One-pot synthesis of elastin-like polypeptide hydrogels with grafted VEGF-mimetic peptides
Lei
Cai
a,
Cong B.
Dinh
a and
Sarah C.
Heilshorn
*ab
aDepartment of Materials Science and Engineering, Stanford University, Stanford, CA, USA
bDepartment of Bioengineering, Stanford University, Stanford, CA, USA. E-mail: heilshorn@stanford.edu; Fax: +1 (650) 498-5596; Tel: +1 (650) 723-3763
First published on 4th February 2014
Abstract
Immobilization of growth factors to polymeric matrices has been a common strategy in the design of tissue engineering scaffolds to promote tissue regeneration, which requires complex cell signaling events with the surrounding matrix. However, the use of large protein growth factors in polymeric scaffolds is often plagued by immunogenicity, short in vivo half-lives, and reduced bioactivity. To address these concerns, we developed a single-step, cell-compatible strategy to tether small, growth-factor-mimetic peptides into a protein-engineered hydrogel with tunable biomaterial properties. Specifically, we covalently immobilized the QK peptide, an angiogenic peptide mimicking the receptor-binding region of vascular endothelial growth factor (VEGF), within tunable elastin-like polypeptide (ELP) hydrogels that include a cell-adhesive RGD sequence. Using a cell-compatible, amine-reactive crosslinker, we conducted a one-pot synthesis to simultaneously encapsulate cells while precisely controlling the QK grafting density (10 nM–100 μM) in the ELP hydrogels without altering other material properties. Fluorescence analysis of fluor-labeled QK peptides demonstrated that the conjugation efficiency to ELP hydrogels was >75% and that covalent immobilization effectively eliminates all QK diffusion. Compared with pristine ELP hydrogels, human umbilical vein endothelial cell (HUVEC) proliferation was significantly enhanced on ELP hydrogels immobilized with 10 nM or 1 μM QK. Moreover, upon encapsulation within tethered QK-ELP hydrogels, HUVEC spheroids maintained near 100% viability and demonstrated significantly more three-dimensional outgrowth compared to those supplemented with soluble QK peptide at the same concentration. These results encourage the further development of protein-engineered scaffolds decorated with growth-factor-mimetic peptides to provide long-term biological signals using this versatile, single-step synthesis.
Introduction
The process of tissue repair is guided by a complex spatiotemporal interplay between cells and their extracellular matrix (ECM), which is a cell-secreted, three-dimensional (3D) matrix containing numerous multifunctional proteins for cell signaling.1,2 Because of these complex signaling events, independent control over individual material parameters is crucial in designing 3D scaffolds that can be optimized to promote tissue regeneration.3 Several strategies exist to design biomaterials with independent tailorability of mechanical cues and integrin binding. These approaches include surface modification of pre-formed biomaterials,4–7 crosslinking or tethering of integrin ligands throughout a polymeric matrix,8–10 and combinatorial copolymer approaches.11–13 Distinct from these approaches, the modular design of protein-engineered materials has emerged as a versatile strategy to create families of tunable ECM-mimetic biomaterials.14–16 Synthesized by recombinant protein technology, multiple peptide modules are combined to form a single biopolymer with control over independent structural, mechanical, bioactive, and degradation profiles.14–16 Our lab previously developed elastin-like polypeptides (ELPs) with high tunability over mechanical stiffness and cell-adhesive ligand density.17–21 For example, ELPs have been designed with a cell-adhesive RGD ligand or non-adhesive, scrambled RDG in their otherwise identical primary amino acid sequences.20 Upon encapsulation of dorsal root ganglia, this system demonstrated the effects of RGD ligand density and matrix stiffness on promoting neurite outgrowth in a decoupled manner.20 Beyond their use as in vitro scaffolds to probe fundamental cell–matrix interactions, ELP-based biomaterials have also been developed for preclinical and clinical studies.22–24In addition to tuning of the scaffold mechanics and integrin binding, several groups have developed techniques to sequester growth factors within 3D biomaterials to promote biochemical cell signaling. Growth factors have been immobilized onto hydrogels by transglutaminase enzyme factors,25 heparin-binding domains,26 and engineered fusion proteins with affinity tags.27,28 However, there are challenges associated with the use of large growth factors within the polymer matrix, including immunogenicity,29 loss of bioactivity,30 and short in vivo half-lives.31,32 An alternative approach is the use of growth-factor-mimetic small peptides that are able to recapitulate the biological functions of full-length proteins. The use of small peptides offers advantages such as ease of synthesis, lower cost, and lower immunogenic potential.33,34 However, small peptides tend to diffuse quickly away from the target site and hence are unable to maintain a sufficiently high concentration for local bioactivity. Current conjugation methods typically involve multiple steps of functionalizing peptides with reactive chemical groups, which limits the yield of the target conjugate and increases the cost and time for production and purification.35 Therefore, it is critical to develop facile methods to sequester these molecules within engineered biomaterials.
While growth-factor-mimetic peptides could be directly encoded into the backbone of protein-engineered biomaterials, this strategy would require genetic engineering of a new recombinant plasmid, the development of a suitable protein expression protocol, and the optimization of a purification scheme for each individual peptide–biomaterial combination. Therefore, we sought to develop a simple and flexible technique to quantitatively graft a specified concentration of bioactive peptide onto protein-engineered biomaterials. Towards this goal, we have designed a cell-compatible, one-pot, single-step synthesis to tether peptides within ELP hydrogels without interfering with the presentation of integrin-binding ligands or the tunable control of mechanical properties. As a proof of concept, we chose a small angiogenic peptide, referred to as QK, which mimics the receptor-binding domain of vascular endothelial growth factor (VEGF). Angiogenesis, the formation of new blood vessels from existing conduits, is a critical requirement for successful tissue engineering.36,37 In soluble form, the QK peptide has demonstrated enhanced endothelial cell attachment and proliferation above a threshold concentration of 1 pM.32,38,39 Moreover, QK has been found to maintain its bioactivity for 24 h in 50% human serum, a stability much better than the full-length VEGF, which has an in vivo half-life of ∼90 min.30 Previously, QK peptides were immobilized on collagen scaffolds via collagen-binding peptides,40 on hydroxyapatite via binding of an osteocalcin sequence,38 and on self-assembled monolayers.41 QK peptides were also covalently bound to polyethylene glycol (PEG) hydrogels via a multi-step conjugation protocol followed by UV light exposure.33
Building on these studies, which demonstrate that the QK peptide retains its bioactivity when tethered, we sought to develop an efficient, single-step method to conjugate QK peptides and simultaneously encapsulate cells within 3D tunable ELP hydrogels. This one-pot strategy enables simultaneous crosslinking of ELP hydrogels of similar mechanical properties while conjugating QK peptides at concentrations ranging from 10 nM to 100 μM. The bioactivity of these matrices was studied using human umbilical vein endothelial cells (HUVECs) in both 2D proliferation and spreading assays and 3D encapsulation and outgrowth models. This one-pot synthesis and cell encapsulation strategy enables rapid, efficient, and reproducible formation of cell-laden hydrogels with tunable material properties. This versatile strategy may be applied easily to the tethering of other bioactive peptide sequences to facilitate further decoupled control of 3D hydrogel material properties, integrin binding, and tethering of growth-factor-mimetic peptides.
Materials and methods
ELP expression and purification
ELP was modularly designed with four repeats of a cell-adhesive extended RGD sequence from fibronectin (TVYAVTG![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) SPASSAA) and a structural, elastin-like domain ((VPGIG)2VPGKG(VPGIG)2)3.17 The polypeptide was expressed and purified using recombinant protein technology as previously described.17 Briefly, the BL21(DE3) strain of Escherichia coli was transformed with a pET15b plasmid containing a T7-lac promoter driven ELP sequence along with ampicillin resistance. ELP production was induced with 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) and allowed to incubate for 5 h. Cell pellets were harvested and lysed in TEN buffer (0.1 M NaCl, 0.01 M Tris, 0.001 M EDTA salt, pH 8) with 100 mM of phenylmethylsulfonyl fluoride and deoxyribonuclease. ELP was purified using a repeated inverse temperature-cycling process, which began with centrifugation at 4 °C in H2O (pH 9) and was followed by agitation at 37 °C in 1 M NaCl and centrifugation to collect the precipitated ELP. Purified ELP was dialyzed against water at 4 °C with a 10
SPASSAA) and a structural, elastin-like domain ((VPGIG)2VPGKG(VPGIG)2)3.17 The polypeptide was expressed and purified using recombinant protein technology as previously described.17 Briefly, the BL21(DE3) strain of Escherichia coli was transformed with a pET15b plasmid containing a T7-lac promoter driven ELP sequence along with ampicillin resistance. ELP production was induced with 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) and allowed to incubate for 5 h. Cell pellets were harvested and lysed in TEN buffer (0.1 M NaCl, 0.01 M Tris, 0.001 M EDTA salt, pH 8) with 100 mM of phenylmethylsulfonyl fluoride and deoxyribonuclease. ELP was purified using a repeated inverse temperature-cycling process, which began with centrifugation at 4 °C in H2O (pH 9) and was followed by agitation at 37 °C in 1 M NaCl and centrifugation to collect the precipitated ELP. Purified ELP was dialyzed against water at 4 °C with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecular weight cut-off membrane and lyophilized. Typical protein yields were 50–100 mg per liter of culture. The purity of ELP was confirmed by gel electrophoresis.
000 molecular weight cut-off membrane and lyophilized. Typical protein yields were 50–100 mg per liter of culture. The purity of ELP was confirmed by gel electrophoresis.
QK-ELP hydrogel preparation
The QK peptide (KKLTWQELYQL[K(Ac)]Y[K(Ac)]GI) was designed with two starting lysine units with primary amines for conjugation, while all other lysines were protected by acetyl groups (represented by Ac). The QK and fluorescein isothiocyanate (FITC)-labeled QK peptides were purchased through custom peptide synthesis from Genscript Corp (Piscataway, NJ, USA) and confirmed to have purity of 92–98% by HPLC. ELP and QK were crosslinked through lysine residues using the crosslinker tetrakis(hydroxymethyl)phosphonium chloride (THPC, Sigma) at a stoichiometry of 1:1 ELP primary amine reactive groups to THPC phosphine reactive groups. The purified ELP was first dissolved at 10 wt% in phosphate-buffered saline (PBS) at 4 °C. THPC stock solution was prepared at 3.38 mg mL−1 in PBS. The QK peptide was dissolved and diluted to the desired concentration in PBS. The precursor solution was prepared by mixing ELP, QK, and THPC together, with a final ELP concentration of 4 wt% and three QK concentrations of 10 nM, 1 μM, or 100 μM. The precursor solution was then transferred to a silicone mold (0.5 mm thickness, 6 mm diameter) affixed to a glass coverslip and allowed to crosslink at room temperature for 20 min. ELP gels crosslinked in the presence of 10 nM, 1 μM, or 100 μM QK are referred to as 10 nM QK-ELP, 1 μM QK-ELP, and 100 μM QK-ELP, respectively.
Characterization of QK grafting to ELP hydrogels
To determine the QK conjugation efficiency, fluorescence recovery after photobleaching (FRAP) measurement was performed on 1 mM QK-ELP gels prepared with the FITC-labeled QK peptide and crosslinked for 20 min. As a control group, ELP gels with unbound QK were prepared by immersing ELP gels in a 1 mM soluble FTIC-labeled QK solution. The total fluorescence intensity was visualized at 37 °C using a Leica TCS SP5 confocal microscope at low light intensity. Photobleaching was conducted by exposing a 100 × 100 μm2 spot in the field of view to high intensity laser light. A series of images were taken every three seconds for 90 seconds to track the recovery of fluorescence. The resulting recovery profiles were modeled by Fickian diffusion according to previously reported methods to calculate diffusivities of peptides in these hydrogels.42 To measure the percentage of bound QK within the gel, the QK-ELP gel was immersed in 1 mL PBS immediately after crosslinking for various times (5–40 min) and agitated for 1 h. The fluorescence intensity of the unbound FITC-labeled QK released into the surrounding PBS was measured and normalized to that of the precursor solution directly diluted in PBS.QK-ELP gel swelling and rheological analysis
Swelling ratios of the ELP gel and 10 nM, 1 μM, and 100 μM QK-ELP gels were measured by immersing them in DI water for 1 day, blotting dry, and weighing to determine the swollen weight (Ws). The swollen hydrogels were then completely dried by lyophilization and weighed again to determine the dry weight (Wd). The swelling ratios were calculated using the equation of (Ws − Wd)/Wd.Rheological properties of the hydrogels were quantified on a stress-controlled rheometer (AR-G2, TA instrument, New Castle, DE) using an 8 mm diameter, parallel-plate geometry. Hydrogels were prepared in silicone molds with 1.0 mm thickness and 8 mm diameter. Linear viscoelastic properties including storage moduli (or shear moduli, G′) and loss moduli (G′′) as functions of frequency from 0.1–1 Hz were measured at 37 °C with a strain of 10%. Samples were measured in triplicate.
HUVEC proliferation on 2D QK-ELP gels
Human umbilical vein endothelial cells (HUVECs, Lonza) were cultured in EBM-2 endothelial basal medium with full EGM-2 bullet kit supplements (Lonza). Cells were kept in a humidified, 5% CO2 environment at 37 °C with media changes every 2 days. Cells were passaged using TrypLE express (Invitrogen), and passages 2–7 were used in subsequent experiments. For all cell studies, the ELP, QK, and THPC were filtered through 0.22 μm pore size syringe filters. The precursor solutions were prepared as described above with QK concentrations of 10 nM, 1 μM, and 100 μM, and then transferred to sterilized silicone molds in a 24-well plate. After crosslinking for 20 min, all the gels were immersed in 1 mL PBS and sterilized under ultraviolet (UV) light for 2–3 h. HUVECs were trypsinized, centrifuged, and suspended in growth factor depleted EBM-2 medium supplemented with 5% heat-inactivated fetal bovine serum (HI-FBS). Cells were seeded at a density of ∼15000 per cm2 on top of each gel (pristine ELP and 10 nM, 1 μM, and 100 μM QK-ELP gels). As a control group, 1 μM soluble QK was added to the media for half of the pristine ELP gels. After culturing for 1, 2, and 4 days, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 solution in PBS, and stained with rhodamine phalloidin (1:300, Life Technologies) and Hoechst (1 μg mL−1, Life Technologies). Cell images were obtained using a Leica TCS SP5 confocal microscope. Cell number was quantified from at least 5 cell images at each time point. Cell area was determined and averaged on 20 non-overlapping cells at day 1 post-seeding using the ImageJ software (National Institutes of Health). The cell doubling time was calculated using the equation of 3 × ln(2)/ln(cell number at day 4/cell number at day 1).
3D HUVEC spheroid encapsulation and outgrowth
HUVEC spheroids consisting of about 500 cells per spheroid were generated in growth factor depleted EBM-2 medium supplemented with 5% HI-FBS using AggreWell™ 400 plates (STEMCELL Technologies) following the manufacturer's protocol. For 3D encapsulation, HUVEC spheroids were mixed with ELP and QK before the addition of THPC. After crosslinking for 20 min at room temperature, hydrogels were immersed in the growth factor depleted media and cultured for 2 and 4 days with media changes every 2 days. As a control group, 1 μM soluble QK was also added to the media for the pristine ELP gels. These samples were either replenished with 1 μM soluble QK or not replenished during media change at day 2. Cell viability was determined using LIVE/DEAD viability/cytotoxicity kit (Invitrogen) at each time point. Confocal z-stack images were collected at ∼0.5 μm intervals to a depth of ∼100 μm into the gels. Z-stacks were processed to obtain a “top view” using a maximum projection, and a “side view”, which is the corresponding projection along the y-axis. Cell viability was calculated as the number of viable green cells divided by the total number of cells (n ≥ 5). The normalized protrusion area was defined as the ratio of the cross-sectional area of all protruded cells in the x–y plane to the cross-sectional area of the compact spheroid. Normalized protrusion area was quantified using ImageJ (n ≥ 5).Statistical analysis
All data are presented as mean ± standard deviation. Statistical comparisons were performed by one-way analysis of variance (ANOVA) followed by Tukey post hoc test. Values were considered to be significantly different when the p value was <0.05.Results and discussion
Single-step QK conjugation to ELP hydrogels
We developed a one-pot synthesis procedure to simultaneously crosslink ELP into a hydrogel while conjugating the angiogenic QK peptide into the same hydrogel network (Fig. 1). This simple, single-step strategy eliminates the multiple chemical functionalization and purification steps that were required in previous photo-reactive tethering protocols.33 THPC, a 4-arm, cell-compatible, amine-reactive crosslinker, efficiently reacts with free primary amines on the ELP and QK in PBS at ambient temperature. The QK peptide was designed with two starting lysine units with primary amines for conjugation, while all other lysines were protected by acetyl groups. Tethering through these extra end-capped lysine linkages minimizes the potential for adverse effects on QK bioactivity. | ||
| Fig. 1 Schematic of QK-ELP design and hydrogel formation. ELPs were designed with four repeats of a cassette containing both cell-binding (orange) and elastin-like (blue) sequences with three lysine amino acids (green) for primary amine-reactive crosslinking. The QK peptide (red) was designed to include two lysine residues at the end for conjugation. Addition of a four-arm crosslinker, THPC (cyan), yielded a 3D ELP hydrogel with tethered QK peptides. Photograph shows a QK-ELP hydrogel in a 6 mm silicone mold (orange) within a 24-well plate. | ||
Left undisturbed at room temperature for 20 min, the reaction created a stable gel in the mold. The conjugation of QK to the ELP hydrogel was confirmed by FRAP analysis, which measures the 2D lateral diffusivity of a probe molecule within the matrix.42 For soluble, FITC-labeled QK that was diffused into the pristine ELP gel, the photobleached spot quickly recovered fluorescence due to diffusion of the surrounding, unbleached, mobile peptides (Fig. 2a). As expected, the high mobility of soluble QK resulted in a near complete recovery of fluorescence 90 s after photobleaching, and its diffusivity was calculated as 61 ± 5 μm2 s−1 (Fig. 2b). In a clear contrast, the photobleached spot in the conjugated QK-ELP gel remained unaltered over 90 s, effectively eliminating all QK diffusion and confirming that QK was immobilized to the hydrogel matrix (Fig. 2b). Based on these diffusivity data, simple encapsulation of soluble QK within ELP hydrogels results in rapid depletion of the peptide. Therefore, to maintain a constant bioactive QK concentration for prolonged signaling, the peptide must be tethered to the biomaterial. In vivo, angiogenesis occurs over many days, and the local angiogenic signals must be temporally regulated over this multi-day time scale.47
 | ||
| Fig. 2 FRAP characterization of QK-ELP hydrogels. (a) Time-lapse fluorescence images of photobleached areas over 90 s in the QK-ELP gel and a control ELP gel with soluble QK. (b) The corresponding fluorescence recovery curves used to derive diffusivity of the QK peptide within the gels. (c) Quantification of percentage of QK bound to ELP gels at various crosslinking times. | ||
Gelation kinetics is an important variable when simultaneously encapsulating cells within chemically crosslinked hydrogels. If the gelation time is too short, it may be difficult to homogeneously distribute the cells throughout the gel prior to gelation. However, if the gelation time is too long, the cells may begin to sink within the solution or may begin to undergo apoptosis due to lack of nutrients, since culture media cannot be added until a stable gel phase has formed. To evaluate the crosslinking kinetics, gels were immediately immersed in PBS after crosslinking for various times to release the unbound QK for quantification (Fig. 2c). No gels were formed after 5 or 10 min of crosslinking. Upon 20 min of crosslinking, we found that 77 ± 3% QK was conjugated to the ELP hydrogel, suggesting a good reaction efficiency and a reaction rate suitable for 3D cell entrapment (Fig. 2c). The 40 min crosslinking time resulted in a higher conjugation efficiency, 93 ± 1% bound QK, but this duration may be too long for efficient cell encapsulation. Therefore, a 20 min crosslinking time was chosen for all further experiments.
These results demonstrate the success of covalently immobilizing a 15-amino acid QK peptide within a protein-engineered matrix. There have not been many reports that use commercially available crosslinkers to efficiently conjugate peptides to hydrogels at physiological temperature and pH. Photocrosslinking is a commonly used method, but this requires additional steps to end-cap peptides with reactive double-bonds, a process which is often difficult and may lead to a loss of bioactivity.33,43 Other strategies include thiol–ene Michael addition,44 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)–N-hydroxysuccinimide (NHS) coupling,45 and Schiff base formation by carbonyl groups.46 Compared to the phosphine-based crosslinker, THPC, presented here, these other chemistries either require much longer crosslinking times or tend to have lower efficiencies.
QK-ELP hydrogel characterization
Mechanical properties of the pristine ELP gel and QK-ELP gels conjugated with 10 nM, 1 μM, and 100 μM QK peptide were determined by rheological measurements in a dynamic frequency sweep mode (Fig. 3a). All hydrogels demonstrated curves characteristic of elastic networks formed by covalent crosslinking, with shear moduli, G′, consistently greater than loss moduli, G′′. Shear moduli of all of the gels were ∼270 Pa, independent of frequency and QK conjugation density (Fig. 3b). As another indicator of hydrogel network structure, swelling ratios were found to be 18.2 ± 0.5, 19.4 ± 3.1, 19.5 ± 1.8, and 18.5 ± 1.9, for pristine ELP gels without QK, and QK-ELP gels conjugated with 10 nM, 1 μM, and 100 μM QK peptide, respectively. No statistical differences were observed among all gels for both measurements (shear moduli and swelling ratio), indicating that the pore size remains the same for all hydrogels with tethered QK concentrations below 100 μM. These results are consistent with our expectations, because even the highest conjugated QK density of 100 μM reacts with less than 1% of the total available lysine residues within the ELP sequence. This minimal rate of reactive site conjugation is unlikely to alter the crosslink density of the bulk ELP gel. Therefore, these QK-ELP hydrogels have similar mechanical properties, identical RGD integrin-binding ligand densities, and controlled QK concentrations, which enables study of the dose-dependent effects of bound QK on endothelial cell behavior. | ||
| Fig. 3 Rheological properties of ELP and 10 nM, 1 μM, and 100 μM QK-ELP gels. (a) Storage moduli (G′, solid symbols) and loss moduli (G′′, open symbols) as a function of frequency for a 10% strain amplitude. (b) Shear moduli at a frequency of 0.2 Hz at 37 °C. | ||
2D HUVEC proliferation responses to tethered QK
To examine the bioactivity of immobilized QK peptides within ELP hydrogels, we performed in vitro 2D cell proliferation studies by seeding HUVECs on the tops of ELP and QK-ELP hydrogels. As a control, HUVECs on pristine ELP gels were exposed to 1 μM soluble QK peptide. In our reaction scheme, the two lysine residues in the middle of the QK peptide sequence, which have been reported to be key for QK bioactivity, were acylated to avoid reaction with the THPC crosslinker. Previous reports have indicated that acylated QK peptides maintain their original bioactivity.33 Furthermore, because the QK peptides are tethered to the ELP gel through the four methylene groups of the lysine side chain, we hypothesized that the QK peptides would maintain sufficient conformational flexibility when tethered to the ELP matrices to retain bioactivity. HUVECs cultured for 1, 2, and 4 days were stained for F-actin and nuclei (Fig. 4). On pristine ELP gels, HUVECs distributed sparsely and proliferated slowly over 4 days. In a clear contrast, treating the cells with 1 μM soluble QK significantly enhanced HUVEC growth, as more cells were dividing already at day 1 and reached ∼70% confluence at day 4, much better than those on ELP gels alone. This result is consistent with previous findings that QK effectively promotes endothelial cell attachment and proliferation.32 Conjugating QK to the ELP gels effectively prohibits all QK diffusion, maintaining a constant QK concentration. On the 10 nM and 1 μM QK-ELP gels, HUVECs showed enhanced proliferation compared to cells cultured on pristine ELP gels. These data suggest that QK retained its bioactivity upon conjugation. In contrast, 100 μM QK-ELP gels did not support cell proliferation, and a loss of cells was observed at longer time points. To confirm the qualitative trends shown in the cell micrographs, we quantified cell number at each time point (Fig. 5a). These data were used to calculate cell doubling times, an indicator of cell proliferation rate (Fig. 5b). Consistent with the cell micrographs, significantly higher cell numbers were observed on ELP gels treated with 1 μM soluble QK, 10 nM QK-ELP gels, and 1 μM QK-ELP gels compared to pristine ELP gels and 100 μM QK-ELP gels. Calculations of cell doubling time indicated that HUVECs proliferate much faster on 10 nM QK-ELP gels and 1 μM QK-ELP gels compared to unmodified ELP gels. Further increasing the QK conjugation concentration to 100 μM eliminated all cell proliferation. This was an interesting observation, since the HUVECs exhibited statistically similar spread area on all of the ELP gels at day 1 (Fig. 5c). This suggested that integrin-engagement with the RGD ligand occurred on all of the gels similarly and that changes in proliferation rate were a direct consequence of QK signaling. | ||
| Fig. 4 2D HUVEC proliferation on QK-ELP gels and control ELP gels with and without the addition of soluble QK. Fluorescence images of HUVEC morphology (F-actin staining with phalloidin in red and nuclei staining with Hoechst in blue) at 1, 2, and 4 days post-seeding. The 200 μm scale bar is applicable to the first three rows and the 50 μm scale bar applies to the last row. | ||
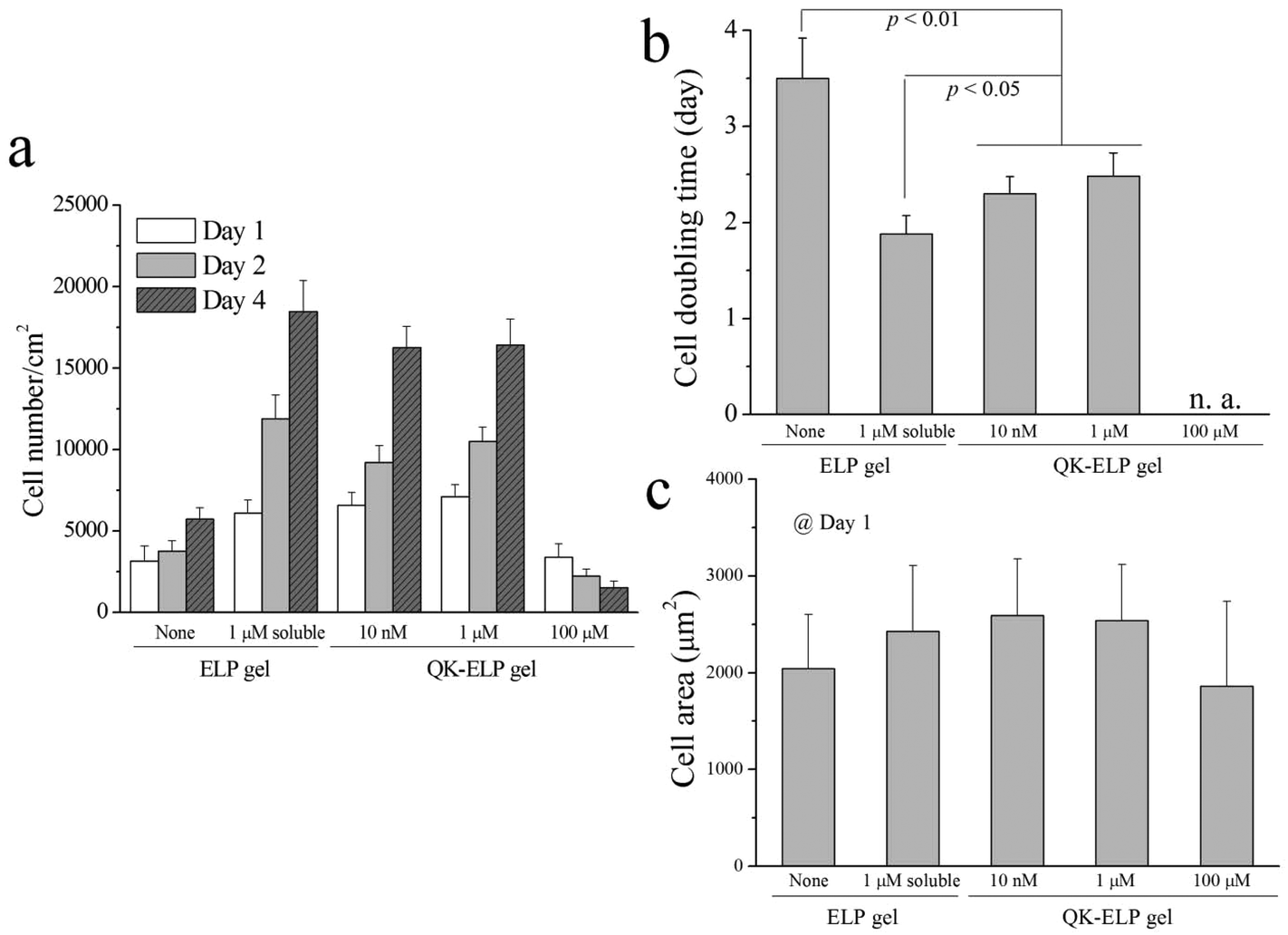 | ||
| Fig. 5 Quantification of 2D HUVEC proliferation on ELP and QK-ELP gels. Comparison of (a) cell number, (b) cell doubling time, and (c) cell area. For panel (a), p < 0.05 between the ELP gel or the 100 μM QK-ELP gel and the other three groups at each time point. Statistical p values are indicated in (b). No statistically significant differences in (c). | ||
Previously, QK has been covalently bound to PEG hydrogels at concentrations of 152 nM and 760 nM.33 Consistent with our results on 10 nM and 1 μM QK-ELP gels, the QK-grafted PEG gels were found to enhance HUVEC proliferation. Similarly, tethering QK combined with a collagen binding domain to collagen surfaces was found to initiate similar levels of signaling through extracellular signal-regulated kinases 1 and 2 (ERK 1/2) as soluble QK peptides.40 Although QK often stimulates endothelial cell proliferation, it is interesting to find that QK appears to exert an antagonistic effect when presented at high densities. This phenomenon was also observed by Koepsel et al. when QK was covalently immobilized at high densities on self-assembled monolayers.41
3D HUVEC spheroid encapsulation and outgrowth within QK-ELP gels
Taking advantage of the cell-compatible reaction conditions, we next simultaneously encapsulated HUVEC spheroids within the ELP hydrogels during the crosslinking and peptide tethering process. Post-encapsulation, the individual spheroids, each containing around 500 cells, maintained near 100% cell viability in all of the ELP hydrogels, as quantified by live/dead staining at 2 and 4 days in vitro (Fig. 6a and b). These data are consistent with the high viability observed for dorsal root ganglia20 and embryonic stem cells18,21 within 3D ELP hydrogels. At day 2, no HUVEC outgrowth from the spheroids was observed within all of the ELP gels tested. However, by day 4, cells exposed either to soluble (1 μM) or tethered (both 10 nM and 1 μM) QK peptides within the ELP gels started to reorganize the matrix and to protrude out of the initial compact spheroid (Fig. 6a and c). Consistent with the 2D HUVEC studies, the 100 μM QK-ELP gel did not stimulate HUVEC outgrowth. The cross-sectional area of cell extension beyond the initial spheroid area was quantified using maximum projections of confocal z-stack slices. The quantitative comparison revealed that the tethered (both 10 nM and 1 μM) QK peptides within the ELP gel resulted in a nearly 10-fold increase in HUVEC protrusions compared to pristine ELP gels (Fig. 6c).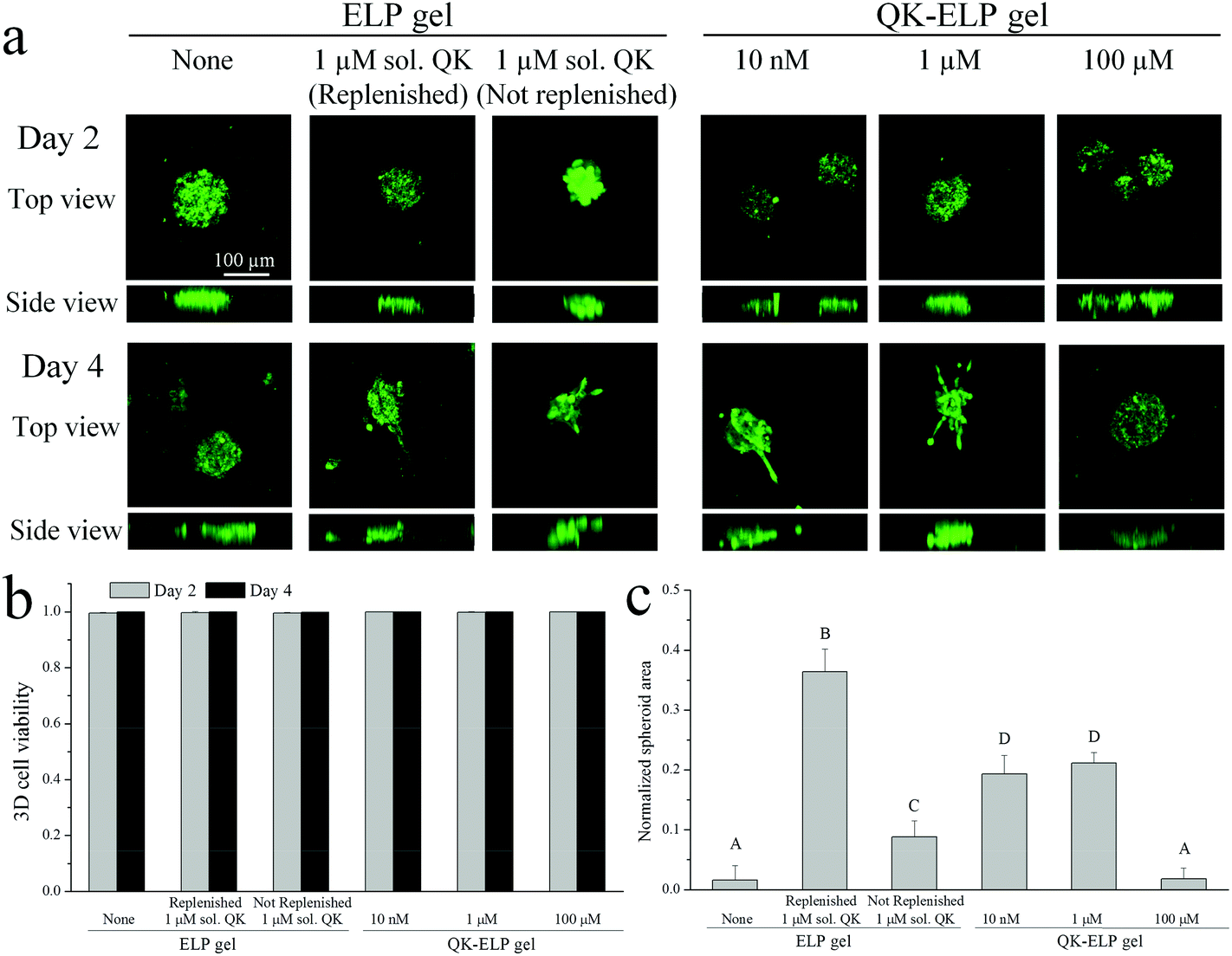 | ||
| Fig. 6 3D HUVEC spheroid encapsulation and outgrowth in ELP and QK-ELP gels. (a) Confocal fluorescence images of encapsulated HUVEC spheroids stained with live/dead markers (green/red, respectively) at 2 and 4 days post-encapsulation. Scale bar of 100 μm is applicable to all panels. Quantification of (b) cell viability on days 2 and 4, and (c) spheroid protrusion area at day 4 normalized to the initially embedded spheroid area. p < 0.05 between any two different capital letters indicated in (c). No statistically significant differences in (b). | ||
As further controls, two different conditions were analyzed using soluble QK added to pristine ELP gels. First, to maximize the bioactivity and availability of the QK peptide for use as a positive control, 1 μM of soluble QK was added to the culture at day 0 and then replenished on day 2. As expected, replenishment of the QK resulted in the highest amount of endothelial cell outgrowth (Fig. 6c). Second, to enable direct comparison with the QK-tethered gels, a single 1 μM dose of soluble QK was provided to the cultures on day 0. As shown in the diffusivity measurements above (Fig. 2a and b), soluble QK can quickly diffuse out of the ELP hydrogel due to its small size (MW = 2.139 kDa). In contrast, tethering the QK peptide to the ELP hydrogel maintains a constant local concentration (Fig. 2a and b). Endothelial outgrowth was significantly enhanced in the 1 μM tethered QK gels compared to the 1 μM soluble QK gels without replenishment (Fig. 6c). Together, these data suggest that tethering the QK peptide to the ELP hydrogel results in sustained concentrations of QK and prolonged angiogenic signaling, while soluble QK diffuses away from the site of action and escapes from the hydrogel.
Unlike 2D cell adhesion, spreading, and proliferation, cell protrusion within a 3D matrix requires local remodeling within the network to enable cell spreading and outgrowth.48,49 Therefore, to further promote HUVEC outgrowth within the hydrogels, future QK-ELP matrices could include proteolytic target sites to enhance cell-mediated matrix degradation. We previously reported the modular design of ELP sequences that contain proteolytic target sites that degrade in response to tissue plasminogen activator (tPA) or urokinase plasminogen activator (uPA) with tunable degradation rates.17 As the tPA and uPA enzymes are both secreted by endothelial cells, these modified ELP hydrogels would be ideal for further optimization of matrix properties to promote QK-induced HUVEC outgrowth.
Conclusions
As new growth-factor-mimetic peptides continue to be identified, tethering these small molecules within biomaterials will become an increasingly important method to improve the bioactivity of tissue engineering scaffolds. Compared to full-length growth factors, small peptides are simpler and more cost-effective to synthesize, have improved stability, and reduce potential immunogenic concerns. In this study, we developed a one-pot, single-step synthesis that eliminates multiple functionalization steps to conjugate QK peptides within ELP hydrogels while simultaneously encapsulating cells with near 100% viability. The efficient conjugation of QK peptides at concentrations ranging from 10 nM to 100 μM was found to not interfere with hydrogel mechanical properties, enabling a reductionist study on the dose–response of HUVECs to immobilized QK peptides. At concentrations of 10 nM and 1 μM, tethered QK was found to enhance 2D adhesion and proliferation and to stimulate more HUVEC protrusions within 3D QK-ELP hydrogels compared to single-dose, soluble QK treatment. In contrast, at a higher concentration of 100 μM, the tethered QK was found to be inhibitory to HUVEC adhesion, proliferation, and outgrowth, although it did not negatively impact cell viability within 3D cultures. This one-step conjugation technique provides a simple, quantitative, and cell-compatible way to optimize protein-based hydrogels with a broad range of bioactive peptides for customized tissue engineering applications.Acknowledgements
Cong Dinh acknowledges support from the Stanford Vice Provost for Undergraduate Education Research Experience for Undergraduates (VPUE-REU) Program. This study was supported by grants from NSF (DMR-0846363) and NIH (R01-DK085720, DP2-OD006477). The authors thank Karen Dubbin for FRAP method optimization and Kyle Lampe for advice on spheroid generation.Notes and references
- T. H. Barker, Biomaterials, 2011, 32, 4211–4214 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- X. Wang, T. C. Boire, C. Bronikowski, A. L. Zachman, S. W. Crowder and H. J. Sung, Tissue Eng., Part B, 2012, 18, 396–404 CrossRef CAS PubMed.
- S. A. DeLong, A. S. Gobin and J. L. West, J. Controlled Release, 2005, 109, 139–148 CrossRef PubMed.
- D. L. Hern and J. A. Hubbell, J. Biomed. Mater. Res., 1998, 39, 266–276 CrossRef CAS.
- D. R. Schmidt and W. J. Kao, J. Biomed. Mater. Res., Part A, 2007, 83, 617–625 CrossRef PubMed.
- C. A. Reinhart-King, M. Dembo and D. A. Hammer, Langmuir, 2002, 19, 1573–1579 CrossRef.
- L. Cai, J. Lu, V. Sheen and S. Wang, Biomacromolecules, 2012, 13, 1663–1674 CrossRef CAS PubMed.
- J. A. Burdick and K. S. Anseth, Biomaterials, 2002, 23, 4315–4323 CrossRef CAS.
- F. Yang, C. G. Williams, D. A. Wang, H. Lee, P. N. Manson and J. Elisseeff, Biomaterials, 2005, 26, 5991–5998 CrossRef PubMed.
- D. M. Brey, I. Erickson and J. A. Burdick, J. Biomed. Mater. Res., Part A, 2008, 85, 731–741 CrossRef PubMed.
- K. Saha, A. J. Keung, E. F. Irwin, Y. Li, L. Little, D. V. Schaffer and K. E. Healy, Biophys. J., 2008, 95, 4426–4438 CrossRef PubMed.
- M. Levy-Mishali, J. Zoldan and S. Levenberg, Tissue Eng., Part A, 2009, 15, 935–944 CrossRef PubMed.
- R. L. DiMarco and S. C. Heilshorn, Adv. Mater., 2012, 24, 3923–3940 CrossRef PubMed.
- K. J. Lampe and S. C. Heilshorn, Neurosci. Lett., 2012, 519, 138–146 CrossRef CAS PubMed.
- L. Cai and S. C. Heilshorn, Acta Biomater., 2014 DOI:10.1016/j.actbio.2013.1012.1028.
- K. S. Straley and S. C. Heilshorn, Soft Matter, 2009, 5, 114–124 RSC.
- C. Chung, E. Anderson, R. R. Pera, B. L. Pruitt and S. C. Heilshorn, Soft Matter, 2012, 8, 10141–10148 RSC.
- C. Chung, K. J. Lampe and S. C. Heilshorn, Biomacromolecules, 2012, 13, 3912–3916 CAS.
- K. J. Lampe, A. L. Antaris and S. C. Heilshorn, Acta Biomater., 2013, 9, 5590–5599 CrossRef CAS PubMed.
- C. Chung, B. L. Pruitt and S. C. Heilshorn, Biomater. Sci., 2013, 1, 1082–1090 RSC.
- L. Boyd and A. Carter, Eur. Spine J., 2006, 15, 414–421 CrossRef PubMed.
- M. Amiram, K. M. Luginbuhl, X. Li, M. N. Feinglos and A. Chilkoti, J. Controlled Release, 2013, 172, 144–151 CrossRef CAS PubMed.
- S. Moktan, E. Perkins, F. Kratz and D. Raucher, Mol. Cancer Ther., 2012, 11, 1547–1556 CrossRef CAS PubMed.
- J. C. Schense and J. A. Hubbell, Bioconjugate Chem., 1999, 10, 75–81 CrossRef CAS PubMed.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 65, 389–402 CrossRef CAS.
- S. E. Sakiyama-Elbert, A. Panitch and J. A. Hubbell, FASEB J., 2001, 15, 1300–1302 CAS.
- K. Vulic and M. S. Shoichet, J. Am. Chem. Soc., 2011, 134, 882–885 CrossRef PubMed.
- H. Schellekens, Clin. Ther., 2002, 24, 1720–1740 CrossRef CAS ; discussion 1719.
- S. M. Eppler, D. L. Combs, T. D. Henry, J. J. Lopez, S. G. Ellis, J. H. Yi, B. H. Annex, E. R. McCluskey and T. F. Zioncheck, Clin. Pharmacol. Ther., 2002, 72, 20–32 CrossRef CAS PubMed.
- A. B. Ennett, D. Kaigler and D. J. Mooney, J. Biomed. Mater. Res., Part A, 2006, 79, 176–184 CrossRef PubMed.
- F. Finetti, A. Basile, D. Capasso, S. Di Gaetano, R. Di Stasi, M. Pascale, C. M. Turco, M. Ziche, L. Morbidelli and L. D. D'Andrea, Biochem. Pharmacol., 2012, 84, 303–311 CrossRef CAS PubMed.
- J. E. Leslie-Barbick, J. E. Saik, D. J. Gould, M. E. Dickinson and J. L. West, Biomaterials, 2011, 32, 5782–5789 CrossRef CAS PubMed.
- G. Santulli, M. Ciccarelli, G. Palumbo, A. Campanile, G. Galasso, B. Ziaco, G. G. Altobelli, V. Cimini, F. Piscione, L. D. D'Andrea, C. Pedone, B. Trimarco and G. Iaccarino, J. Transl. Med., 2009, 7, 41 CrossRef PubMed.
- J. Y. Shu, B. Panganiban and T. Xu, Annu. Rev. Phys. Chem., 2013, 64, 631–657 CrossRef CAS PubMed.
- J. Folkman, Nat. Rev. Drug Discovery, 2007, 6, 273–286 CrossRef CAS PubMed.
- B. H. Annex, Nat. Rev. Cardiol., 2013, 10, 387–396 CrossRef CAS PubMed.
- J. S. Lee, A. J. Wagoner Johnson and W. L. Murphy, Adv. Mater., 2010, 22, 5494–5498 CrossRef CAS PubMed.
- L. D. D'Andrea, G. Iaccarino, R. Fattorusso, D. Sorriento, C. Carannante, D. Capasso, B. Trimarco and C. Pedone, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14215–14220 CrossRef PubMed.
- T. R. Chan, P. J. Stahl and S. M. Yu, Adv. Funct. Mater., 2011, 21, 4252–4262 CrossRef CAS.
- J. T. Koepsel, E. H. Nguyen and W. L. Murphy, Integr. Biol., 2012, 4, 914–924 RSC.
- P. Jonsson, M. P. Jonsson, J. O. Tegenfeldt and F. Hook, Biophys. J., 2008, 95, 5334–5348 CrossRef PubMed.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- E. A. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. A. Sulchek, T. H. Barker and A. J. Garcia, Adv. Mater., 2012, 24, 64–70 CrossRef CAS PubMed , 62.
- L. Calderon, E. Collin, D. Velasco-Bayon, M. Murphy, D. O'Halloran and A. Pandit, Eur. Cell. Mater., 2010, 20, 134–148 CAS.
- J. L. Moreau, D. Kesselman and J. P. Fisher, J. Biomed. Mater. Res., Part A, 2007, 81, 594–602 CrossRef PubMed.
- E. A. Silva and D. J. Mooney, Biomaterials, 2010, 31, 1235–1241 CrossRef CAS PubMed.
- J. Patterson and J. A. Hubbell, Biomaterials, 2010, 31, 7836–7845 CrossRef CAS PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |