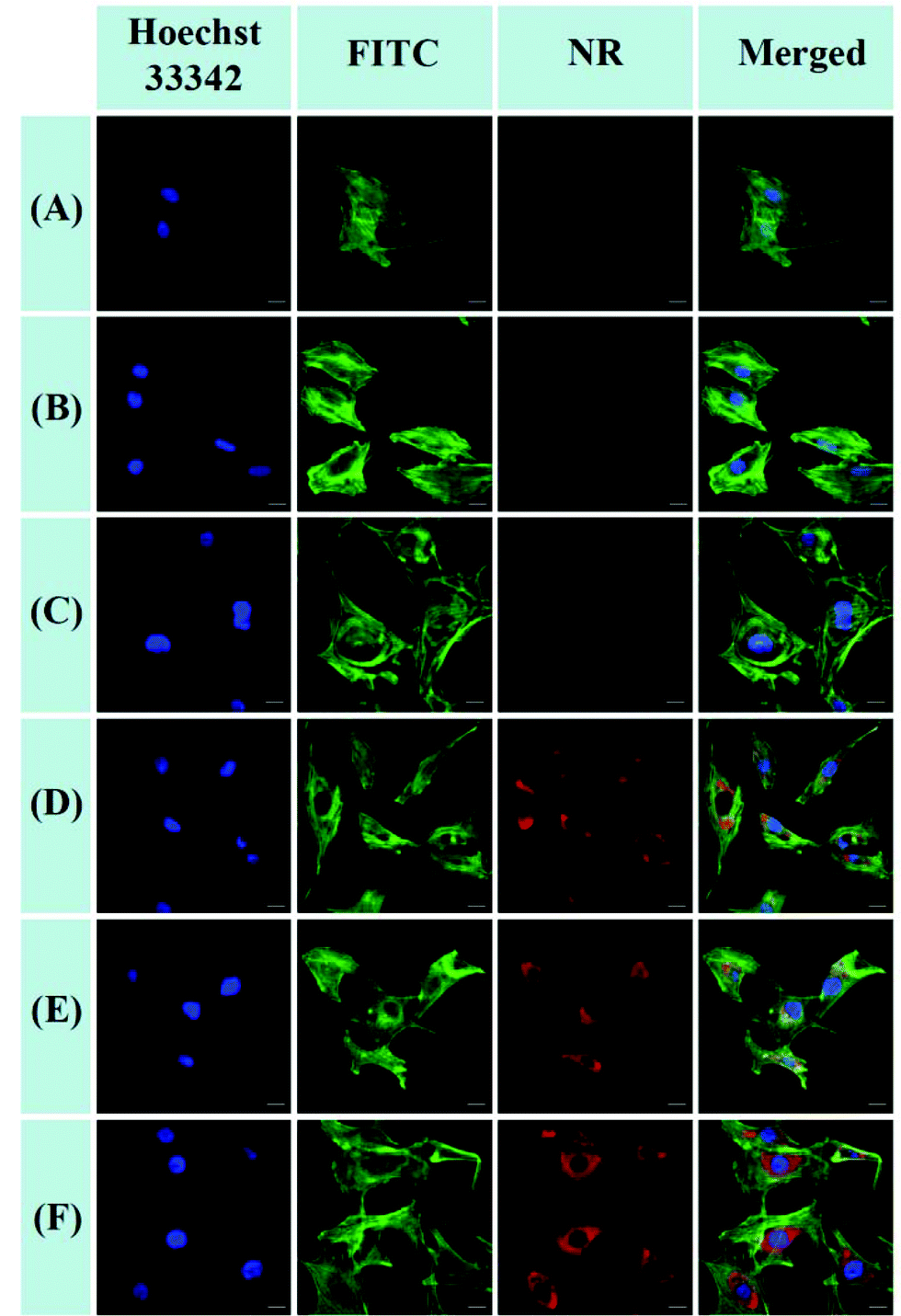
Cellular uptakes intracellular NR release of NR-loaded POEGSSMIBu and POEGMIBu micelles were observed on CLSM (BX61W1-FV1000, OLYMPUS, Japan). Cellular uptake of free NR was performed as a control. Autoclave sterilized coverslips were placed in a 6-well plate. The MC3T3-E1 cells were seeded with a concentration of 2 × 104 cells per well in 1.8 mL of complete α-MEM and cultured for 24 h. Then NR-loaded micelles or free NR were added, and cultured for 2 h and 4 h, respectively. Then the culture media was removed, followed by rinsing of the cells three times with PBS (0.01 M, pH 7.4) and fixing with 4% paraformaldehyde at room temperature for 15 min. The cells were stained with fluorescein isothiocyanate (FITC)-labeled phalloidin (Sigma-Aldrich) for cell cytoskeletons and Hoechst 33342 (Sigma Aldrich) for cell nucleus. After being mounted with neutral balsam, the samples were studied using a 60× magnification microscope.
The amount of NR in the cells was determined using a Spectramax M5 plate reader (Molecular Devices, LLC, USA). 180 μL of a suspension of the MC3T3-E1 cells was seeded into each well of a 96-well plate (5 × 103 cells) and equal culture mediate was added into the wells without cells as the blank. After pre-incubation under the same conditions for 24 h, NR-loaded micelles or free NR in 20 μL of PBS (0.01 M, pH 7.4) were added. An equivalent amount of PBS was added into the negative control and blank. After 2 or 4 h incubation, the cells were washed with PBS twice, and finally 100 μL of PBS was added. The content of NR inside the cells was determined by fluorescence intensity (Spectramax M5, ex: 550 nm, em: 605 nm). Each group was tested in quintuplicate.
Results and discussion
Synthesis and characterization of POEGSSM and POEGM
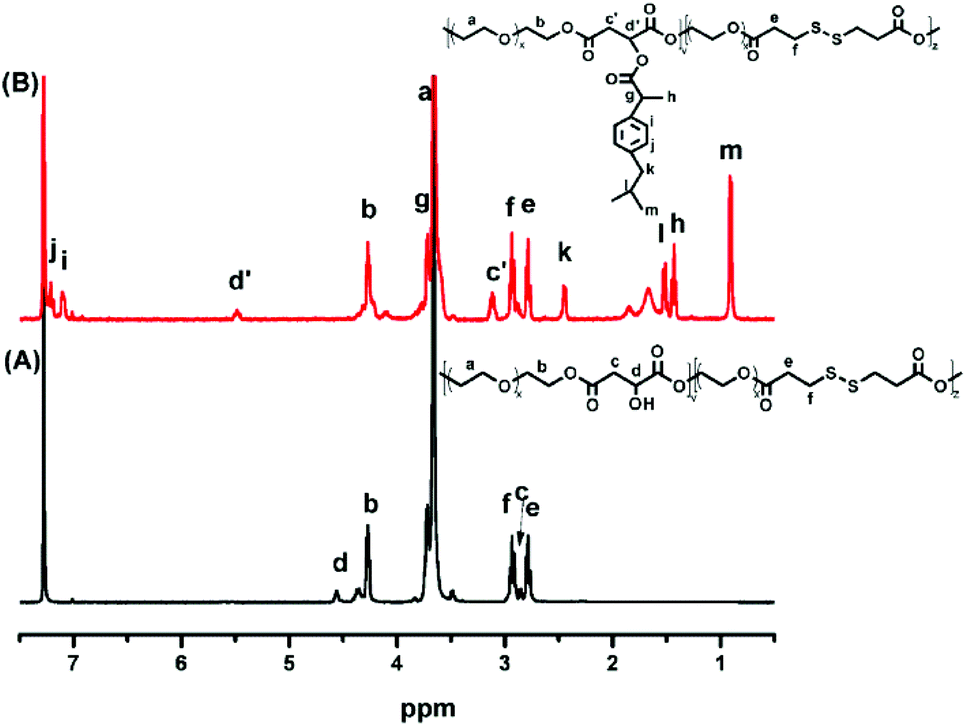
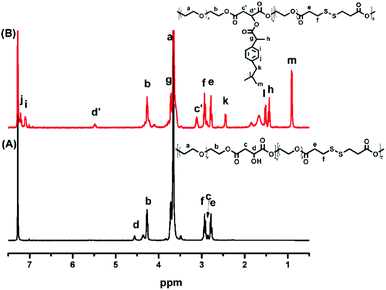
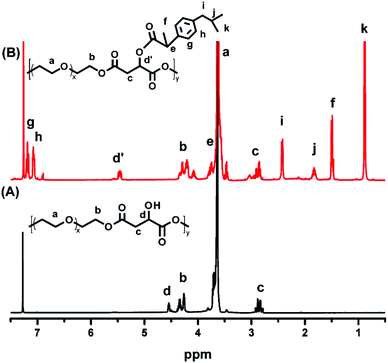
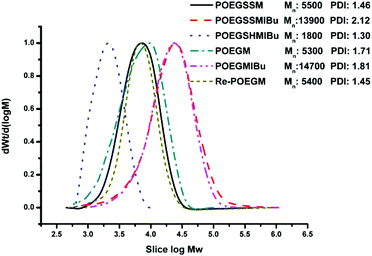
POEGSSM was prepared via the direct polycondensation of OEG diol with malic acid and 3,3′-dithiodipropionic acid, which was carried out under reduced pressure in bulk using Sc(OTf)3 as the chemoselective catalyst. The 1H NMR spectrum of POEGSSM is shown in Fig. 1A with all the relevant signals well labelled, especially for the signals ascribed to DTPA, which clearly implies the introduction of a disulfide bond into the backbone of the polyester. Notably, the appearance of the signal centered at 4.55 ppm (Hd), attributed to the protons of the methylidyne group in the malicate unit, indicated the synthesis of polyesters with pendent secondary hydroxyl groups by chemoselective esterification of primary hydroxyl groups with carboxyl groups. Meanwhile, the molar ratio of OEG–MA–DPTA in the copolymer is in agreement with the feeding molar ratio (2:1:1) according to the integrals of the corresponding protons. As a control group, another PEG derivative without disulfide bonds (POEGM) was also synthesized in the same manner by adding OEG diol with an equimolar amount of malic acid. The 1H NMR spectrum of POEGM, as well as the signal assignments, are presented in Fig. 2A, which also clearly verified the synthesis of a PEG derivative with pendant hydroxyl groups. The molecular weights of the copolymers were determined by GPC (Fig. 3), which were unimodal, further confirming the successful synthesis of POEGSSM and POEGM via polycondensation.
 |
| | Fig. 1
1H NMR spectra of POEGSSM (A) and POEGSSMIBu (B). | |
 |
| | Fig. 2
1H NMR spectra of POEGM (A) and POEGMIBu (B). | |
 |
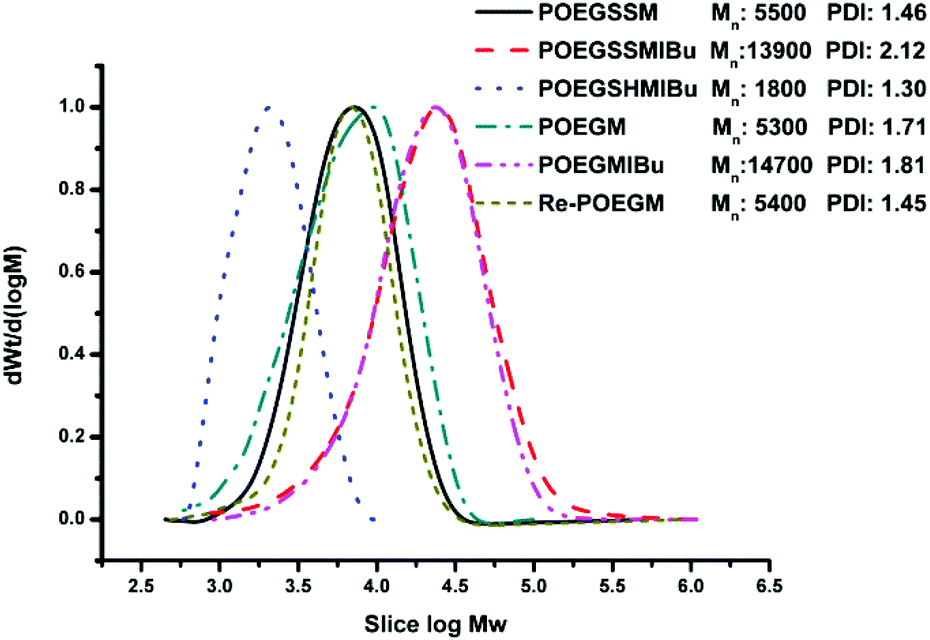
| | Fig. 3 GPC curves of POEGSSM, POEGSSMIBu, POEGSHMIBu, POEGM, POEGMIBu, Re-POEGM. | |
Synthesis and characterization of POEGSSMIBu and POEGMIBu
The synthesis of a reductive-sensitive polymeric prodrug via the acylation reaction of IBu-COCl to POEGSSM was performed at 0 °C in the presence of TEA. A representative 1H NMR spectrum of POEGSSMIBu is shown in Fig. 1B, clearly exhibiting the variation of the chemical structure compared with its precursor (POEGSSM). The shift of the signal from δ 4.55 ppm (Hd) to δ 5.46 ppm (Hd′), as well as the presence of the signals assigned to IBU moieties, implies the successful conjugation of IBu to POEGSSM by an ester bond, resulting in a polymeric prodrug with a disulfide bond in the backbone. Meanwhile, the non-reducing cleavable prodrug POEGMIBu was prepared in a similar way and the 1H NMR spectrum is presented in Fig. 2B. Moreover, the POEGSSMIBu and POEGMIBu were further characterized by GPC measurements. As shown in Fig. 3, compared with their corresponding precursors, the curves of the polymeric prodrugs significantly shifted to a high molecular weight region, but still remained unimodal, signifying the successful synthesis of the polymeric prodrugs.
The disulfide bonds of POEGSSMIBu were completely cleaved by treatment with DTT (10 mM) at 37 °C for 10 h, resulting in a reductively degraded product (POEGSHMIBu), which was measured by GPC. Compared with POEGSSMIBu, the GPC curve of POEGSHMIBu shifted to the low molecular weight region, which clearly implies the reductive degradation of POEGSSMIBu owing to the cleavage of disulfide bonds in the backbone.
Preparation of micelles from POEGSSMIBu and POEGMIBu
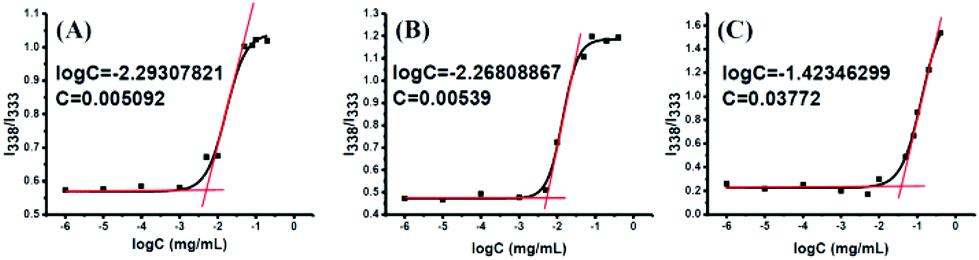
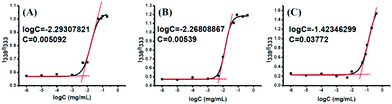
Amphiphilic polymeric prodrugs can self-assemble into micelles in an aqueous solution by the dialysis method, and have been characterized by measuring the CMC using pyrene as a fluorescent probe.46 The intensity ratio of bands at 338 and 333 nm (I338/I333) was calculated and plotted versus the polymer concentrations, presenting a sigmoid curve as shown in Fig. 4. The sharp rise in the intensity ratio of the peaks at 338 and 333 nm of pyrene in the excitation spectra points the on-set of micellization (CMC) for the amphiphilic copolymer. The CMC values of POEGSSMIBu and PEOGMIBu were about 5.09 mg L−1 (Fig. 4A) and 5.39 mg L−1 (Fig. 4B), respectively. The rather low CMC indicates that the amphiphilic polymeric prodrugs can form stable micelles under such a low micellar concentration. The CMC of POEGSHMIBu was also evaluated by fluorescence spectroscopy. After treatment with 10 mM DTT at 37 °C for 10 h, the CMC value of the polymeric prodrug showed a tremendous increase from 5.09 mg L−1 to 38.0 mg L−1 (Fig. 4C), which indicates that the micelles formed by the amphiphilic polymeric prodrug containing disulfide bonds becomes unstable in a reductive environment due to cleavage of the disulfide bonds.
 |
| | Fig. 4 Plots of fluorescence intensity ratio I338/I333 from pyrene excitation spectra vs. log C for POEGSSMIBu (A), POEGMIBu (B) and POEGSHMIBu (C). | |
Preparation of IBu-loaded micelles based on polymeric prodrugs POEGSSMIBu and POEGMIBu
The polymeric prodrug micelles can steadily encapsulate IBu, and were prepared in an aqueous solution by the dialysis method. The initial weight ratio of IBu to polymer was 3/10. The drug loading efficiency was surprisingly high, that is 75.2% for POEGSSMIBu micelles and 77.2% for POEGMIBu micelles. The total drug loading content can reached as high as 38.9 wt% for POEGSSMIBu micelles and 46.6 wt% for POEGMIBu micelles, respectively. These high drug loading capacities of the POEGSSMIBu and POEGMIBu micelles might be attributed to the π–π aromatic stacking force between the covalently bonded IBu moieties and free IBu molecules, which results in more free IBu molecules being incorporated into the micellar core.47
Size and morphology of micelles
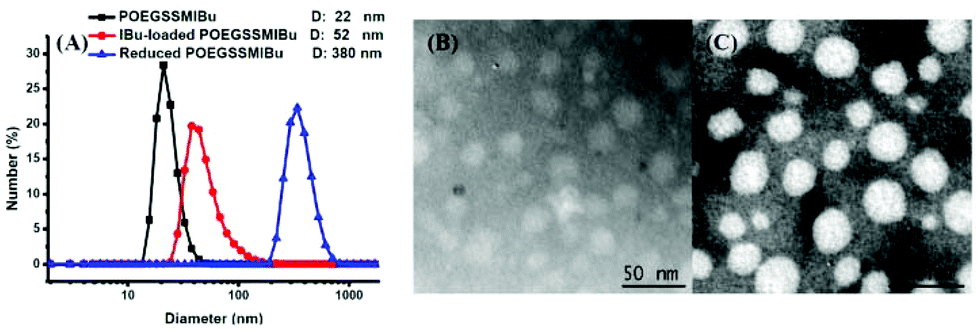
The size and morphology of the POEGSSMIBu micelles were determined by DLS and TEM, as shown in Fig. 5. The diameter of the micelles was measured by DLS and were found to be approximately 22 nm, which is in good accordance with that detected by TEM. Interestingly, after loading free IBu into the micelles, the diameter of the micelles was significantly enlarged to 52 nm, but the morphology of the micelles still maintained a spherical shape according to the TEM image. The increase of the micelle size after drug loading is probably assigned to the entrapment of the drug in the core of the micelles. The size change of the POEGSSMIBu micelles in response to 10 mM DTT was also determined by DLS. After treating with DTT for 10 h, large stable aggregates with a broad size distribution were observed. This result indicates the rapid response of POEGSSMIBu micelles in a reductive environment, which is in good accordance with GPC and CMC data.
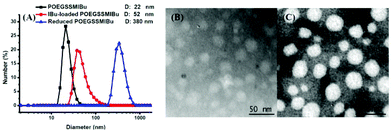 |
| | Fig. 5 Particle size and distribution of POEGSSMIBu micelles, IBu-loaded POEGSSMIBu micelles and reduced POEGSSMIBu micelles measured by DLS (A); TEM images of POEGSSMIBu micelles (B) and IBu-loaded POEGSSMIBu micelles (C). | |
Similarly, the size and morphology of POEGMIBu micelles were also determined by DLS and TEM as shown in Fig. 6. The typical TEM images show the spherical morphology of the micelles with a uniform size distribution. Compared to the blank micelles, the size of the IBu-loaded micelles also showed a significant increase.
 |
| | Fig. 6 Particle size and distribution of POEGMIBu micelles and IBu-loaded POEGMIBu micelles measured by DLS (A); TEM images of POEGMIBu micelles (B) and IBu-loaded POEGMIBu micelles (C). | |
In vitro IBu release from micelles
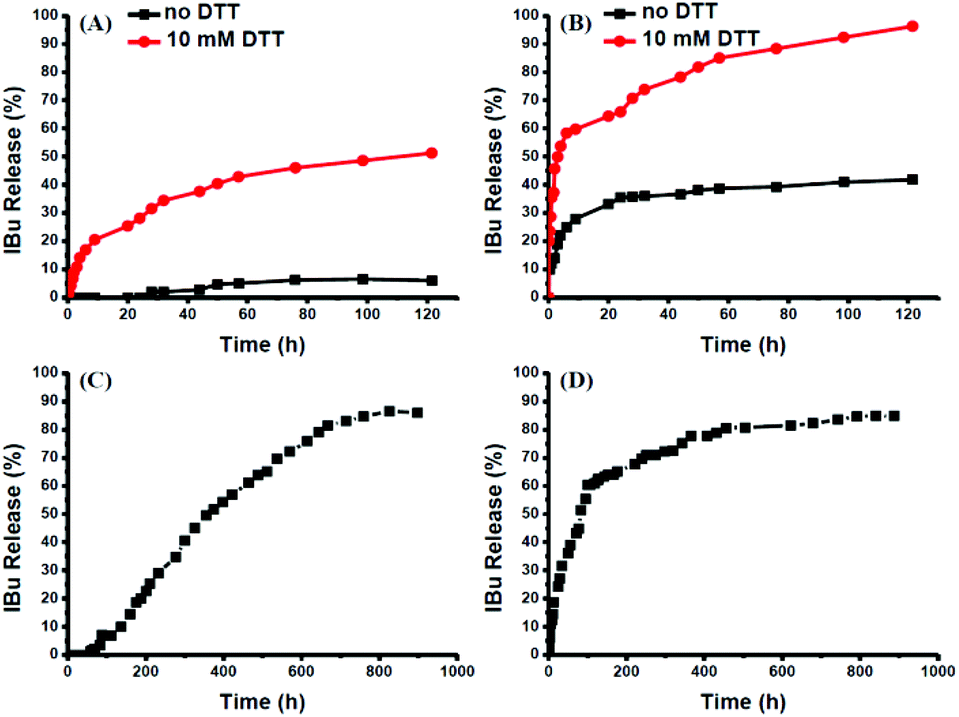
The intracellular concentration of GSH (0.5–10 mM) is significantly higher than that outside cells (2–20 μM).48,49 The huge difference may selectively trigger the cleavage of the disulfide bonds of the polymeric prodrug, resulting in a controlled release of the loaded drugs into cells. Here, 10 mM DTT was chosen to mimic the reductive environment in a cytoplasmic environment.50–52 DTT-induced IBu release from POEGSSMIBu micelles was investigated by dialysis. The solutions taken out at predetermined intervals were characterized by UV-vis spectrophotopmetry at 222 nm depending on the calibration curves, which were established from known concentrations of IBu in a corresponding medium. For POEGSSMIBu micelles, almost 35% of the conjugated IBu was released within 30 h in the presence of DTT (10 mM), whereas little release of IBu was observed from the POEGSSMIBu micelles in pure PBS even after 100 h (Fig. 7A), which reveals that the release rates of conjugated IBu had a great dependence on the reducing reagent. Meanwhile, for IBu-loaded POEGSSMIBu micelles, as shown in Fig. 7B, a burst release of IBu was observed in the presence of DTT within the first 10 h, and a relatively delayed drug release was observed during the same period without DTT. After 10 h, the release behaviors of IBu-loaded POEGSSMIBu micelles are similar to those of POEGSSMIBu micelles. It could be concluded that the release of conjugated IBu was slow and sustained owing to the gradual hydrolysis of the ester bonds, while the release rate of the encapsulated IBu from the micelles was much faster, but both of them clearly showed reducing-dependent release behaviors. The reductive cleavage of disulfide bonds can not only accelerate the release of encapsulated free IBu with the disassociation of the micelles, but also induces the fast hydrolysis of ester bonds to release conjugated IBu by reducing the steric hindrance that water molecules encounter in the hydrolysis of the ester bonds. The IBu release behaviors of POEGMIBu micelles (Fig. 7C) and IBu-loaded POEGMIBu micelles (Fig. 7D) are similar to those of POEGSSMIBu micelles and IBu-loaded POEGSSMIBu micelles in pure PBS without DTT. The release of conjugated IBu could be sustained for more than 40 days. After all of the IBu moieties were hydrolyzed and released from the POEGMIBu micelles, the remaining polymer (Re-POEGM) was recovered by lyophilization and measured by GPC. As shown in Fig. 3, the GPC curve and molecular weight of Re-POEGM are similar to those of POEGM, which indicates that the hydrolysis of ester bonds formed from the pendent secondary hydroxyl groups was relatively faster than those in the polymer backbone based on primary hydroxyl groups, and the polymer backbone is hardly hydrolyzed even after 40 days.
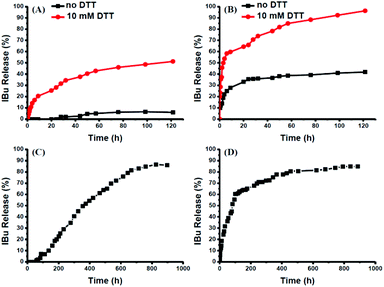 |
| | Fig. 7
In vitro IBu release profiles from POEGSSMIBu micelles (A); IBu-loaded POEGSSMIBu micelles (B); POEGMIBu micelles (C) and IBu-loaded POEGMIBu micelles (D). | |
In vitro cytotoxicities of POEGSSMIBu and POEGMIBu
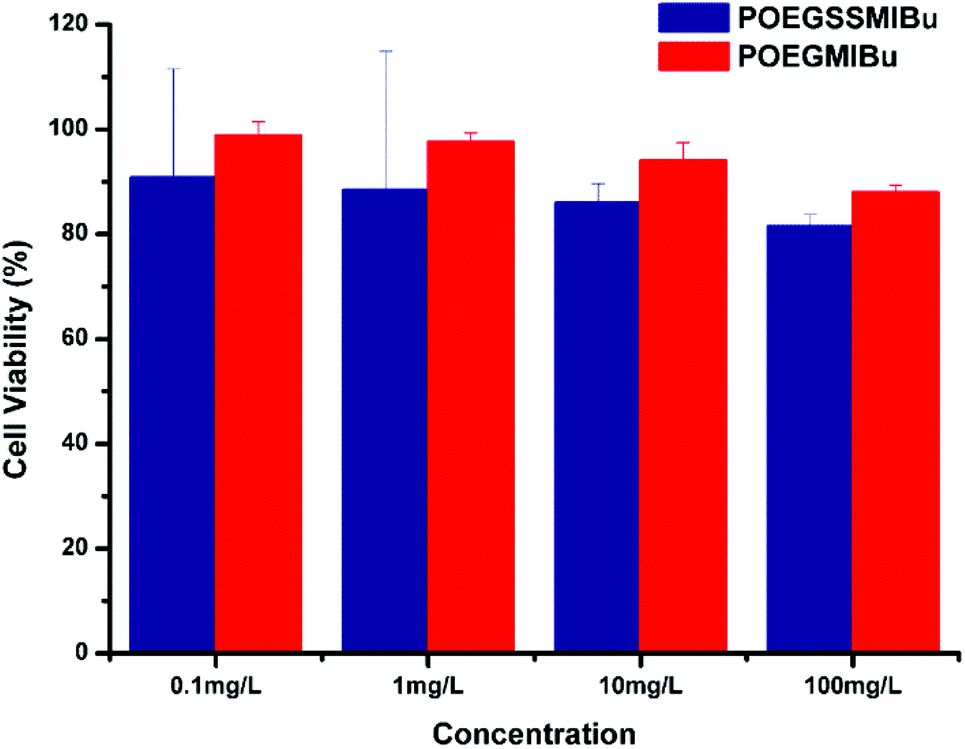
The cytotoxicities of POEGSSMIBu and POEGMIBu were both determined by CCK-8 assays against MC3T3-E1 cells. A series of polymeric prodrug solutions with various concentrations were used to investigate the biocompatibility of PEG-IBu copolymer. The results indicate that both POEGSSMIBu and POEGMIBu showed great biocompatibility to MC3T3-E1 cells (Fig. 8). IBu itself might be toxic,53,54 however, the great biocompatibility of the PEG-IBu copolymer shown in Fig. 8 indicated that the toxicity of IBu could be greatly reduced by conjugating with PEG derivatives.
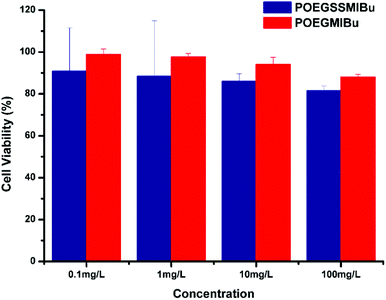 |
| | Fig. 8 Relative cell viability of POEGSSMIBu and POESMIBu against normal cultivated MC3T3-E1 cells for 24 h. | |
Cellular uptake and intracellular drug release of POEGSSMIBu and POEGMIBu micelles
The cellular uptake and intracellular release of NR-loaded micelles in response to cellular GSH against MC3T3-E1 cells were investigated using CLSM and a Spectramax M5 plate reader with NR (red fluorescence) as a hydrophobic fluorescence probe. The MC3T3-E1 cells were incubated with NR-loaded micelles for 2 or 4 h. Meanwhile, the MC3T3-E1 cells incubated with equivalent amounts of free NR for 2 or 4 h were used as the control. As shown in Fig. 9A and B, little fluorescence of NR can be detected in the cytoplasm of the cells when the cells were treated with free NR for 2 h and 4 h, indicating that free hydrophobic molecules could hardly be taken up by the cells. In contrast, the fluorescence of NR could be clearly observed in the cytoplasm of the cells after treatment with NR-loaded POEGMIBu and POEGSSMIBu micelles for both 2 h and 4 h (Fig. 9C–F), which implied that POEGMIBu and POEGSSMIBu micelles can be internalized into the cells efficiently. Moreover, the fluorescence intensity increased with the incubation time, owing to the continuous internalization of the NR loaded micelles. To sum up, the hydrophobic agents can be well transported into cells by encapsulated them into the hydrophobic core of the micelles, which is very important and shows the potential of their use as ideal drug carriers. More importantly, cells incubated with NR-loaded POEGSSMIBu micelles emitted an obviously stronger fluorescence of NR than those treated with NR-loaded POEGMIBu micelles for the same time. This phenomenon clearly confirms the efficient cellular uptake and successive destabilization of POEGSSMIBu micelles by high intracellular GSH concentrations in the cytoplasm of cells, which accelerates the release of encapsulated NR with the cleavage of disulfide bonds in response to the reductive environment (Scheme 3). These results are in accordance with the results for the in vitro release of encapsulated IBu from POEGSSMIBu and POEGMIBu micelles in the presence of DTT.
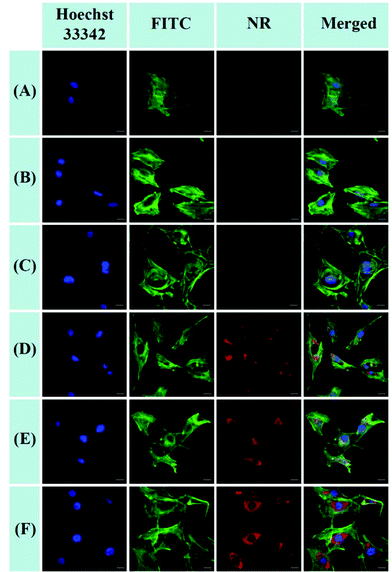 |
| | Fig. 9 CLSM images of MC3T3-E1 cells after 2 or 4 h incubation with NR-loaded micelles and free NR. For each panel, the images from left to right showed cell nuclei stained by Hoechst 33342 (blue), cytoskeletons stained by fluorescein isothiocyanate (FITC)-labeled phalloidin (green), NR fluorescence in cells (red), overlays of three left images. The scale bars correspond to 20 μm in all the images. (A) free NR, 2 h incubation; (B) free NR, 4 h incubation; (C) NR-loaded POEGMIBu micelles, 2 h incubation; (D) NR-loaded POEGMIBu micelles, 4 h incubation; (E) NR-loaded POEGSSMIBu micelles, 2 h incubation; (F) NR-loaded POEGSSMIBu micelles, 4 h incubation. | |
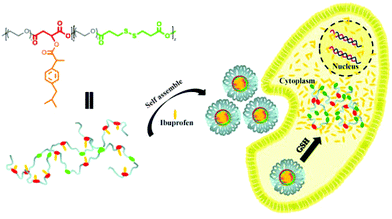 |
| | Scheme 3 Schematic illustration of the fabrication of IBu-loaded prodrug micelles for their efficient cellular uptake and intracellular IBu release triggered by GSH. | |
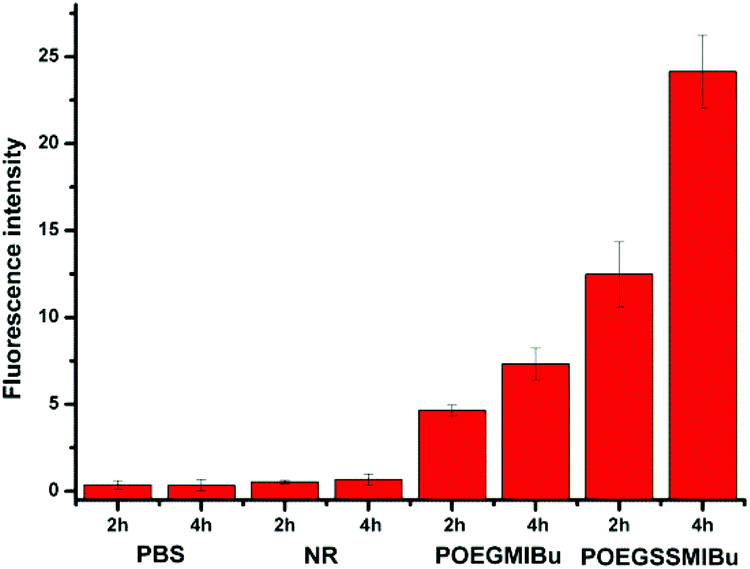
The quantitative fluorescence intensities in the MC3T3-E1 cells were determined using a Spectramax M5. The MC3T3-E1 cells were incubated in a similar manner to that for CLSM measurements. As showed in Fig. 10, analogous to the control group (cells treated with pure PBS), almost no fluorescence signals were detected in the cells after treatment with free NR, clearly indicating that free hydrophobic NR could hardly be taken up by the cells. Nevertheless the MC3T3-E1 cells cultured in NR-loaded POEGMIBu and POEGSSMIBu micellar solutions all showed a fairly high fluorescence intensity of NR. Furthermore, the MC3T3-E1 cells treated with POEGSSMIBu micelles showed an enhanced intracellular fluorescence of NR compared to those incubated with POEGMIBu micelles under the same conditions, which clearly implied the rapid and complete intracellular NR release from POEGSSMIBu micelles under a reductive environment. Additionally, of course, the fluorescence intensity also increased with the increase of incubation time. These results quantify the CLSM measurements quite well.
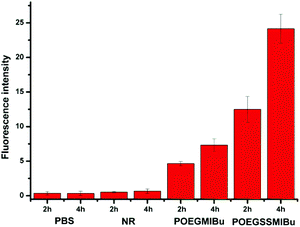 |
| | Fig. 10 Fluorescence intensity in MC3T3-E1 cells after incubation with PBS, free NR, NR-loaded POEGMIBu and POEGSSMIBu micelles for 2 h and 4 h. | |
Conclusions
In this study, an amphiphilic polymeric prodrug with pendent hydrophobic IBu moieties and a hydrophilic backbone containing multiple disulfide bonds was easily synthesized by a combination of polycondensation and esterification, forming stable nanosized micelles in aqueous solution with a low CMC and a high IBu loading content (38.9 wt%). The conjugated IBu could be released from the nanocarriers by the hydrolysis of the ester bonds, presenting a slow and sustained release. While the encapsulated IBu molecules could be released by diffusion, showing a relatively fast release. Importantly, this kind of redox responsive nanocarriers could accelerate the release of encapsulated and conjugated IBu simultaneously under reductive environment because of the cleavage of disulfide bonds in the polymeric backbone. The micelles based on this amphiphilic polymeric prodrug show excellent biocompatibility, and can be efficiently taken up by cells, realizing a redox-responsive release because of the high intracellular GSH concentration. This study provides a facile and universal strategy for the efficient preparation of a smart drug delivery system for controlled drug release. Meanwhile, amphiphilic polymeric prodrugs for cancer therapeutics could also be synthesized based on the PEG derivative with pendant hydroxyl groups and anticancer drugs within one or two synthetic steps. Furthermore, other stimuli-responsive bonds, such as pH-responsive bonds, thermal-responsive bonds and light-responsive bonds, could be facilely introduced between the polymeric backbone and the anticancer drug moiety. The strategy developed in our study provides an efficient and smart drug delivery system with multiple-responses which could be fabricated easily and at low cost on a large scale for cancer chemotherapy.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (21274121 and 51173163), the Major State Basic Research Project (2011CB606001), the National Science-Technology Support Plan Pproject of China (2012BAI07B01) and the Fundamental Research Funds for the Central Universities (2012QNA7043).
Notes and references
- N. Abdullah Al, H. Lee, Y. S. Lee, K. D. Lee and S. Y. Park, Macromol. Biosci., 2011, 11, 1264–1271 CrossRef PubMed.
- X. Feng, F. Lv, L. Liu, H. Tang, C. Xing, Q. Yang and S. Wang, ACS Appl. Mater. Interfaces, 2010, 2, 2429–2435 CAS.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2007, 120, 131–148 CrossRef CAS PubMed.
- K. T. Oh, H. Yin, E. S. Lee and Y. H. Bae, J. Mater. Chem., 2007, 17, 3987–4001 RSC.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- R. Savic, L. B. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615–618 CrossRef CAS PubMed.
- L. Jabr-Milane, L. van Vlerken, H. Devalapally, D. Shenoy, S. Komareddy, M. Bhavsar and M. Amiji, J. Controlled Release, 2008, 130, 121–128 CrossRef CAS PubMed.
- W. Chen, P. Zhong, F. H. Meng, R. Cheng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef CAS PubMed.
- X. Chen, S. S. Parelkar, E. Henchey, S. Schneider and T. Emrick, Bioconjugate Chem., 2012, 23, 1753–1763 CrossRef CAS PubMed.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, Polym. Chem., 2010, 1, 1205–1214 RSC.
- G. L. Li, J. Y. Liu, Y. Pang, R. B. Wang, L. M. Mao, D. Y. Yan, X. Y. Zhu and J. Sun, Biomacromolecules, 2011, 12, 2016–2026 CrossRef CAS PubMed.
- M. Talelli, K. Morita, C. J. F. Rijcken, R. W. M. Aben, T. Lammers, H. W. Scheeren, C. F. van Nostrum, G. Storm and W. E. Hennink, Bioconjugate Chem., 2011, 22, 2519–2530 CrossRef CAS PubMed.
- F. Zhan, W. Chen, Z. Wang, W. Lu, R. Cheng, C. Deng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2011, 12, 3612–3620 CrossRef CAS PubMed.
- J. X. Ding, W. G. Xu, Y. Zhang, D. K. Sun, C. S. Xiao, D. H. Liu, X. J. Zhu and X. S. Chen, J. Controlled Release, 2013, 172, 444–455 CrossRef CAS PubMed.
- L. Y. Tang, Y. C. Wang, Y. Li, J. Z. Du and J. Wang, Bioconjugate Chem., 2009, 20, 1095–1099 CrossRef CAS PubMed.
- J. Chen, X. Qiu, J. Ouyang, J. Kong, W. Zhong and M. M. Q. Xing, Biomacromolecules, 2011, 12, 3601–3611 CrossRef CAS PubMed.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef CAS.
- Y. L. Wu, W. Chen, F. H. Meng, Z. J. Wang, R. Cheng, C. Deng, H. Y. Liu and Z. Y. Zhong, J. Controlled Release, 2012, 164, 338–345 CrossRef CAS PubMed.
- H. S. Yoo, K. H. Lee, J. E. Oh and T. G. Park, J. Controlled Release, 2000, 68, 419–431 CrossRef CAS.
- R. Tong and J. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4830–4834 CrossRef CAS PubMed.
- E. S. Lee, K. Na and Y. H. Bae, J. Controlled Release, 2005, 103, 405–418 CrossRef CAS PubMed.
- V. R. Caiolfa, M. Zamai, A. Fiorino, E. Frigerio, C. Pellizzoni, R. d'Argy, A. Ghiglieri, M. G. Castelli, M. Farao, E. Pesenti, M. Gigli, F. Angelucci and A. Suarato, J. Controlled Release, 2000, 65, 105–119 CrossRef CAS.
- K. Ulbrich and V. Subr, Adv. Drug Delivery Rev., 2010, 62, 150–166 CrossRef CAS PubMed.
- X. Q. Li, H. Y. Wen, H. Q. Dong, W. M. Xue, G. M. Pauletti, X. J. Cai, W. J. Xia, D. L. Shi and Y. Y. Li, Chem. Commun., 2011, 47, 8647–8649 RSC.
- P. F. Gou, W. P. Zhu, N. Xu and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6962–6976 CrossRef CAS.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, Biomacromolecules, 2010, 11, 934–943 CrossRef CAS PubMed.
- J. A. MacKay, M. Chen, J. R. McDaniel, W. Liu, A. J. Simnick and A. Chilkoti, Nat. Mater., 2009, 8, 993–999 CrossRef PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- Y. Wang, H. Du, L. L. Gao, H. G. Ni, X. D. Li, W. P. Zhu and Z. Q. Shen, Polym. Chem., 2013, 4, 1657–1663 RSC.
- K. Zhang, Y. Wang, W. P. Zhu, X. D. Li and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2045–2052 CrossRef CAS.
- W. Li, P. Zhan, E. De Clercq, H. Lou and X. Liu, Prog. Polym. Sci., 2013, 38, 421–444 CrossRef CAS PubMed.
- W. P. Zhu, Y. Wang, Q. J. Zhang and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4886–4893 CrossRef CAS.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- X. L. Hu, S. Liu, Y. B. Huang, X. S. Chen and X. B. Jing, Biomacromolecules, 2010, 11, 2094–2102 CrossRef CAS PubMed.
- X. L. Hu, L. S. Yan, H. H. Xiao, X. Y. Li and X. B. Jing, J. Appl. Polym. Sci., 2013, 127, 3365–3373 CrossRef CAS.
- G. Mo, J. Yue, P. A. Ma, X. S. Chen, Y. B. Huang and X. B. Jing, Polym. Int., 2011, 60, 1269–1276 CrossRef CAS.
- H. H. Xiao, H. Q. Song, Q. Yang, H. D. Cai, R. G. Qi, L. Yan, S. Liu, Y. H. Zheng, Y. B. Huang, T. J. Liu and X. B. Jing, Biomaterials, 2012, 33, 6507–6519 CrossRef CAS PubMed.
- Y. Shibata and A. Takasu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5747–5759 CrossRef CAS.
- A. Takasu, Y. Shibata, Y. Narukawa and T. Hirabayashi, Macromolecules, 2006, 40, 151–153 CrossRef.
- L. L. Gao, Q. J. Luo, Y. Wang, H. Du, X. D. Li, Z. Q. Shen and W. P. Zhu, RSC Adv., 2014, 4, 4177–4180 RSC.
- W. P. Zhu, L. L. Gao, Q. J. Luo, C. Gao, G. Y. Zha, Z. Q. Shen and X. D. Li, Polym. Chem., 2014, 5, 2018–2026 RSC.
- W. P. Zhu, X. W. Tong, W. H. Xie and Z. Q. Shen, J. Appl. Polym. Sci., 2010, 118, 1943–1948 CAS.
- J. Y. Liu, Y. Pang, J. Chen, P. Huang, W. Huang, X. Y. Zhu and D. Y. Yan, Biomaterials, 2012, 33, 7765–7774 CrossRef CAS PubMed.
- J. Wang, G. Yang, X. Guo, Z. M. Tang, Z. D. Zhong and S. B. Zhou, Biomaterials, 2014, 35, 3080–3090 CrossRef CAS PubMed.
- A. Gallardo, J. L. Eguiburu, M. J. F. Berridi and J. San Roman, J. Controlled Release, 1998, 55, 171–179 CrossRef CAS.
- M. Wilhelm, C. L. Zhao, Y. C. Wang, R. L. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- J. B. Liu, Y. H. Xiao and C. Allen, J. Pharm. Sci., 2004, 93, 132–143 CrossRef CAS PubMed.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS.
- G. Y. Wu, Y. Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 489–492 CAS.
- B. Khorsand, G. Lapointe, C. Brett and J. K. Oh, Biomacromolecules, 2013, 14, 2103–2111 CrossRef CAS PubMed.
- H. L. Sun, B. N. Guo, X. Q. Li, R. Cheng, F. H. Meng, H. Y. Liu and Z. Y. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS PubMed.
- L. Wu, Y. Zou, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomaterials, 2013, 34, 5262–5272 CrossRef CAS PubMed.
- J. Li and P. Yao, Langmuir, 2009, 25, 6385–6391 CrossRef CAS PubMed.
- R. Rosario-Melendez, W. L. Yu and K. E. Uhrich, Biomacromolecules, 2013, 14, 3542–3548 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The determination of the loading content of POEGSSMIBu and POEGMIBu micelles and the calibration curves of IBu in PBS with and without DTT. See DOI: 10.1039/c4bm00065j |
| ‡ These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.