Decreased tissue factor expression with increased CD11b upregulation on elastin-based biomaterial coatings
M. K.
Rooney
and
K. A.
Woodhouse
*
Department of Chemical Engineering, Queen's University, Ontario, Canada. E-mail: kim.woodhouse@queensu.ca
First published on 24th June 2014
Abstract
Elastin-like polypeptide (ELP) coatings have been shown to have non-thrombogenic properties both in vitro and in vivo. In this work, we expand our understanding of this phenomenon by investigating the interaction of these coatings with leukocytes. Citrated whole blood was exposed to a shear rate of 300 s−1 for 2 hours at 37 °C on ELP1- and ELP4-coated polyethylene terephthalate (Mylar™) surfaces in a cone and plate device. Scanning electron microscopy and flow cytometry were used to measure leukocyte activation and platelet–leukocyte aggregation in response to the ELP1 and ELP4 coatings on the surface and in the bulk, respectively. Surface analysis showed little leukocyte activity on the surface of uncoated positive controls. Both the tissue factor (TF) expression (indicative of leukocyte activation) and CD61 expression (indicative of platelet–leukocyte aggregates), in the bulk were decreased by 40% and 20%, respectively, with the ELP coating of Mylar™, while a two- to three-fold increase in CD11b upregulation (indicative of leukocyte activation) for ELP1 and ELP4 was determined. Two of three bulk markers indicated that ELP-coated Mylar™ decreased the leukocyte response compared to the uncoated Mylar™, while the third, CD11b, indicated an increase in leukocyte response to the ELP coatings.
1. Introduction
Synthetic large diameter vascular grafts of polyethylene terephthalate (PET, Dacron™) and expanded polytetrafluoroethylene (ePTFE, Gore-Tex™) are successful in the clinical setting.1 However, their small diameter counterparts (inner diameter, ID < 6 mm) frequently fail 5–10 years post implantation2,3 due to luminal narrowing caused by thrombosis4 and intimal hyperplasia.3 Currently, autografts are the principal treatment in small ID applications, but in about 30% of the cases, this treatment is unavailable to the patient due to previous surgery, disease, or individual anatomy.5 The need for synthetic small diameter vascular grafts that are resistant to both thrombosis and intimal hyperplasia continues to be high.Blood compatibility is a complex process. In this work, we are focused specifically on adding insight into the non-thrombogenic characteristics of elastin polypeptides. Research suggests that the process of thrombosis is not only a dynamic interaction between platelets and plasma proteins, but leukocytes and complement as well.6 The response of monocytes and neutrophils is of particular interest in this study.
The initial indication of leukocyte activation is manifested in changes in membrane surface receptors, such as tissue factor (TF) and CD11b, followed by degranulation, release of inflammatory mediators, oxidative burst and adhesion.7 TF expression and CD11b upregulation impact thrombus formation. TF directly participates in the coagulation cascade of the TF-dependent pathway, forming the TF-factor VIIa complex which activates factor X (FX). CD11b indirectly affects the coagulation cascade through activation of FX and conversion of fibrinogen to fibrin via surface binding.7
The concept of using platelet–leukocyte aggregates as an indication of thrombosis is relatively new.6 Activated platelets express P-selectin, an adhesion molecule that forms aggregates with monocytes and neutrophils via the P-selectin glycoprotein ligand-1 (PSGL-1) on the leukocytes.7,8 Platelet–leukocyte aggregates can also be formed between GPIIb/IIIa (CD41/CD61) on platelets and Mac-1 (CD11b/CD18) on leukocytes, bridged by fibrinogen.8
The extracellular matrix protein, elastin, has been found to be one of the least thrombogenic components of blood vessels and has been investigated for its potential as a biomaterial in purified and recombinant forms.9,10 Coatings based on elastin proteins have been shown to mitigate the thrombogenic response to biomaterials.11,12
Woodhouse et al.13 conducted an in vivo rabbit study on elastin-like polypeptide-2 (ELP2)-coated polyurethane catheters in which the coated catheters remained patent for approximately three times as long as the uncoated catheters. To better understand the lower thrombogenic response associated with elastin polypeptides, several different polypeptides of differing lengths (ELP1, ELP2 and ELP4) were coated on Mylar™ and evaluated in vitro. Srokowski et al.,14 investigated these coatings in reconstituted whole blood, and Srokowski & Woodhouse,15 in a fibrinogen solution and in whole blood. These studies indicated reduced fibrinogen adsorption, platelet adhesion and platelet activation with ELP-coated Mylar™ under shear compared to uncoated Mylar™. The most pronounced differences, including an altered adsorbed fibrinogen conformation, were noted for the ELP4 coating, the longer polypeptide.14,15 In order to further understand the underlying mechanism governing the low thrombogenicity of these ELPs, the focus of this work was to study the leukocyte response of ELP1- and ELP4-coated Mylar™ in vitro.
2. Materials and methods
2.1. Materials
All chemical reagents were purchased from Sigma-Aldrich Canada (Oakville, CA) and used as received, unless otherwise specified. Mylar™ (300 Å) sheets were purchased from Active Industries (Clifton Park, USA). Low density polyethylene (LDPE) (90 μm) sheets were purchased from KMac Plastics (Wyoming, USA). Circular Mylar™ and LDPE surfaces (33 mm in diameter) were pre-treated with triplicate 30 minute methanol rinses (Fisher Scientific, Whitby, CA), a 30 minute rinse in plain Tyrode's buffer (0.14 M NaCl, 0.0025 M KCl, 0.012 M NaHCO3, 0.0004 M Na2HPO4, pH 7.4), and equilibrated overnight at room temperature in 3 mL of plain Tyrode's buffer, to ensure a clean initial surface.2.2. Elastin-like polypeptide coatings
Elastin-like polypeptides (ELPs) based on the native elastin protein precursor, tropoelastin, are produced recombinantly in this work. These ELPs consist of alternating hydrophobic domains, coded for by exons 20 and 24 of the human gene, and cross-linking domains, coded for by exons 21 and 23 of the human gene, in the form of ELP20-(21-23-24)n, or ELPn, where n is the number of repeated 21-23-24 domains. A representation can be seen in Fig. 1.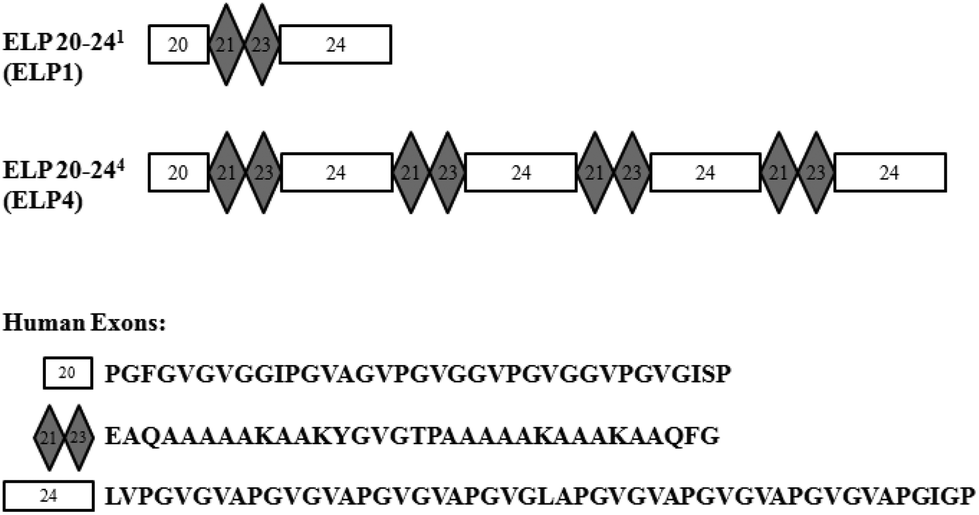 | ||
| Fig. 1 Representations of elastin-like polypeptide (ELP) sequences for ELP1 and ELP4. Human exons are indicated with their respective amino acid sequences. | ||
Elastin-like polypeptides ELP1 and ELP4 were expressed, produced, and purified using previously established methods.16 Briefly, transfected BL21 Escherichia coli cells were cultured in a 10.5 L BioFlo 110 Benchtop Modular Fermentor (New Brunswick Scientific Co., Enfield, USA) for up to 5 hours, then induced with isopropyl-α-D-thiogalactopyranoside (IPTG) (final concentration of 0.5 mM). Culture was continued for up to 3 hours after induction, or until cell growth reached a plateau. The cell pellet was retrieved by centrifugation, cells were lysed with formic acid and the ELP1 and ELP4 proteins were cleaved with cyanogen bromide before dialysis in distilled deionized water and then in sodium acetate. Further purification was undertaken using a SP sepharose cationic ion exchange column. The ELPs were then dialyzed in acetic acid before they were lyophilized.
Protein adsorption was carried out as previously described.14 Pre-treated Mylar™ films in 40 mm diameter disc form were affixed to the bases of the wells of a cone and plate device, where a 33 mm diameter film was exposed in each well. ELP1 and ELP4 were dissolved overnight at 1 mg mL−1 in Dulbecco's phosphate buffered saline (PBS) (Fisher Scientific, Whitby, CA) and then were statically adsorbed to the pre-treated Mylar™ surfaces for 2 hours at room temperature. Surfaces were rinsed 3 times with plain Tyrode's buffer before shear treatment.
2.3. Shear treatment in cone and plate device
Approximately 20 mL of human whole blood was collected into buffered 3.2% (0.109 M) sodium citrate vacutainers (BD Bioscience, Mississauga, CA) with a 21-gauge needle (BD Bioscience, Mississauga, CA) from healthy volunteers, free of medications for 10 days and in accordance with ethics approval from Queen's University (Queen's University ethics protocol #CHEM-003-11). The first collection tube was discarded (∼2 mL) and the rest were stored at room temperature under constant gentle mixing. All samples were used and fixed within 8 hours of collection. Citrate anticoagulant was chosen for consistency with previous work14,15 and to avoid false CD11b upregulation by heparin.17The cone and plate device used in this work is modified from Skarja et al.18 in order to accommodate 4 wells of 32.5 mm diameter, with cone diameter of 31 mm and cone angle of 4°. During each run, the wells were loaded with ELP1-coated Mylar™, ELP4-coated Mylar™, uncoated Mylar™ and LDPE films in a randomized order to mitigate the impact of confounding variables. LDPE was used as a negative control.19
Citrated whole blood (0.8 mL) was added to each well, cones were lowered such that the tip barely touched the bottom surface of the well, and then the cones were rotated at a shear rate of up to 300 s−1 for 2 hours at 37 °C. This shear rate of 300 s−1 was chosen to mimic that of small diameter vessels.20 Static controls of 1 mL resting citrated whole blood (WB) and 5 μg endotoxin in 1 mL of WB (LPS-activated-WB) were carried out at 37 °C for 2 hours. A thrombin control of 0.25 U in 1 mL of citrated whole blood was used to qualitatively confirm that the blood demonstrated the ability to clot.
2.4. Surface leukocyte evaluation using scanning electron microscopy
Uncoated samples of Mylar™ and LDPE from shear rates of 100 s−1, 200 s−1, and 300 s−1 were used to evaluate the surface reaction of leukocytes. Each shear rate for each material was tested in triplicate. The sample preparation procedure for scanning electron microscopy (SEM) was modified from Flynn.21 After 2 hours of citrated whole blood exposure to shear, each surface was rinsed with plain Tyrode's buffer, and then fixed for 1 hour at room temperature and then overnight at 4 °C with 2.5% glutaraldehyde. Samples were then rinsed again in plain Tyrode's buffer and were exposed to a series of ethanol dehydrations (70%, 90%, 95%, and 100%) before chemical drying with graduations of hexamethyldisilazane (HDMS) in ethanol (33%, 66%, and 100%). Samples were left overnight to completely dry before being cut into 8 mm discs with a biopsy punch (VWR, Mississauga, CA), gold sputter-coated and imaged on a JEOL JSM-840 scanning electron microscope at an accelerating voltage of 10 kV.2.5. Bulk leukocyte evaluation with flow cytometry
ELP1- and ELP4-coated Mylar™, uncoated Mylar™, and LDPE surfaces were used to evaluate the bulk leukocyte response at the shear rate of interest of 300 s−1. After shear exposure, 6–100 μL samples of citrated whole blood were taken from each well and added to blocking solution (final concentration 5% BSA, 0.05% Tween-20 in PBS, pH 7.5) and incubated at room temperature for 15 minutes to reduce background staining. Each set of samples from the wells were single stained for 30 minutes in the dark at room temperature or at 4 °C in accordance with the manufacturer's instructions with undiluted (a) 10 μL Alexafluor488-conjugated mouse anti-human tissue factor (R&D Systems, Minneapolis, USA), (b) 10 μL phycoerythrin (PE)-conjugated mouse anti-human CD11b (R&D Systems, Minneapolis, USA), (c) 20 μL FITC mouse anti-human CD61 (BD Bioscience, Mississauga, CA), and (d) one sample per well was left unstained for gating purposes. Then, 2 mL of a proprietary erythrocyte lysing buffer, BD FACS lysing solution (BD Bioscience, Mississauga, CA), was added to each sample and incubated for 10 minutes. Samples were centrifuged twice for 5 minutes at 500g, re-suspended in 2 mL of HEPES Tyrode's buffer (HTB) (0.14 M NaCl, 0.0025 M KCl, 0.012 M NaHCO3, 0.0004 M Na2HPO4, 0.001 M D-glucose, 0.35% BSA, 0.001 M MgCl2, 0.01 M HEPES, pH 7.4), and then fixed with 0.5 mL of 1% paraformaldehyde for 30 minutes at room temperature. Samples were re-suspended in 1 mL of HTB for analysis on a Cytomics FC500 flow cytometer at the Cytometry and Imaging Facility at the Cancer Research Institute at Queen's University. Unstained samples were used to determine fluorescence compensation settings.Raw flow cytometry data were analyzed using FlowJo vX flow cytometry analysis software (Tree Star Inc., Ashland, USA). Scatter plots were used to gate for and identify monocyte and granulocyte populations. TF and CD61 expression were determined by the number of positive events against unstained controls. CD11b upregulation was determined using the fluorescence expression ratio, calculated using the median fluorescence intensities of positive events against unstained controls. There was a total of 15–16 samples, where N = 6 donors with n = 1–4 samples per donor.
2.6. Statistical analysis
Statistical analysis was undertaken using Prism 6 statistical software (GraphPad, La Jolla, USA). Surfaces were compared using a repeated measures one-way analysis of variance (ANOVA), with Dunnett's multiple comparison post hoc testing against uncoated Mylar™. All results are reported as mean ± 1 standard deviation (SD), with statistical significances reported based on p < 0.05.3. Results and discussion
3.1. Surface evaluation – scanning electron microscopy
SEM was used to evaluate the surface leukocyte activation and platelet leukocyte aggregates on the uncoated Mylar™ and LDPE. Lower shear rates of 100 s−1 and 200 s−1 in addition to the shear rate of interest, 300 s−1, which best represents the shear flow environment of small diameter vessels,20 were used to evaluate leukocyte adhesion to the biomaterial surfaces. Fig. 2 illustrates the scanning electron micrographs of LDPE and Mylar™ samples when exposed to citrated whole blood at 37 °C at shear rates of 100 s−1, 200 s−1, and 300 s−1 for 2 hours. Leukocytes are distinguished by their large size, ∼10 μm diameter with smooth or rough surface morphology.22,23 The activated platelets are recognized by their small size, a few microns in diameter, and characteristic long spider-like extensions.7 Finally, the morphology of red blood cells is generally biconcave, but it can also appear crenated or spherical with spicules as a result of changes in osmotic pressure during SEM preparation.25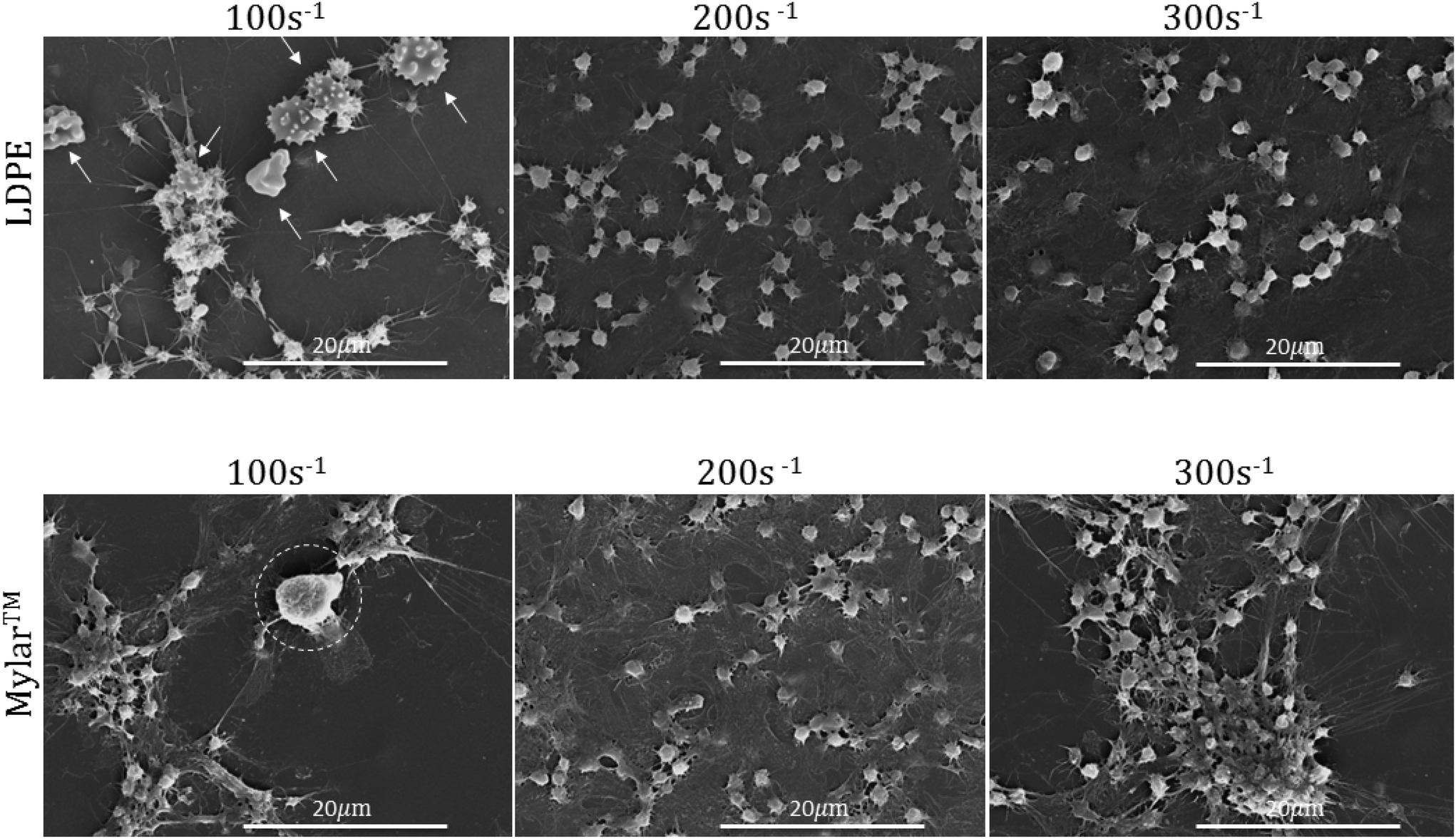 | ||
| Fig. 2 Representative scanning electron micrographs of low density polyethylene (LDPE) and Mylar™ at shear rates of 100 s−1, 200 s−1, and 300 s−1. Red blood cells are indicated by arrows and leukocytes are encircled. Test performed in triplicate. Scale bars are 20 μm. | ||
In Fig. 2 at 100 s−1, there appears to be both a leukocyte, encircled, and activated platelets on the Mylar™ surface, suggesting the possibility of platelet–leukocyte aggregation. In addition, the platelets are spreading in a manner associated with the presence of surface fibrinogen.24 Additionally, on the LDPE surface, there are what appear to be crenated red blood cells, indicated by arrows, forming aggregates with the activated platelets. Although activated platelets are still present on both surfaces, there is no evidence of leukocyte adhesion when the shear rate is increased to 200 s−1, and 300 s−1. These results are consistent with studies by Weber & Springer26 and Kuijper et al.27 in which lower shear rates increase the tendency for leukocytes to adhere to surfaces because the cells have a greater chance of contacting the surface and remaining.
Increases in platelet activation with shear rate have been reported by Turitto et al.28 and the current results at 200 s−1 and 300 s−1 for both Mylar™ and LDPE surfaces are consistent with these findings, as demonstrated by their higher aggregation and clustering at 300 s−1. Platelet activation is also more pronounced at the lowest shear rate (100 s−1); however, the increase platelet activation at 100 s−1 on Mylar™ could be due to inflammatory mediators released by the leukocytes present.6 Regardless of shear rate, the platelet activation appears to be higher on the Mylar™ than the LDPE surface, indicated by the extent of platelet grouping and fibrinogen/spread platelet film, and is consistent with previous work.14 This finding supports the hypothesis that fibrinogen adsorption is required for platelet adhesion to occur,29 where the GPIIb platelet surface receptor binds to fibrinogen's RGD sequence.30
Since leukocytes did not adhere to the uncoated Mylar™ at the shear rate of interest of 300 s−1, and previous work showed that there was a decrease in fibrinogen and platelet adsorption on ELP-coated Mylar™,14 leukocyte adhesion on the ELP-coated surfaces at this shear rate is unlikely. Moreover, Gorbet & Sefton31,32 have shown that monocyte adhesion is dependent on platelet adhesion, which further supports the unlikelihood of leukocyte adhesion to ELP-coated surfaces. The study was therefore not expanded to include these surfaces in favour of a more detailed evaluation of the cellular response within the bulk whole blood.
3.2. Bulk evaluation – flow cytometry
Leukocyte activation is first indicated by a change in membrane receptors,7 including the expression of TF and upregulation of CD11b. Citrated whole blood exposed to the ELP1- and ELP4-coated Mylar™, uncoated Mylar™, and LDPE surfaces under 300 s−1 shear rate for 2 h at 37 °C in the cone and plate device was tested for these membrane receptors. The percentage of cells which express TF in the monocyte and granulocyte populations in response to each surface is summarized in Fig. 3, while the degree of CD11b upregulation for these populations in response to the surfaces is presented in Fig. 4.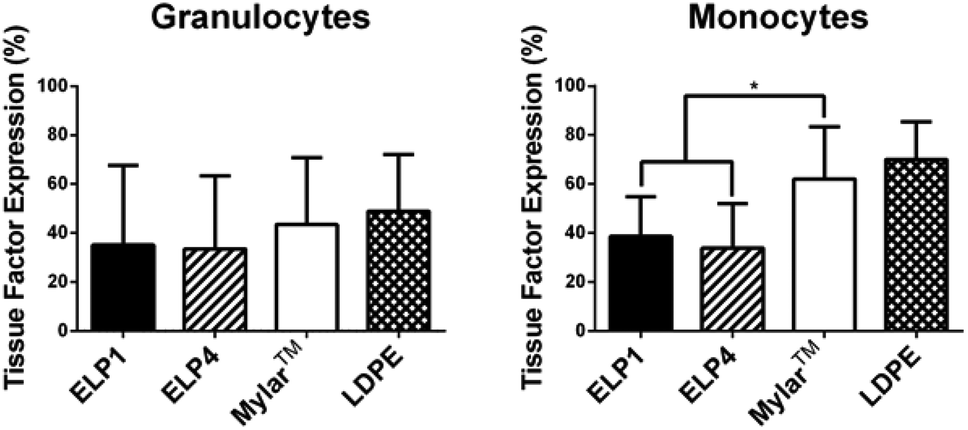 | ||
| Fig. 3 Tissue factor expression on granulocytes and monocytes for ELP-coated Mylar™ compared to controls, at 300 s−1. Static controls are LPS-activated-WB and WB. * indicates significantly different compared to Mylar™, p < 0.05. Mean ± SD, 15 samples total (N = 6 donors with n = 1–4 samples per donor). | ||
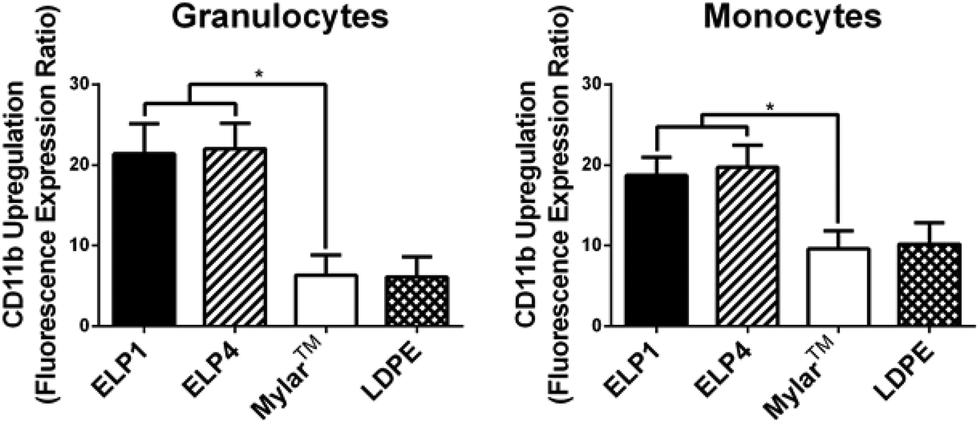 | ||
| Fig. 4 CD11b upregulation on granulocytes and monocytes for ELP-coated Mylar™ measured by the fluorescence expression ratio and compared to controls, at 300 s−1. Static controls are LPS-activated-WB and WB. * indicates significantly different compared to Mylar™, p < 0.05. Mean ± SD, 15 samples total (n = 1–4 samples per donor from N = 6 donors). | ||
Our work found no significant differences in granulocyte TF expression for the ELP coatings compared to the uncoated Mylar™. It is unknown whether granulocytes express TF33 and these results may be an indication that they do not. Work by Egorina et al.34 showed a significant transfer of TF from monocytes to granulocytes and Gorbet & Sefton31 attributed significant changes in biomaterial-induced leukocyte TF expression primarily to monocytes.
In the monocytes, the TF expression was significantly lower for the ELP1 (38.6 ± 16.3%) and ELP4 (33.9 ± 18.1%) coatings compared to the uncoated Mylar™ (62.0 ± 21.5%), indicating that the ELP coatings reduced monocyte activation by a factor of about 40%. The monocyte response to ELP1 and ELP4 coatings is very similar, which suggests that the sequence length does not have a large influence on monocyte TF expression. Although it is possible that the citrate anticoagulant could attenuate TF expression which is calcium dependent,35 we believe the data strongly suggest that ELP is influencing monocyte activation.
In Fig. 4, both ELP1- and ELP4-coated materials show elevated granulocyte (ELP1 = 21.4 ± 3.7, ELP4 = 22.0 ± 3.2) and monocyte responses (ELP1 = 18.7 ± 2.2, ELP4 = 19.7 ± 2.7), with a significantly higher CD11b upregulation (2–3 fold) compared to uncoated Mylar™ (granulocytes: 6.4 ± 2.5, monocytes: 9.6 ± 2.2). This finding is intriguing because it indicates that the ELP coatings activate leukocytes more than the uncoated Mylar™, contrary to the TF expression results. Other research has indicated that TF expression and CD11b upregulation follow similar patterns upon leukocyte activation.31,36,37 The reasons for this result are still unclear. The use of citrate has not been reported to likely have an effect on CD11b upregulation38 while it has been suggested that heparin may falsely upregulate CD11b on leukocytes.17
In the granulocyte and monocyte populations, platelet–leukocyte aggregates were classified as positive CD61 expression (a platelet marker). The percentages reported in Fig. 5 indicate the fraction of the cells positive for expression within the populations. Fig. 5 shows that CD61 expression for the ELP coatings in both the granulocytes (ELP1 = 62.7 ± 17.0%, ELP4 = 60.5 ± 20.1%) and monocytes (ELP1 = 63.1 ± 17.1%, ELP4 = 61.8 ± 16.8%) is significantly lower (∼20%) compared to uncoated Mylar™ (granulocytes: 80.7 ± 16.5%, monocytes: 75.0 ± 14.1%), which is consistent with previous findings for ELP4 under shear at 300 s−1 for 1 h.15 This suggests that ELP1 and ELP4 coatings reduce platelet–leukocyte aggregates compared to uncoated Mylar™. While it is known that the mechanism for platelet–leukocyte aggregation requires calcium,39 the use of a calcium chelating anticoagulant in this work did not appear to prevent the ability to discern differences between surfaces.
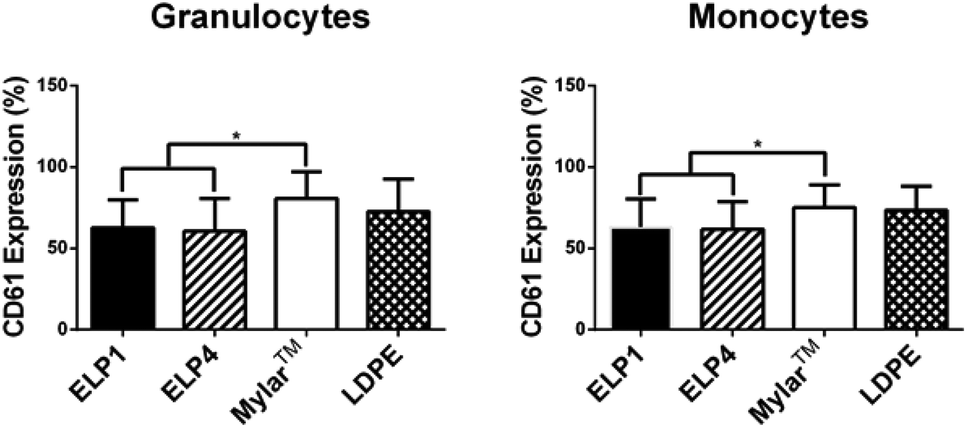 | ||
| Fig. 5 CD61 expression on granulocytes and monocytes for ELP-coated Mylar™ compared to controls, at 300 s−1. Static controls are LPS-activated-WB and WB. * indicates significantly different compared to Mylar™, p < 0.05. Mean ± SD, 16 samples total (n = 1–4 samples per donor from N = 6 donors). | ||
Two out of three markers in this work indicate that ELP coatings reduce leukocyte response compared to the uncoated Mylar™. Fig. 3 and 5 show that both the TF expression on monocytes and the platelet–leukocyte aggregates, respectively, are significantly decreased relative to the uncoated Mylar™, whereas Fig. 4 shows that CD11b upregulation is not. This suggests that there is a correlation between monocyte TF expression and platelet–leukocyte aggregates. Studies have shown that TF expression is dependent on P-selectin expression31,40 and that TF mediates platelet–leukocyte aggregates.40 Furthermore, studies done by Srokowski et al.,15 showed a significant reduction in the P-selectin expression for ELP2- and ELP4-coated Mylar™ compared to uncoated Mylar™, reflected by their significant reduction in platelet–leukocyte aggregates in ELP2 and ELP4. This trend is consistent with the current findings, where the platelet–leukocyte aggregates are lower for the ELP coatings. TF expression, platelet–leukocyte aggregates and P-selectin expression appear to follow similar trends and are in agreement with the literature. This further supports the idea that platelet activation, by P-selectin, affects leukocyte activation, through TF expression, which in turn mediates platelet–leukocyte aggregates. The results also suggest that in this experimental system, the platelets and leukocytes aggregate prominently through the P-selectin/PSGL-1 and not GPIIb/IIIa/Mac-1, where Mac-1 is CD11b/CD18 on leukocytes, because TF expression and P-selectin are reduced with the coated surfaces and CD11b upregulation is increased.
The increase in CD11b upregulation in Fig. 4 is complemented by the decrease in TF expression shown in Fig. 3 for the ELP-coated surfaces compared to controls. These results show an interesting difference from previous studies. It has been found that TF expression is dependent on the presence of platelets while platelets do not appear to influence CD11b upregulation.31,41 This suggests that CD11b can be upregulated by a mechanism different from that of TF expression. Possible contributing factors include complement and exposure time.
Complement proteins C3a and C5a attract leukocytes and C3b promotes leukocyte adhesion and activation.42,43 Leukocyte activation, indicated by CD11b upregulation, has been shown to be highly influenced by complement activation, whereas TF expression is not.44 Gorbet & Sefton44 compared the CD11b upregulation of non-complement activating polystyrene beads to complement activating PEG-modified polystyrene beads and determined that CD11b upregulation via complement is material dependent. Additionally, complement can activate platelets with the complement protein C1q, causing them to express P-selectin and GPIIb/IIIa,45 which are known to induce platelet–leukocyte aggregates.31,40 This suggests that investigations which evaluate the role of complement should be considered in future work.
Material exposure time may also contribute to the increased CD11b upregulation. TF and CD11b are presented differently on the cell surface and as a result, the time to expression differs with these surface receptors. TF is internally synthesized before expression once cell activation has occurred46 and requires hours to be significantly expressed.6 Small amounts of CD11b are naturally present on the cell membrane, but upon cell activation, CD11b present in internal granules is translocated to the cell membrane to increase surface concentration.47 CD11b upregulation requires less time than TF expression because transcription and translation do not need to occur upon leukocyte activation. The experimental time scale in this work as well as previous in vitro and in vivo studies with recombinant ELPs did not exceed 4 hours.12–15 The increased CD11b upregulation of the current results suggests that perhaps leukocytes are in fact activated and that TF expression may not be a good indicator of leukocyte activation in the short term.
The low thrombogenicity of these ELP coatings was shown in the initial in vivo studies where an ELP-coated polyurethane catheter remained patent for the duration of the experiment (4 hours), while an uncoated polyurethane catheter was completely occluded within 2 hours.13 The mechanism for the non-thrombogenic nature of the ELPs was then further investigated in vitro on Mylar™ where fibrinogen adsorption,14,15 platelet adhesion,13,14 and platelet activation13,15 were all reduced compared to uncoated Mylar™. It is unexpected that leukocytes may be activated by the coatings when there was low thrombogenicity overall in the in vivo model. This suggests that there is a factor in the in vivo model, such as endothelial cells, that may reduce the possible leukocyte activation. TFPI, which inhibits TF,48 as well as CD46, CD55, and CD59, which inhibit complement via C3 convertase and MAC,49 are expressed on endothelial cells and may impact TF expression and CD11b upregulation in vivo. The impact of these molecules on ELP-coated Mylar™ should be further investigated.
4. Conclusions
The current and previous investigations show that ELP coatings may reduce the thrombogenicity of Mylar™. Bulk studies demonstrate a 40% reduction in leukocyte activation in terms of tissue factor expression as well as a 20% reduction in platelet–leukocyte aggregates for the elastin-like polypeptide coatings ELP1 and ELP4. An unexpected 2–3 fold increase in CD11b upregulation for both coatings merits further investigation. Both ELP1 and ELP4 coatings showed similar results, independent of sequence length. The results of this study show that ELP1 and ELP4 surface coatings continue to demonstrate characteristics of low thrombogenicity and have potential for use in vascular grafts.Acknowledgements
Funding for this project was provided by Science Foundation Ireland (SFI) (07/SRC/B1163), Natural Sciences and Engineering Research Council (NSERC), and Queen's University. The authors would like to thank Dr Lillicrap's research group (Queen's University) for their assistance in blood collection and Mr Matt Gordon (Queen's University) for his expertise in flow cytometry. They would also like to thank Dr Brian Amsden for his support.Notes and references
- B. H. Walpoth and G. L. Bowlin, Expert Rev. Med. Devices, 2005, 2, 647–651 CrossRef PubMed.
- S. E. Greenwald and C. L. Berry, J. Pathol., 2000, 190, 292–299 CrossRef CAS.
- B. W. Lytle, N. Engl. J. Med., 2004, 351, 2262–2264 CrossRef CAS PubMed.
- S. Ito, S. Ishimaru and S. E. Wilson, Angiology, 1998, 49, 289–297 CrossRef CAS PubMed.
- R. C. Darling and R. F. Linton, Surgery, 1971, 472–479 Search PubMed.
- M. B. Gorbet and M. V. Sefton, Biomaterials, 2004, 25, 5681–5703 CrossRef CAS PubMed.
- M. B. Gorbet and M. V. Sefton, in Hemostasis and Thrombosis, ed. R. Colman, A. Clowes, S. Goldhaber, V. Marder, and J. George, Lippincott Williams & Wilkins, Philadelphia, 5th edn, 2006, pp. 751–759 Search PubMed.
- C. Eriksson and H. Nygren, J. Lab. Clin. Med., 2001, 137, 296–302 CrossRef CAS PubMed.
- A. Waterhouse, S. G. Wise, M. K. C. Ng and A. S. Weiss, Tissue Eng., Part B, 2011, 17, 93–99 CrossRef CAS PubMed.
- S. W. Jordan and E. L. Chaikof, J. Vasc. Surg., 2007, 45, 104A–115A CrossRef PubMed.
- K. M. Defife, K. M. Hagen, D. L. Clapper and J. M. Anderson, J. Biomater. Sci., Polym. Ed., 1999, 10, 1063–1074 CrossRef CAS PubMed.
- S. W. Jordan, C. A. Haller, R. E. Sallach, R. P. Apkarian, S. R. Hanson and E. L. Chaikof, Biomaterials, 2007, 28, 1191–1197 CrossRef CAS PubMed.
- K. A. Woodhouse, P. Klement, V. Chen, M. B. Gorbet, F. W. Keeley, R. Stahl, J. D. Fromstein and C. M. Bellingham, Biomaterials, 2004, 25, 4543–4553 CrossRef CAS PubMed.
- E. M. Srokowski, P. H. Blit, W. G. McClung, J. L. Brash, J. P. Santerre and K. A. Woodhouse, J. Biomater. Sci., 2011, 22, 41–57 CrossRef CAS PubMed.
- E. M. Srokowski and K. A. Woodhouse, J. Biomed. Mater. Res., Part A, 2013, 1–12 Search PubMed.
- C. M. Bellingham, K. A. Woodhouse, P. Robson, S. J. Rothstein and F. W. Keeley, Biochim. Biophys. Acta, 2001, 1550, 6–19 CrossRef CAS.
- M. H. El Habbal, L. Smith, M. J. Elliot and S. Strobel, Cardiovasc. Res., 1995, 30, 676–681 CrossRef CAS.
- G. A. Skarja, R. L. Kinlough-Rathbone, D. W. Perry, F. D. Rubens and J. L. Brash, J. Biomed. Mater. Res., 1997, 34, 427–438 CrossRef CAS.
- M.-C. Belanger and Y. Marois, J. Biomed. Mater. Res., 2001, 58, 467–477 CrossRef CAS PubMed.
- J. J. Hathcock, Arterioscler., Thromb., Vasc. Biol., 2006, 26, 1729–1737 CrossRef CAS PubMed.
- L. E. Flynn, Biomaterials, 2010, 31, 4715–4724 CrossRef CAS PubMed.
- M. H. Ross and P. Wojciech, in Histology, Lippincott Williams & Wilkins, Philadelphia, 5th edn, 2006, pp. 247–279 Search PubMed.
- S. Jaisson, S. Lorimier, S. Ricard-Blum, G. D. Sockalingum, C. Delevallée-Forte, G. Kegelaer, M. Manfait, R. Garnotel and P. Gillery, Chem. Biol., 2006, 13, 149–159 CrossRef CAS PubMed.
- J. I. Sheppard, W. G. Mcclung and I. A. Feuerstein, J. Biomed. Mater. Res., 1994, 28, 1175–1186 CrossRef CAS PubMed.
- D. Zhong, Y. Jiao, Y. Zhang, W. Zhang, N. Li, Q. Zuo, Q. Wang, W. Xue and Z. Liu, Biomaterials, 2013, 34, 294–305 CrossRef CAS PubMed.
- C. Weber and T. A. Springer, J. Clin. Invest., 1997, 100, 2085–2093 CrossRef CAS PubMed.
- P. H. M. Kuijper, H. I. Gallardo Torres, J. A. M. van der Linden, J.-W. J. Lammers, J. J. Sixma, J. J. Zwaginga and L. Koenderman, Blood, 1997, 89, 2131–2138 CAS.
- V. T. Turitto, H. J. Weiss and H. R. Baumgartner, Microvasc. Res., 1980, 19, 352–365 CrossRef CAS.
- L. Vroman, A. L. Adams, G. C. Fischer and P. C. Munoz, Blood, 1980, 55, 156–159 CAS.
- W.-B. Tsai, J. M. Grunkemeier, C. D. Mcfarland and T. A. Horbett, J. Biomed. Mater. Res., Part A, 2002, 60, 348–359 CrossRef CAS PubMed.
- M. B. Gorbet and M. V. Sefton, J. Biomed. Mater. Res., Part A, 2003, 67, 792–800 CrossRef CAS PubMed.
- M. B. Gorbet and M. V. Sefton, J. Lab. Clin. Med., 2001, 137, 345–355 CrossRef CAS PubMed.
- K.-E. Eilertsen and B. Østerud, Blood Coagul. Fibrinolysis, 2004, 15, 521–538 CAS.
- E. M. Egorina, M. A. Sovershaev, J. O. Olsen and B. Østerud, Blood, 2008, 111, 1208–1216 CrossRef CAS PubMed.
- R. Bach and D. B. Rifkin, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 6995–6999 CrossRef CAS.
- X. Chang and M. Gorbet, J. Biomater. Appl., 2012, 0, 1–9 Search PubMed.
- S. T. Fan and T. S. Edgington, J. Clin. Invest., 1991, 87, 50–57 CrossRef CAS PubMed.
- H. Repo, S.-E. Jansson and M. Leirisalo-Repo, J. Immunol. Methods, 1995, 185, 65–79 CrossRef CAS.
- S. Bournazos, J. Rennie, S. P. Hart and I. Dransfield, Arterioscler., Thromb., Vasc. Biol., 2008, 28, e2–e3 CrossRef CAS PubMed.
- A. Celi, G. Pellegrini, R. Lorenzet, A. De Blasi, N. Ready, B. C. Furie and B. Furie, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8767–8771 CrossRef CAS.
- D. E. Lorant, M. K. Topham, R. E. Whatley, R. P. McEver, T. M. McIntyre, S. M. Prescott and G. A. Zimmerman, J. Clin. Invest., 1993, 92, 559–570 CrossRef CAS PubMed.
- M. Kazatchkine and M. Carreno, Biomaterials, 1988, 9, 30–35 CrossRef CAS.
- B. P. Morgan, Eur. J. Clin. Invest., 1994, 24, 219–228 CrossRef CAS PubMed.
- M. B. Gorbet and M. V. Sefton, J. Biomed. Mater. Res., Part A, 2005, 74, 511–522 CrossRef CAS PubMed.
- E. I. Peerschke and B. Ghebrehiwet, Immunobiology, 1998, 199, 239–249 CrossRef CAS.
- N. Semeraro, A. Biondi, R. Lorenzet, D. Locati, A. Mantovani and M. B. Donati, Immunology, 1983, 50, 529–535 CAS.
- D. F. Bainton, L. J. Miller, T. K. Kishimoto and T. A. Springer, J. Exp. Med., 1987, 166, 1641–1653 CrossRef CAS.
- B. A. Lwaleed and P. S. Bass, J. Pathol., 2006, 208, 327–339 CrossRef CAS PubMed.
- F. Tedesco, F. Fischetti, M. Pausa, A. Dobrina, R. B. Sim and M. R. Daha, Mol. Immunol., 1999, 36, 26–268 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |