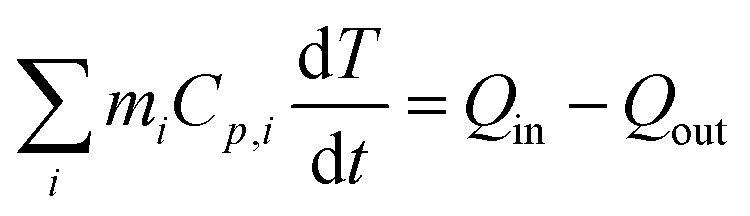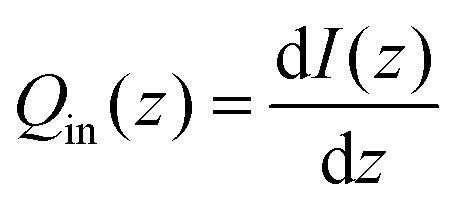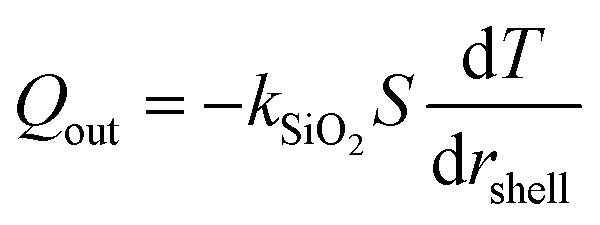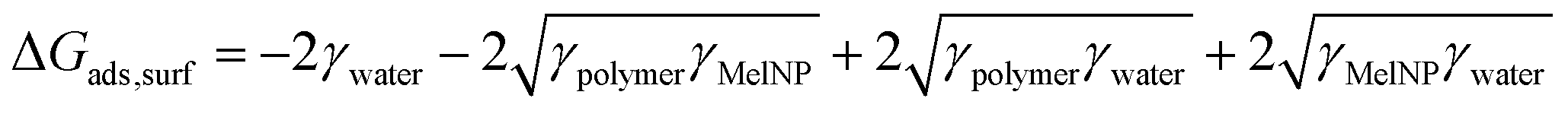DOI:
10.1039/C3BM60321K
(Paper)
Biomater. Sci., 2014,
2, 766-774
Photoresponsive hydrogel networks using melanin nanoparticle photothermal sensitizers†
Received
16th December 2013
, Accepted 11th February 2014
First published on 24th February 2014
Abstract
Photoreconfigurable and photodegradable polymeric networks have broad utility as functional biomaterials for many applications in medicine and biotechnology. The vast majority of these functional polymers are synthesized using chemical moieties that may be cytotoxic in vivo. Materials synthesized from these substituents also pose unknown risk upon implantation and thus will encounter significant regulatory challenges prior to use in vivo. This work describes a strategy to prepare photodegradable hydrogel networks that are composed of well-characterized synthetic polymers and natural melanin pigments found within the human body. Self-assembled networks of poly(L-lactide-co-glycolide)-poly(ethylene glycol) ABA triblock copolymers are doped with melanin nanoparticles to produce reconfigurable networks based on photothermal phase transitions. Self-assembled hydrogel networks with melanin nanoparticles exhibit a storage modulus ranging from 1.5 ± 0.6 kPa to 8.0 ± 7.5 kPa as measured by rheology. The rate of UV-induced photothermal heating was non-monotonic and varied as a function of melanin nanoparticle loading. A maximum steady state temperature increase of 20.5 ± 0.30 °C was measured. Experimental heating rates were in close agreement with predictions based on attenuation of light in melanins via photothermal absorption and Mie scattering. The implications of melanin nanoparticles on hydrogel network formation and light-induced disintegration were also characterized by rheology and dynamic light scattering. Taken together, this class of photoreconfigurable hydrogels represents a potential strategy for photodegradable polymers with increased likelihood for clinical translation.
1. Introduction
Photodegradable polymers are an emerging class of biomaterials that exhibit broad utility in many applications including regenerative medicine and controlled release.1–3 Light can serve as a mild, benign stimulus to manipulate polymeric networks that are loaded with bioactive agents such as peptides, proteins, and cells.4,5 Hydrogel networks with photolabile chemistries permit spatiotemporal control of in situ structure formation and actuation.5,6 There are several established strategies to confer photodegradability in polymeric networks. Hydrogels can be synthesized with photolabile functional groups as pendant groups or within the polymer backbone.2,4 Photolabile chemistries include nitrobenzyl derivatives,2,5,7 coumarins,8,9 and triazine-polyacrylates.10 Many photolabile chemistries leverage photorearrangement reactions.11,12 The composition of these functional polymeric biomaterials is suitable for applications as model in vitro environments for elucidating cell–materials interactions.13,14 However, the evolution of potentially cytotoxic by-products through reactions including uncaging may limit widespread use as a functional medical material for use in clinical applications.15 Towards this end, progress in the field is motivated by: (1) designing photolabile polymers with photoresponsive groups that exhibit reduced cytotoxicity both in vitro and in vivo;13,16 (2) synthesis of polymer-nanoparticle networks composed of thermoresponsive hydrogel networks doped with photothermal sensitizers.17,18 Incorporating photothermal sensitizers can provide increased functionality including reconfigurability.17,19 Hydrogels with lower critical solution temperature (LCST) behavior loaded with gold nanomaterials have been used in light-controlled actuation of microstructures and controlled release of biomolecules.20–22 Physically crosslinked hydrogel networks composed of poly(L-lactide-co-glycolide)-b-poly(ethylene glycol)-b-poly(L-lactide-co-glycolide) (PLGA-b-PEG-b-PLGA) exhibit LCST behavior,23,24 are thermally reconfigurable, and its cytotoxicity has been well studied in numerous contexts.25–29 This report describes a strategy to transform self-assembled PLGA-b-PEG-b-PLGA hydrogels into photoreconfigurable networks by incorporating biologically-derived melanin nanoparticles (MelNP) as efficient photothermal sensitizers.
2. Materials and methods
2.1. Preparation of aqueous melanin nanoparticle dispersions
Natural eumelanin extracts from Sepia officinalis (Sigma-Aldrich, Milwaukee, WI, USA) were used as received. Feature dimensions were measured from scanning electron micrographs (Philips XL-30 FEG, FEI, Hillsboro, OR, USA). Samples were coated with 4 nm layer of platinum before imaging (Emtech K575X, Quorum Technologies, Guelph, ON, Canada). Aqueous dispersions of MelNP were prepared by ultrasonication of eumelanins in Milli-Q water (EMD Millipore Corporation, Billerica, MA, USA) at 30% amplitude, pulsing every 15 seconds for a total of 20 minutes with the tapered 0.125′′ microtip (Cole-Parmer, Vernon Hills, IL, USA). The size distribution of MelNP was measured immediately after ultrasonication via dynamic light scattering (DLS; Zetasizer Nano ZS, Malvern, Worcestershire, UK).
2.2. Photothermal heating of aqueous melanin dispersions
The concentration-dependent heating rate of aqueous dispersions of MelNP was measured using thermal imaging. Briefly, aqueous solutions of MelNP at prescribed concentrations were loaded into scintillation vials and the solution depth was recorded. The apical surfaces of dispersions were continuously exposed to incident UV light (Imax at λ = 365 nm) at total intensities of 10.8 mW m−2 (Black-Ray UV, UVP, Upland, CA). Solution temperatures were recorded using a thermal camera (IRXP-5000, SPi infrared, Las Vegas, NV, USA) at predetermined time points until a steady-state temperature was attained. Control samples were composed of aqueous solutions without MelNP.
2.3. Preparation and rheological characterization of physically crosslinked hydrogels
Poly(lactide-co-glycolide)-b-poly(ethylene glycol)-b-poly(lactide-co-glycolide) (PLGA-PEG-PLGA, 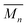 ∼ 1000-1000-1000 g mol−1; lactide
∼ 1000-1000-1000 g mol−1; lactide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide ratio 25:75) (Sigma-Aldrich, Milwaukee, WI, USA) was used as received. Aqueous hydrogel precursor solutions (20% w/v) were prepared by dissolving 200 mg of PLGA-PEG-PLGA into 1 mL of Milli-Q H2O. The solution was incubated at 4 °C for 18 hours. PLGA-PEG-PLGA hydrogel networks were formed in situ on a Peltier plate of a rheometer in parallel plate configuration (TA Instruments DHR-3, New Castle, DE, USA). Hydrogels were prepared from 0.25 mL of the solution within a gap of 1000 μm at a preset temperature of 37 °C. A temperature sweep was performed from 5–75 °C with a step interval of 2 °C and equilibration time of 180 seconds at each measurement point. Rheological experiments were performed using a strain amplitude of γ = 0.5% and a frequency of ω = 5 rad s−1. These parameters ensured that mechanical properties of hydrogels were measured in the linear viscoelastic regime (see ESI†). The data was processed using a local regression using weighted linear least squares and a 2nd degree polynomial model with a MATLAB function (LOESS).30
:glycolide ratio 25:75) (Sigma-Aldrich, Milwaukee, WI, USA) was used as received. Aqueous hydrogel precursor solutions (20% w/v) were prepared by dissolving 200 mg of PLGA-PEG-PLGA into 1 mL of Milli-Q H2O. The solution was incubated at 4 °C for 18 hours. PLGA-PEG-PLGA hydrogel networks were formed in situ on a Peltier plate of a rheometer in parallel plate configuration (TA Instruments DHR-3, New Castle, DE, USA). Hydrogels were prepared from 0.25 mL of the solution within a gap of 1000 μm at a preset temperature of 37 °C. A temperature sweep was performed from 5–75 °C with a step interval of 2 °C and equilibration time of 180 seconds at each measurement point. Rheological experiments were performed using a strain amplitude of γ = 0.5% and a frequency of ω = 5 rad s−1. These parameters ensured that mechanical properties of hydrogels were measured in the linear viscoelastic regime (see ESI†). The data was processed using a local regression using weighted linear least squares and a 2nd degree polynomial model with a MATLAB function (LOESS).30
2.4. Preparation and characterization of photosensitized physically crosslinked hydrogels
Physically crosslinked hydrogels photosensitized with MelNP were prepared in a similar manner as previously described. Briefly, aqueous dispersions of MelNP at prescribed concentrations (50 μg mL−1 to 20 mg mL−1) were prepared via ultrasonication and homogenized with aqueous solutions of PLGA-PEG-PLGA. Polymer and MelNP size distribution were characterized by DLS at three distinct temperatures, 25, 37, and 45 °C. Hydrogel formation was induced by heating to 37 °C in a water bath. The gel was transferred to a quartz plate on the rheometer equipped with UV irradiation accessory (Exfo Omnicure S2000, TA Instruments, New Castle, DE, USA). PLGA-PEG-PLGA hydrogels loaded with MelNP was then exposed to UV light (maximum intensity, Imax at λ = 365 nm; total intensity of 70.4 mW m−2). The storage (G′) and loss (G′′) modulus were measured as a function of irradiation time. Frequency sweeps were conducted at strain amplitudes of γ = 0.5% for values of ω between 0.1 and 100 rad s−1 at a temperature of 37 °C. All data presented as mean ± standard deviation where appropriate.
3. Results and discussion
3.1 Melanin nanoparticles are efficient biologically-derived photothermal sensitizers
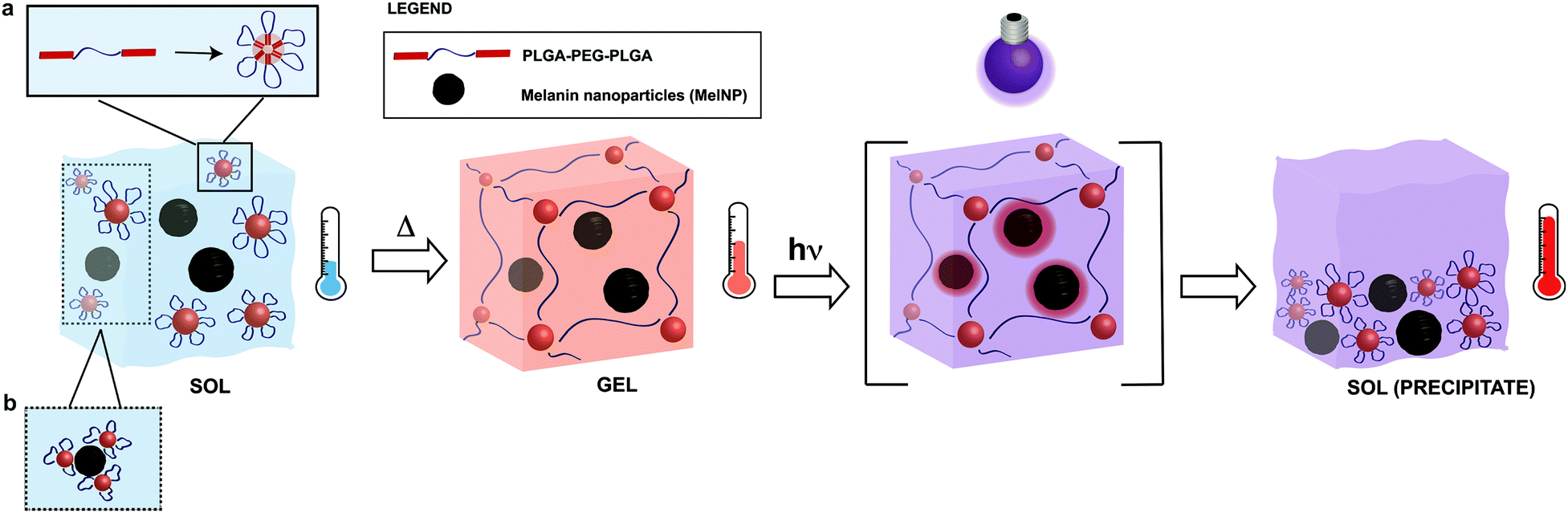
Melanins are a broad class of ubiquitous pigments that are found in many organisms in the fungi, plant, and animal kingdoms.31–34 Eumelanins, a sub-class of melanins, are composed of polyaromatic indole derivatives and heterogeneous superstructures that condense into nanoparticles.31,32,35 The combination of chemical and structural properties produce many unique physical properties including heavy metal ion chelation,36 hydration-dependent semiconducting electron transport behavior37 and broadband UV absorption.38,39 Another unique consequence of this structure is photon–phonon conversion with near ideal efficiency.38,40,41 The abundance of eumelanins in biological systems coupled with the native nanoscale morphology and unique photon–phonon interactions suggest it will serve as an efficient dopant to confer photoreconfigurability in thermally sensitive hydrogel networks. This material may be suitable as an in situ forming hydrogel for applications in wound healing, tissue regeneration, or endovascular applications. The toxicity of melanins used in these applications has been previously evaluated.42,43 The proposed mechanism for UV-induced photothermal phase transitions in MelNP-loaded PLGA-PEG-PLGA hydrogels is shown in Fig. 1. Micelle formation of pristine PLGA-PEG-PLGA has been characterized previously.23,24,28,44 Here, the adsorption process of micelles in the presence of MelNP is highlighted. Physical crosslinks are formed as the temperature increases above the sol–gel transition.11,17,18 UV irradiation of MelNP-doped hydrogels generates local heating and network disruption via precipitate formation.
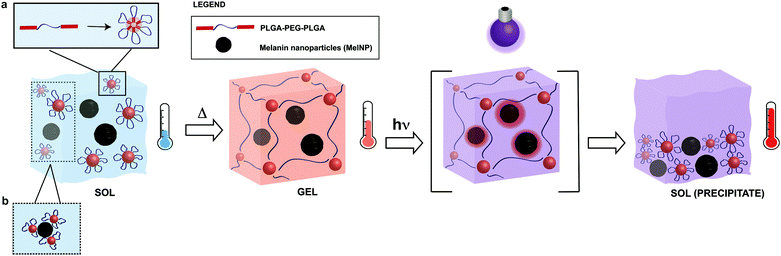 |
| | Fig. 1 Schematic of phase transitions of photoreconfigurable hydrogel networks. PLGA-PEG-PLGA solutions (sol) are incubated with melanin nanoparticles (MelNP) prior to the formation of MelNP-doped physically crosslinked hydrogels (gel). Micelles are formed from triblock PLGA-PEG-PLGA in the sol phase (a). Adsorption process of some micelles on MelNP is hypothesized (b). Physical crosslinks are formed as the temperature increases above the sol–gel transition. Irradiation of hydrogels with UV light rapidly increases the temperature of the network through efficient photon–phonon conversion, eventually leading to disintegration and precipitation (sol-precipitate). | |
Natural eumelanin (hereby referred to as melanins for simplicity) is composed of polydisperse nanoparticles with diameters between 80 and 270 nm, as determined by SEM and analyzed by NIH ImageJ (Fig. 2). Aqueous dispersions of melanin nanoparticles (MelNP) indicate a narrow size distribution centered about DMelNP = 200 nm as determined by DLS. Size distributions determined by DLS were largely independent of MelNP concentration, which suggests that MelNP dispersions were resistant to transient flocculation (Fig. 2b). Particle diameters measured by DLS may represent as overestimation due to rotation diffusion of non-spherical melanin nanoparticles to the correlation function. This phenomenon has been observed previously in pristine gold nanoparticles or carbon nanotubes.45,46
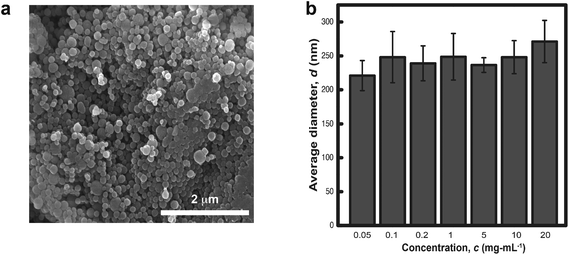 |
| | Fig. 2 Average size of the melanin nanoparticles were measured in (a) the dehydrated neat state via scanning electron microscopy and in (b) ultrasonicated aqueous dispersions via dynamic light scattering. | |
3.2 Photothermal heating rates of aqueous melanin suspensions is non-monotonic
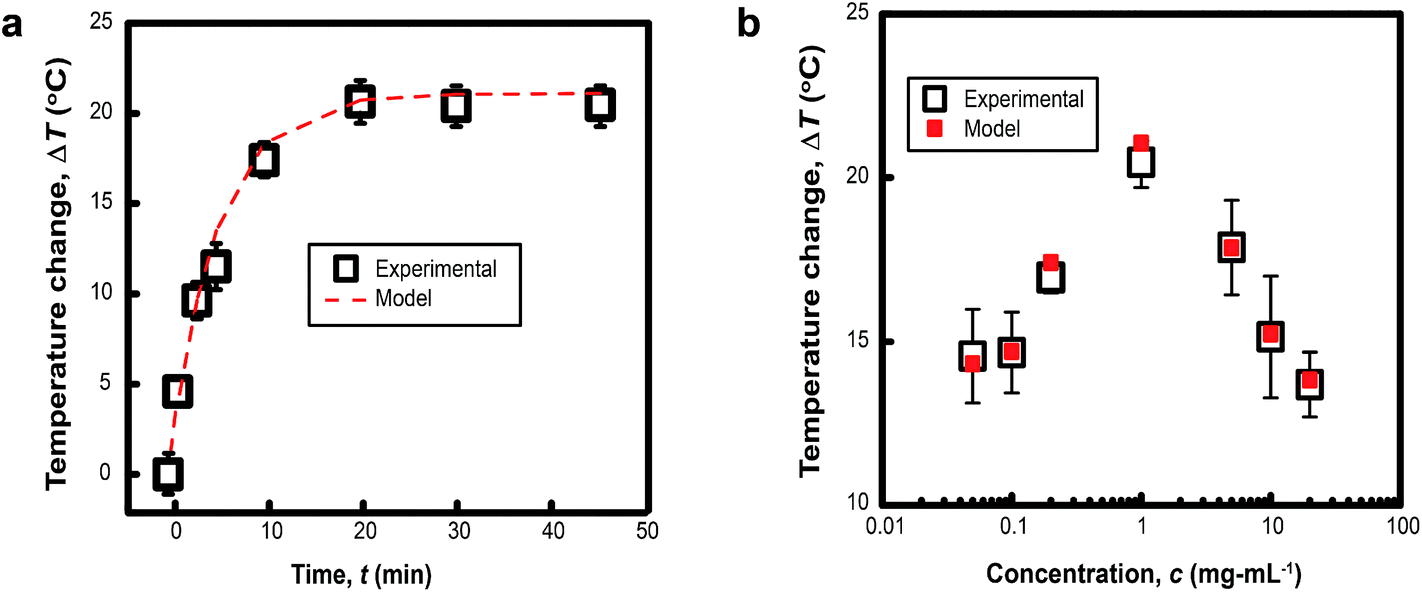
The characteristic size of MelNP suggests that attenuation of incident UV irradiation is governed by two processes. Absorption contributes to heating of optically transparent PLGA-PEG-PLGA hydrogel networks, while scattering reduces the incident intensity without contributing to local heating. MelNP can participate in both of these processes by virtue of the high photon–phonon conversion efficiency40 and the characteristic particle dimensions.47 Aqueous dispersions of MelNP achieve steady-state temperature profiles within 20 min of continuous irradiation (Fig. 3a). The increase in steady state temperature of aqueous MelNP dispersions varied non-monotonically with the concentration of MelNP (Fig. 3b). The largest increase in steady state dispersion temperature (ΔT = +20.5 °C) was observed for MelNP concentrations of 1 mg mL−1, which represents a relative increase of +14.0 °C compared to UV-induced heating of pristine hydrated PLGA-PEG-PLGA (Fig. S8, ESI†). The steady state temperature of aqueous MelNP dispersions was predicted by balancing heating from UV absorption (Qin) with heat loss that is dominated by conduction (Qout).| | 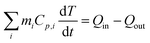 | (1) |
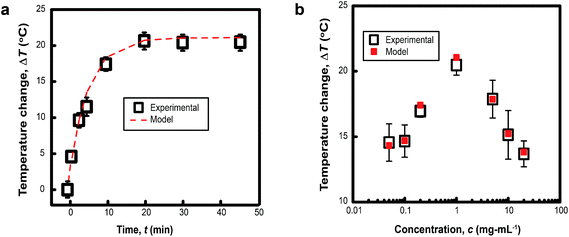 |
| | Fig. 3 (a) The temporal evolution of the temperature of aqueous dispersions of MelNP at cMelNP = 1 mg mL−1 under continuous UV irradiation is shown. These data indicate that steady state temperatures are achieved after 20 min. The heating rate is in close agreement with predictions based on optical absorption and scattering from nanoparticles (see text). (b) The steady state temperature increase of aqueous dispersions under continuous UV irradiation varies non-monotonically with particle concentration. Maximum heating rates are observed for MelNP concentration of 1 mg mL−1. | |
In this equation, mi and Cp,i represent the mass and heat capacity of component i, T represents the temperature of the aqueous dispersion. The rate of energy supplied was calculated as the one-dimensional differential of the absorbed light intensity.
| | 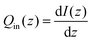 | (2) |
In this equation, I(z) represents the intensity of light at a given depth z and Qin(z) represents the local heating rate at a given depth z. It was assumed that none of the light was transmitted, which is a reasonable conclusion based on the macroscopic observations that MelNP dispersions are non-transparent. The absorbed light intensity is calculated using the following relationship derived from the Beer–Lambert law.48,49
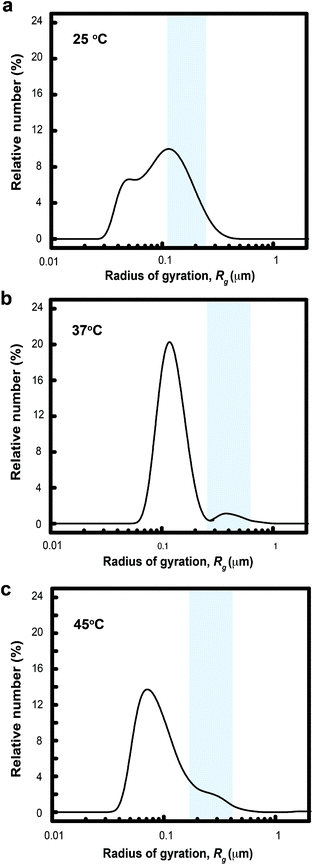
In the expression, I0 is the incident light intensity at z = 0 measured experimentally by a radiometer and β is the scattering coefficient estimated from Mie scattering.50 The value of β was estimated using concentration-dependent parameters from DLS measurements (Fig. 2b). The mass density and refractive index of MelNP were set at 1.68 g cm−3 and 1.3, respectively.35,38,41 The rate of heat loss Qout is governed by one-dimensional steady state conduction via the following equation.
| | 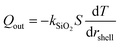 | (4) |
In this expression, kSiO2 represents the heat transfer coefficient of silicon oxide (1 W m−1 K−1),51S is the surface area of conduction and rshell is the coordinate within the silicon oxide shell between the inner (Rin) and outer radii (Rout) of the conduction path where |Rout − Rin| ≪ Rin. The resulting prediction for steady state temperatures of aqueous MelNP dispersions matched experimental data for all concentrations of MelNP in this study (Fig. 3; ESI†). A representative plot of temperature profiles is shown in Fig. 3a. The predicted transient temperature profiles of aqueous MelNP dispersions more closely matched that of experimentally determined values at low concentrations of MelNP (≤1 mg mL−1). However, the temporal temperature profiles deviated significantly from experimental observations as the MelNP concentration was increased. This may be attributed to an increase in natural convection that may evolve both thermal and density gradients (ρMelNP = 1.68 g cm−3 > ρH2O = 1 g cm−3).
3.3. Melanin nanoparticle photosensitizers can alter the phase transition behavior of physically crosslinked hydrogels
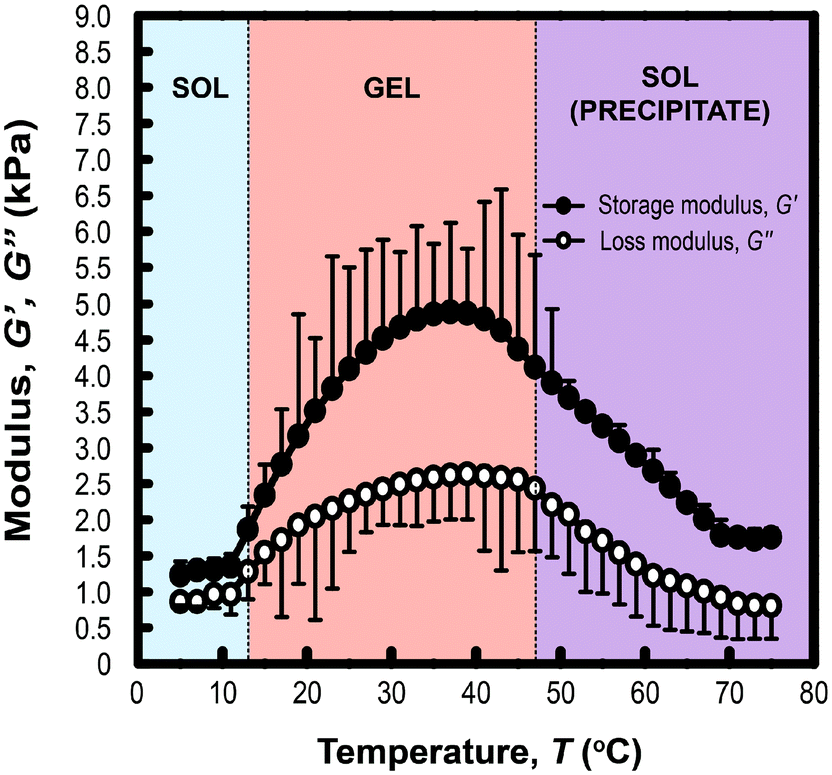
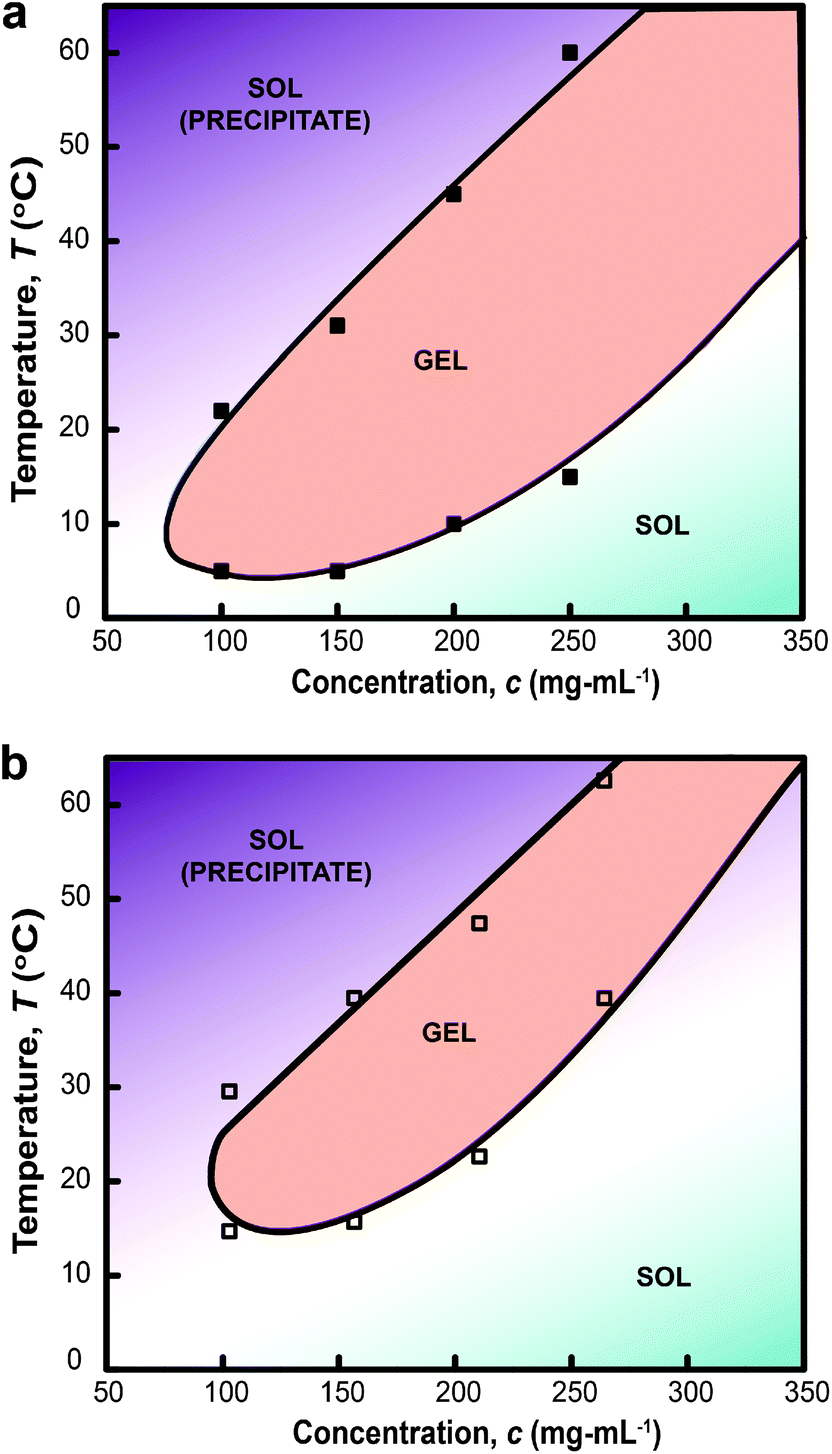
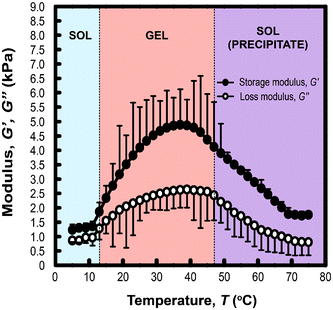
Rheological measurements of PLGA-PEG-PLGA triblock copolymer hydrogels (200 mg mL−1) formed with MelNP (1 mg mL−1) across temperatures ranging from 5 to 75 °C is shown in Fig. 4. The three phases of interest are defined as sol, gel, and precipitate. The increase in storage modulus and loss modulus was inferred as the sol–gel point, while the decrease indicated the gel-precipitate transition.52–54 These experiments were repeated at several other polymer concentrations to construct the phase diagram for hydrogel with 1 mg mL−1 MelNP (Fig. 5a). The phase diagram for pristine PLGA-PEG-PLGA hydrogel without MelNP was also constructed in a similar method (Fig. 5b) and was comparable to phase behaviors observed in other studies.24,28,44 The addition of MelNP impacts the temperature of sol–gel transitions. Sol–gel transition temperatures of PLGA-PEG-PLGA solutions (200 mg mL−1) are reduced when MelNP are added at concentrations of 1 mg mL−1. The observed offset in sol–gel transition temperatures was a strong function of the PLGA-PEG-PLGA concentration (ESI; Fig. S2†). Including MelNP into PLGA-PEG-PLGA networks generally expands the temperature range of the sol phase compared to pristine networks. This shift in the phase diagram can be attributed to the hydrophobic nature of the aromatic substituents.32,40 MelNP can act as hydrophobic nodes to accelerate gelation and retard precipitation. The driving force for phase transitions of triblock PLGA-PEG-PLGA is driven by the hydrophobicity of the system, provided by the PLGA blocks.24,28 The temperature range of the gel phase was expanded by increasing PLGA content in the triblock copolymers28 by reducing (elevating) the temperature of sol–gel (gel-precipitate) transitions. Similar phase behavior was observed in the MelNP loaded PLGA-PEG-PLGA system in this study. The addition of MelNP may serve as hydrophobic nodes that increase micelle bridging.
 |
| | Fig. 4 Phase transition behavior of hydrogel formed from 20 mg mL−1 PLGA-PEG-PLGA with 1 mg mL−1 MelNP. Temperature sweeps indicate the onset of sol–gel transitions as measured by the rapid increase of storage modulus G′ and loss modulus G′′. Reduction in G′ and G′′ corresponds to triblock copolymer precipitation. | |
 |
| | Fig. 5 Phase diagram of hydrogel formation and disassembly in aqueous environments as a function of PLGA-PEG-PLGA concentration (cpoly) (a) with MelNP and (b) doped without MelNP at a constant concentration of 1 mg mL−1. Loading MelNP into PLGA-PEG-PLGA polymer networks expands the observed temperature range for the gel transition due to the presence of additional hydro-phobic residues. This secondary component can both accelerate gelation and retard precipitation condensation (see text). | |
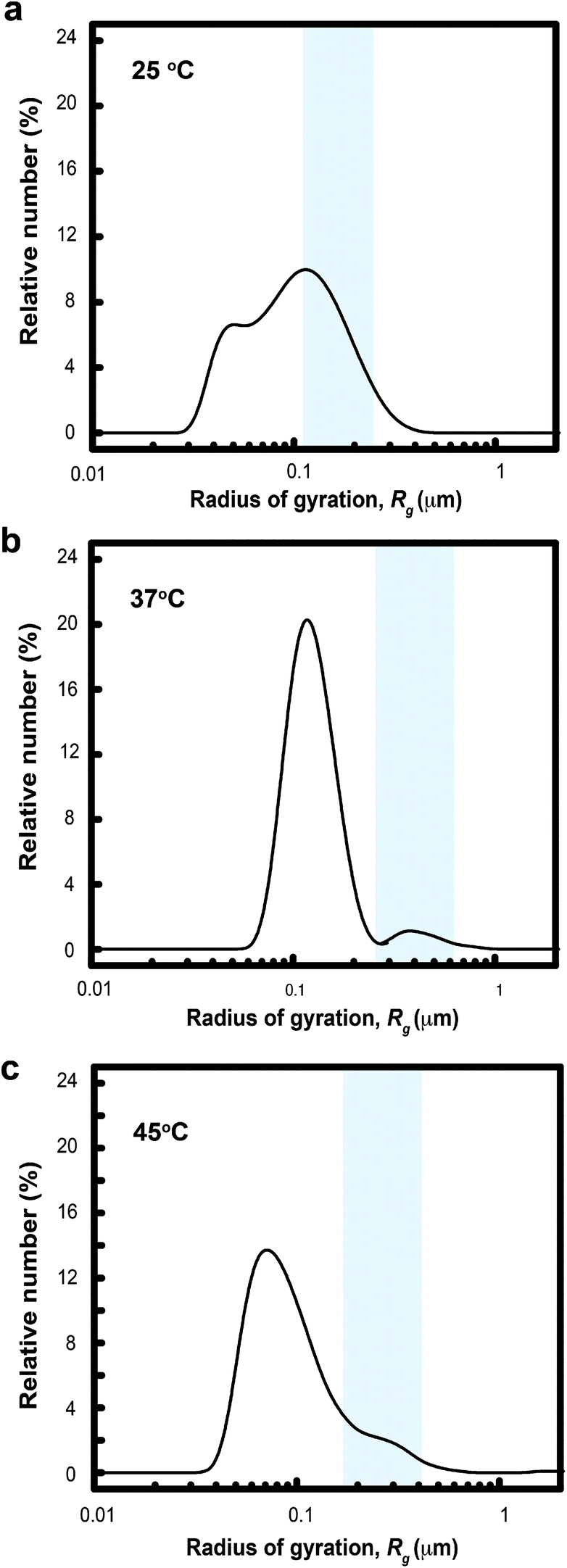
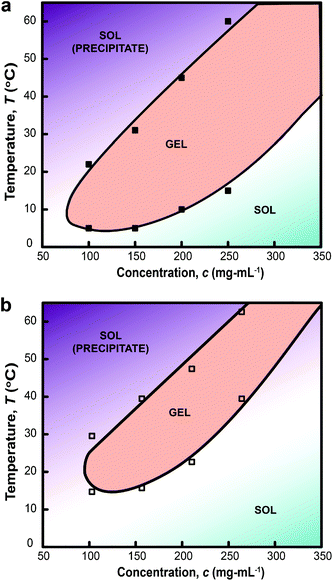
The gelation mechanism through micelle bridging was also inferred from DLS measurements. Analysis of the correlation function estimated the number fraction of two populations of colloids structures: (1) Rg between 34–52 nm, PLGA-PEG-PLGA micelles (ESI†); and (2) Rg between 61–412 nm (Fig. 6). Increasing the temperature to 37 °C abolishes population (1) and induces the formation of aggregates with larger Rg in the range of 600–1000 nm (Fig. 6). These data suggest that both micelle bridges and possible flocculated MelNP are present.23,24,28 This peak shifted towards smaller values of Rg as the temperature is increased to 45 °C. This peak shift can be attributed to micelle consolidation due to overhydrophobicity that has been previously observed in pristine PLGA-PEG-PLGA hydrogel networks.23,24,28
 |
| | Fig. 6 Size distribution of dilute aqueous two-phase dispersions of melanin nanoparticles and PLGA-PEG-PLGA micelles as a function of temperature as assessed by dynamic light scattering. The combined scattering signatures reveal two distinct particles at all three temperatures: (1) pristine PLGA-PEG-PLGA particles; and (2) melanin nanoparticles. Particle size distributions at 25 °C reveal populations of particles with Rg centered about 43 and 200 nm. Increasing the temperature to 37 °C reduces the presence of pristine PLGA-PEG-PLGA micelles and generates a consolidated population with a particle size distribution centered about Rg ∼ 400 nm (shaded region). The characteristic size of aggregates composed of both pristine PLGA-PEG-PLGA micelles and MelNP complexed with PLGA-PEG-PLGA shift to smaller Rg as the temperature is increased to 45 °C. These peak shifts correspond to PLGA-PEG-PLGA micelle consolidation due to thermally induced overhydrophobicity (see text). | |
3.4. Photothermal modulation of hydrogel networks with loaded melanin nanoparticle photosensitizers
The temporal evolution of G′ and G′′ of MelNP-loaded PLGA-PEG-PLGA hydrogel networks under UV irradiation is shown in Fig. 7. The storage modulus declines rapidly within the first 200 seconds of UV irradiation and reach plateau. This was inferred as the phase transition53,54 as also observed in Fig. 4. These data suggest that MelNP-loaded PLGA-PEG-PLGA networks undergo a rapid sol-precipitate phase transition. Light-induced sol-precipitate phase transitions are made possible through the addition of MelNP photosensitizers. Disintegration of pristine PLGA-PEG-PLGA hydrogels by UV irradiation is unattainable otherwise at this intensity.
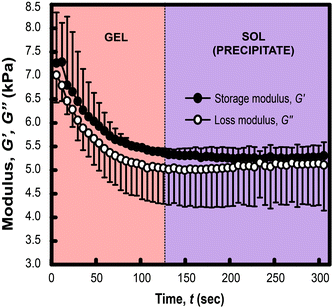 |
| | Fig. 7 Photothermal response of hydrogel networks prepared from aqueous solution of 200 mg mL−1 PLGA-PEG-PLGA loaded with 1 mg mL−1 MelNP. The hydrogel was set at 37 °C initially followed by irradiation with UV light. The storage G′ and loss G′′ modulus were measured as a function of UV irradiation time. The decrease in G′ and G′′ followed by a plateau region indicate a phase transition from gel to sol (precipitate). This phase transition is not achievable through UV-irradiation of pristine PLGA-PEG-PLGA hydrogels (ESI†). | |
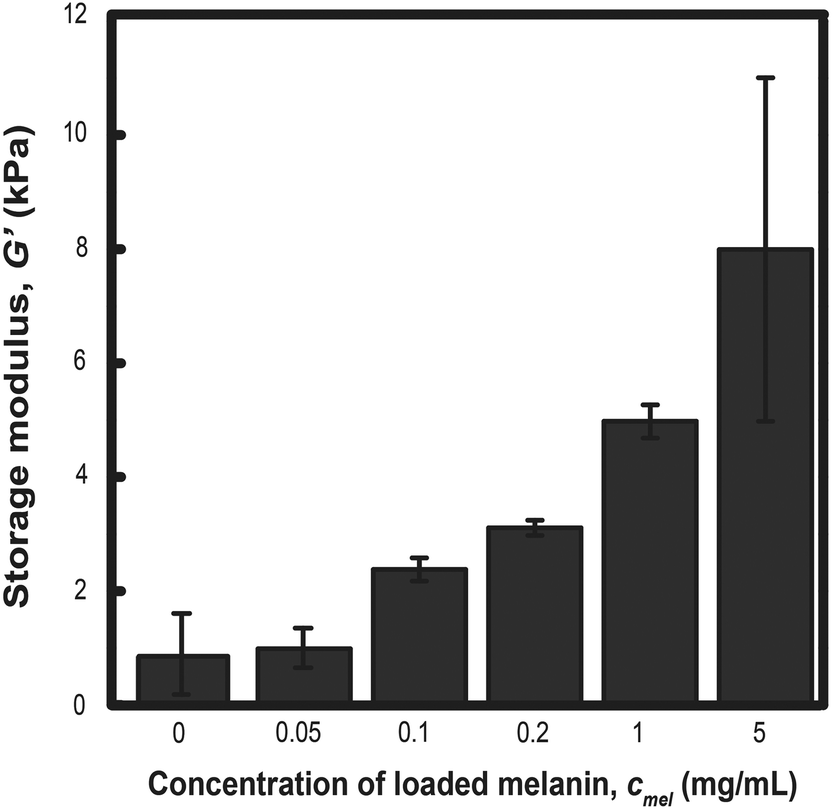
The storage modulus was measured for PLGA-PEG-PLGA hydrogels prepared from MelNP concentrations ranging from 50 μg mL−1 to 5 mg mL−1 and compared with the modulus of hydrogels without MelNP (1.0 ± 1.8 kPa). The storage modulus increases from 1.5 ± 0.6 kPa to 8.0 ± 7.5 kPa with increasing MelNP concentrations (Fig. 8). Melanins exhibit a Young's modulus of 1.57 GPa.55 Although the observed increase in G′ may be due to the inclusion of a rigid component as demonstrated in other nanoparticle-polymer composites,56 the volume fraction (estimated at ϕMelNP = 0.0036% to 0.3% for cMelNP from 50 μg mL−1 to 5 mg mL−1) of melanins in this system is not sufficient to impact G′ by this mechanism. MelNP concentrations ≥10 mg mL−1 prevented gel formation in PLGA-PEG-PLGA networks that are heated to 37 °C. Hydrophobic MelNP are suspected to influence network formation in aqueous environments via competitive adsorption of PLGA-PEG-PLGA triblock copolymers. The relative partitioning of triblocks between hydrogel formation and MelNP adsorption are estimated using thermodynamic calculations. The Gibb's free energy of PLGA-PEG-PLGA gelation ΔGgel was estimated to be −6.1 J mL−1 on a volume basis.57 The free energy of adsorption of PLGA-PEG-PLGA to MelNP in aqueous environments on a per area basis (ΔGads) was estimated by the Good–Girifalco–Fowkes equation.58
| | 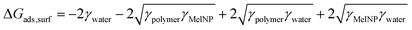 | (5) |
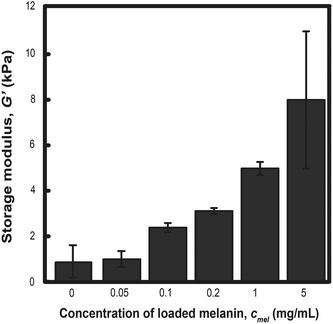 |
| | Fig. 8 The storage modulus of hydrogels prepared from aqueous solutions of 200 mg mL−1 PLGA-PEG-PLGA increases proportionally with increased concentration of MelNP. This increase can be attributed to increased bridging between hydrophobic domains upon gelation. Melanin nanoparticle concentrations of ≥10 mg mL−1 prohibited network gelation (see text). | |
The values of γpolymer, γMelNP, and γwater were 40, 44, and 71.4 mJ m−2, respectively.59 The value of ΔGads,surf was calculated to be −9.0 mJ m−2. The free energy of adsorption ΔGads,vol per volume was calculated as a function of MelNP concentrations.
| | | ΔGads,vol = ΔGads,surfSAMelNPNMelNP | (6) |
This value was calculated by estimating the surface area of a single MelNP (SAMelNP) and the number density of MelNP in solution (NMelNP), where the average melanin diameter (DMelNP) was taken as 200 nm (Fig. 2b). Spontaneous gelation of physically crosslinked PLGA-PEG-PLGA networks was observed for MelNP concentrations of 5 mg mL−1 or lower since gelation of PLGA-PEG-PLGA networks is thermodynamically favorable to MelNP adsorption (ΔGads,vol < ΔGgel). As the concentration of MelNP was increased to ≥10 mg mL−1, MelNP adsorption becomes energetically favorable compared to network gelation (ΔGads,vol> ΔGgel). This calculation is consistent with the trends in gelation versus MelNP concentration (ESI; Table S2†).
Physically crosslinked PLGA-PEG-PLGA hydrogels sensitized with MelNP represent a photoreconfigurable hydrogel composition with the potential for rapid clinical translation. PLGA-PEG-PLGA has been used as a material in many medical devices.24,26,27,44 Melanin is a naturally occurring compound that also exhibits many beneficial qualities including chelation of heavy metals and free radical scavenging.60 Photoreconfigurable hydrogel compositions described in this work may be suitable for use as injectable hydrogels that can be manipulated after gelation using UV irradiation with optical instruments, although it still faces challenges associated with this class of light-responsive materials including the lack of UV penetration and damage from UV light. Although biocompatibility profiles of the individual components have been characterized in various contexts, the combination of MelNP and PLGA-PEG-PLGA may face additional regulatory challenges.
Hydrogels with embedded MelNP exhibit increased photosensitivity, thereby enabling light-inducible gel-precipitate phase transitions. MelNP may be used to confer photoreconfigurability of other hydrogel network compositions that exhibit LCST behavior including poly(N-isopropylacrylamide).61–63 Transient hydrogel networks with MelNP may also serve as temporary materials to protect tissue from harmful UV irradiation during surgical procedures that use high intensity light. The temperature range needed to achieve phase transition of these hydrogels (Fig. 5) is comparable to widely accepted range for hyperthermia (40 °C–50 °C).64–66 The condition for photothermal heating can also be altered by varying concentrations of MelNPs, the composition of PLGA-PEG-PLGA, and the intensity of UV irradiation for appropriate applications.
4. Conclusions
Photoreconfigurable physically crosslinked PLGA-PEG-PLGA hydrogels doped with biologically-derived melanin nanoparticle photosensitizers represent a strategy to achieve light-inducible disintegration of polymer networks with benign chemistries. The heating rate and steady state temperatures of aqueous dispersions of MelNP irradiated with UV is non-monotonic and can be predicted by considering Beers law and Mie scattering. Aqueous MelNP dispersions impact sol–gel phase transitions and network formation due to the presence of hydrophobic domains that can preferentially adsorb amphiphilic PLGA-PEG-PLGA network precursors. These considerations determine the relative parameter space for polymer and MelNP concentrations that can form photoreconfigurable hydrogel networks. Taken together, this work provides a framework for engineering a new class of light-responsive polymeric biomaterials for potential use in biomedical applications.
Acknowledgements
Funding provided by the following organizations: the Berkman Foundation; the Shurl and Kay Curci Foundation; the American Chemical Society Petroleum Research Fund (ACS PRF #51980-DNI7); the National Institutes of Health (NIH; R21EB015165); the American Heart Association (AHA 12SDG12050297). Thermal and mechanical characterization was performed at the Carnegie Mellon University Thermomechanical Characterization Suite. The authors would also like to thank the Colloids, Polymers, and Sciences Laboratory at Carnegie Mellon University for assistance in light scattering measurements.
References
- D. Han, X. Tong and Y. Zhao, Macromolecules, 2011, 44, 437–439 CrossRef CAS.
- G. Pasparakis, T. Manouras, P. Argitis and M. Vamvakaki, Macromol. Rapid Commun., 2012, 33, 183–198 CrossRef CAS PubMed.
- R. P. Narayanan, G. Melman, N. J. Letourneau, N. L. Mendelson and A. Melman, Biomacromolecules, 2012, 13, 2465–2471 CrossRef CAS PubMed.
- J. Kim and R. C. Hayward, Trends Biotechnol., 2012, 30, 426–439 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- S. W. Thomas, Macromol. Chem. Phys., 2012, 213, 2443–2449 CrossRef CAS.
- V. V. Ramanan, J. S. Katz, M. Guvendiren, E. R. Cohen, R. A. Marklein and J. A. Burdick, J. Mater. Chem., 2010, 20, 8920–8926 RSC.
- M. V. S. N. Maddipatla, D. Wehrung, C. Tang, W. Fan, M. O. Oyewumi, T. Miyoshi and A. Joy, Macromolecules, 2013, 46, 5133–5140 CrossRef CAS.
- C. Zhu and C. J. Bettinger, Macromol. Rapid Commun., 2013, 34, 1446–1451 CrossRef CAS PubMed.
- E. C. Buruiana, T. Buruiana, H. Lenuta, T. Lippert, L. Urech and A. Wokaun, J. Polym. Sci. Part A: Polym. Chem., 2006, 44, 5271–5282 CrossRef CAS.
- O. D. Fedoryak and T. M. Dore, Org. Lett., 2002, 4, 3419–3422 CrossRef CAS PubMed.
- I. Aujard, C. Benbrahim, M. Gouget, O. Ruel, J.-B. Baudin, P. Neveu and L. Jullien, Chem.–Eur. J., 2006, 12, 6865–6879 CrossRef CAS PubMed.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS PubMed.
- C. Siltanen, D.-S. Shin, J. Sutcliffe and A. Revzin, Angew. Chem., Int. Ed., 2013, 125, 9394–9398 CrossRef.
- A. P. Pelliccioli and J. Wirz, Photochem. Photobiol. Sci., 2002, 1, 441–458 Search PubMed.
- A. M. Kloxin, M. W. Tibbitt and K. S. Anseth, Nat. Protocols, 2010, 5, 1867–1887 CAS.
- M. Das, N. Sanson, D. Fava and E. Kumacheva, Langmuir, 2006, 23, 196–201 CrossRef PubMed.
- V. Viswanath, S. Maity, J. R. Bochinski, L. I. Clarke and R. E. Gorga, Macromolecules, 2013, 46, 8596–8607 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- S. R. Sershen, S. L. Westcott, N. J. Halas and J. L. West, J. Biomed. Mater. Res., 2000, 51, 293–298 CrossRef CAS.
- J. L. West and N. J. Halas, Annu. Rev. Biomed. Eng., 2003, 5, 285–292 CrossRef CAS PubMed.
- M. B. Charati, I. Lee, K. C. Hribar and J. A. Burdick, Small, 2010, 6, 1608–1611 CrossRef CAS PubMed.
- M. S. Shim, H. T. Lee, W. S. Shim, I. Park, H. Lee, T. Chang, S. W. Kim and D. S. Lee, J. Biomed. Mater. Res., 2002, 61, 188–196 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae and S. W. Kim, Macromolecules, 1999, 32, 7064–7069 CrossRef CAS.
- G. Bonacucina, M. Cespi, G. Mencarelli, G. Giorgioni and G. F. Palmieri, Polymers, 2011, 3, 779–811 CrossRef CAS.
- L. Chen, Z. Xie, J. Hu, X. Chen and X. Jing, J. Nanopart. Res., 2007, 9, 777–785 CrossRef CAS.
- B. Jeong, Y. H. Bae and S. W. Kim, J. Controlled Release, 2000, 63, 155–163 CrossRef CAS.
- D. S. Lee, M. S. Shim, S. W. Kim, H. Lee, I. Park and T. Chang, Macromol. Rapid Commun., 2001, 22, 587–592 CrossRef CAS.
- M. K. Nguyen and D. S. Lee, Macromol. Biosci., 2010, 10, 563–579 CrossRef CAS PubMed.
- W. S. Cleveland and S. J. Devlin, J. Am. Stat. Assoc., 1988, 83, 596–610 CrossRef.
- C.-T. Chen, V. Ball, J. J. de Almeida Gracio, M. K. Singh, V. Toniazzo, D. Ruch and M. J. Buehler, ACS Nano, 2013, 7, 1524–1532 CrossRef CAS PubMed.
- A. A. R. Watt, J. P. Bothma and P. Meredith, Soft Matter, 2009, 5, 3754–3760 RSC.
- M. Rajadhyaksha, S. Gonzalez, J. M. Zavislan, R. R. Anderson and R. H. Webb, J. Invest. Dermatol., 1999, 113, 293–303 CrossRef CAS PubMed.
- A. R. Young, Phys. Med. Biol., 1997, 42, 789 CrossRef CAS.
- L. Zeise, R. B. Addison and M. R. Chedekel, Pigment Cell Res., 1990, 3, 48–53 CrossRef.
- C. C. Felix, J. S. Hyde, T. Sarna and R. C. Sealy, J. Am. Chem. Soc., 1978, 100, 3922–3926 CrossRef CAS.
- A. B. Mostert, B. J. Powell, F. L. Pratt, G. R. Hanson, T. Sarna, I. R. Gentle and P. Meredith, Proc. National Acad. Sci. U. S. A, 2012, 109, 8943–8947 CrossRef CAS PubMed.
- D. K. Sardar, R. D. Glickman and M. L. Mayo, J. Biomed. Optics, 2001, 6, 404–411 CrossRef CAS PubMed.
- J. D. Simon, Acc. Chem. Res., 2000, 33, 307–313 CrossRef CAS PubMed.
- S. I. C. Vieira, M. Araujo, R. Andre, P. Madeira, M. Humanes, M. J. V. Lourenco and N. C. A. De Castro, J. Nanofluids, 2013, 2, 104–111 CrossRef CAS PubMed.
- I. A. Vitkin, J. Woolsey, B. C. Wilson and R. R. Anderson, Photochem. Photobiol., 1994, 59, 455–462 CrossRef CAS.
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384–9395 CrossRef CAS PubMed.
- C. J. Bettinger, J. P. Bruggeman, A. Misra, J. T. Borenstein and R. Langer, Biomaterials, 2009, 30, 3050–3057 CrossRef CAS PubMed.
- E. Khodaverdi, F. Hadizadeh, F. Tekie, A. Jalali, S. Mohajeri and F. Ganji, Polym. Bull., 2012, 69, 429–438 CrossRef CAS.
- B. Krause, M. Mende, P. Potschke and G. Petzold, Carbon, 2010, 48, 2746–2754 CrossRef CAS PubMed.
- B. N. Khlebtsov and N. G. Khlebtsov, Colloid J., 2011, 73, 118–127 CrossRef CAS.
- M. A. van Dijk, A. L. Tchebotareva, M. Orrit, M. Lippitz, S. Berclaud, D. Lasne, L. Cognet and B. Lounis, Phys. Chem. Chem. Phys, 2006, 3486–3495 RSC.
- B. Mohlenhoff, M. Romeo, M. Diem and B. R. Wood, Biophys. J., 2005, 88, 3635–3640 CrossRef CAS PubMed.
- A. J. Welch, IEEE J. Quantum Electron., 1984, 20, 1471–1481 CrossRef.
-
S. Prahl, Mie Scattering Calculator, http://omlc.ogi.edu/calc/mie_calc.html Search PubMed.
-
N. P. Bansal and R. H. Doremus, Handbook of glass properties, 1986 Search PubMed.
- L. Yu, G. Chang, H. Zhang and J. Ding, J. Polym. Sci. Part A: Polym. Chem., 2007, 45, 1122–1133 CrossRef CAS.
- C. Pratoomsoot, H. Tanioka, K. Hori, S. Kawasaki, S. Kinoshita, P. J. Tighe, H. Dua, K. M. Shakesheff and F. R. A. J. Rose, Biomaterials, 2008, 29, 272–281 CrossRef CAS PubMed.
- K. Nagahama, Y. Imai, T. Nakayama, J. Ohmura, T. Ouchi and Y. Ohya, Polymer, 2009, 50, 3547–3555 CrossRef CAS PubMed.
- V. Ball, D. Frari, M. Michel, M. Buehler, V. Toniazzo, M. Singh, J. Gracio and D. Ruch, BioNanoScience, 2012, 2, 16–34 CrossRef.
- M. Surve, V. Pryamitsyn and V. Ganesan, Phys. Rev. Lett., 2006, 96, 177805 CrossRef.
-
B. Jeong, Biodegradable thermoreversible hydrogels for injectable drug delivery systems, University of Utah, 1999 Search PubMed.
- R. J. Good, Nature, 1966, 212, 276–277 CrossRef CAS.
- J. Jiang, L. Zhu, L. Zhu, B. Zhu and Y. Xu, Langmuir, 2011, 27, 14180–14187 CrossRef CAS PubMed.
- K.-Y. Ju, Y. Lee, S. Lee, S. B. Park and J.-K. Lee, Biomacromolecules, 2011, 12, 625–632 CrossRef CAS PubMed.
- Y. Zhang, Y. Guan and S. Zhou, Biomacromolecules, 2006, 7, 3196–3201 CrossRef CAS PubMed.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- C. Wang, N. T. Flynn and R. Langer, Adv. Mater., 2004, 16, 1074–1079 CrossRef CAS.
- M. W. Dewhirst, B. L. Viglianti, M. Lora-Michiels, M. Hanson and P. J. Hoopes, Int. J. Hyperthermia, 2003, 19, 267–294 CrossRef CAS PubMed.
- J. A. Dickson and S. K. Calderwood, Ann. N. Y. Acad. Sci., 1980, 335, 180–205 CrossRef CAS.
- P. Wust, B. Hildebrandt, G. Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix and P. M. Schlag, Lancet Oncol., 2002, 3, 487–497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60321k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 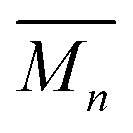 ∼ 1000-1000-1000 g mol−1; lactide
∼ 1000-1000-1000 g mol−1; lactide