Polymeric micelles with π–π conjugated moiety on glycerol dendrimer as lipophilic segments for anticancer drug delivery†
Yuanlin
Li
ab,
Ting
Su
a,
Sai
Li
*b,
Yusi
Lai
a,
Bin
He
*a and
Zhongwei
Gu
a
aNational Engineering Research Center for Biomaterials, Sichuan University, Chengdu, China. E-mail: bhe@scu.edu.cn
bSchool of Chemical Engineering, Sichuan University, Chengdu, China. E-mail: lisai@scu.edu.cn
First published on 3rd March 2014
Abstract
Polymeric micelles are important nanovehicles for anticancer drug delivery. The lipophilic segment in polymeric micelles is an important factor to affect the drug loading properties. In our previous work, we found that small molecules with π–π conjugated structures could be used to replace hydrophobic polymeric chains as lipophilic segments for anticancer drug delivery. Herein, we report a novel polymeric micelle with π–π conjugated cinnamate moiety on glycerol dendrimer as lipophilic segment, the modified dendritic segment was connected to poly(ethylene glycol) (PEG) via click chemistry. The received amphiphiles self-assembled into micelles in aqueous medium. The properties of the polymeric micelles such as critical micelle concentration (CMC), mean size and morphology were investigated. Anticancer drug doxorubicin (DOX) was loaded in the polymeric micelles. The π–π interaction, drug release profile and in vitro anticancer efficiency of the DOX loaded micelles were studied. The results showed that the micelles with more cinnamate moieties exhibited a lower CMC. The drug loading content and release rate of the micelles increased with increasing generation of glycerol dendrimer. Strong π–π stacking interaction was detected between DOX and carriers. The DOX loaded polymeric micelles exhibited efficient anticancer activity in vitro.
Introduction
In recent decades, the study of polymeric micelles for drug delivery has become a hot topic in pharmaceutics and biomaterials research due to the unique properties of polymeric micelles and their potential in optimizing the efficacy of chemotherapy.1 Polymeric micelles have typical sizes within 20 to 250 nanometres to enhance accumulation2–4 in tumor sites via the enhanced permeability and retention (EPR) effect,5,6 in which the nanoparticles are extravasated from the highly permeable blood vessels into tumor tissues and trapped there owing to the lack of lymphatic drainage.7,8Anticancer drugs such as doxorubicin (DOX), paclitaxel (PTX) and camptothecin (CPT) are widely used in cancer chemotherapy. These anticancer drugs have the same drawbacks such as poor water solubility, serious toxicity to normal tissues and inescapable multi-drug resistance.9,10 The encapsulation of hydrophobic drugs in polymeric micelles could not only improve the solubility of anticancer drugs but also reduce the side effects, meanwhile improving drug tolerance and enhancing bioavailability.11,12 In traditional polymeric micelles, most of the hydrophobic segments are biodegradable macromolecules. The drugs are trapped in the hydrophobic cores via hydrophobic interaction. Other non-covalent weak interactions including hydrogen bonds,13 π–π interactions14 and host–guest interactions15 were introduced in polymeric micelles to improve the drug loading properties.
In our previous work, we proposed a new strategy to fabricate polymeric micelles.16–20 Small molecules with π–π conjugated structures were used as lipophilic segments to replace hydrophobic polymeric chains, these small lipophilic molecules evoked additional π–π interaction as well as hydrophobic interaction between anticancer drugs and polymeric micelles to improve drug loading content and stability. The small π–π conjugated molecules were immobilized on the terminal group of PEG chains directly or through lysine linkers. DOX and 9-nitro-20(s)-camptothecin (9-NC) were encapsulated in these micelles, optimistic results in drug loading and release were received.
In this paper, glycerol dendrimers were used as linkers to connect PEG chains and small molecules with π–π conjugated structures. Cinnamate moieties were immobilized on the peripheral groups of glycerol dendrimers.21,22 Click chemistry was carried out to link PEG and cinnamate moiety modified glycerol dendrimers. The synthetic route is shown in Scheme 1. The amphiphiles self-assembled into micelles, and the size and morphology of the micelles were tested by dynamic laser scattering (DLS) and transmission electron microscopy (TEM). DOX was loaded in the micelles and the release profiles were explored. The cytotoxicity of the blank micelles and the in vitro anticancer activity of the DOX loaded micelles were investigated.
 | ||
| Scheme 1 The synthetic route of mPEG-2K-[G1]-CIN and mPEG-2K-[G2]-CIN amphiphiles. | ||
Experimental
Materials and measurements
Poly(ethylene glycol) methyl ether (Mn = 2000; mPEG-2K), Nile red, ion-exchange resin (Dowex 50W), triglycerol (a-[G2]-OH), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI), N,N-diisopropylethylamine (DIPEA), doxorubicin hydrochloride (DOX·HCl) and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich. Sodium azide (NaN3), methanesulfonyl chloride (MsCl), cinnamic acid (CIN), sodium ascorbate, triethylamine (TEA), 2,2-dimethoxypropane, copper sulfate pentahydrate (CuSO4·5H2O), bicarbonate (NaHCO3), p-toluenesulfonic acid monohydrate (PTSA), tetrabutylammonium iodide (TBAI), DL-1,2-isopropylideneglycerol (b-[G1]-OH), propargyl bromide (80 wt% in toluene), ammonium chloride (NH4Cl), sodium hydride (60 wt% in mineral oil; NaH) and sodium sulfate (Na2SO4) were purchased from Aladdin (Shanghai, China) and used as received. N,N-Dimethylformamide (DMF), methanol, dichloromethane (DCM), tetrahydrofuran (THF), diethyl ether, n-hexane, ethyl acetate (EA), petroleum ether (PE) and dimethyl sulfoxide (DMSO) were purchased from Kelong Chemical Co. (Chengdu, China). All the solvents were purified before use.The 1H NMR spectra were recorded on a Bruker AVANCE-400 MHz NMR spectrometer at 25 °C using CDCl3 as solvent and the chemical shift was reported in ppm on the δ scale. Fourier transform infrared (FTIR) spectra were recorded on a Thermo Scientific Nicolet iS10 spectrophotometer. The mass spectra (MS) of the amphiphiles were recorded on a MALDI-TOF spectrometer (Bruker, autoflex III smartbeam). The differential scanning calorimetry (DSC) analysis was performed on a Q2000 (TA Instruments) under nitrogen atmosphere. All samples were firstly heated to 100 °C with a heating rate of 10 °C min−1 and held at 100 °C for 5 minutes to erase the thermal history, then the samples were cooled to −80 °C with a cooling rate of 10 °C min−1 and held at −80 °C for 5 minutes. The samples were finally heated to 100 °C with a heating rate of 10 °C min−1. UV measurements were carried out on a UV-vis spectrometer (Perkin-Elmer) at 25 °C. Fluorescence measurements were performed on a fluorescence detector (F700, HITACHI, Japan). Dynamic laser light scattering (DLS) measurements were carried out on a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK) at 25 °C. Transmission electron microscopy (TEM) images were performed on a JEM-100CX (JEOL) transmission electron microscope with the samples stained by phosphotungstic acid on a carbon-coated copper grid. In the cell biology experiment, the absorbance was detected with a Thermo scientific MK3 (Thermo Fisher, USA) at the wavelength of 450 nm. The confocal laser scanning microscopy (CLSM) tests were performed on Leica TCS SP5 with the excitation at 485 nm. For the flow cytometry measurements, the fluorescence intensity was measured (excitation: 480 nm; emission: 590 nm) on a BD FACS Calibur flow cytometer (Beckton Dickinson).
Synthesis of b-[G2]-OH
Triglycerol (7.22 g, 30 mmol) was heated to 60 °C under vacuum in an oil bath and kept for 5 h to remove the water in triglycerol. After cooling to room temperature, 10.0 mL of 2,2-dimethoxypropane was added under nitrogen atmosphere. A solution of PTSA (570.66 mg, 3 mmol) in 5 mL of 2,2-dimethoxypropane was added dropwise into the mixture. The reaction was carried out at 35 °C overnight and a yellow-orange solution resulted. The solution was neutralized by the addition of TEA (420 μL, 3 mmol) and subsequently stirred at room temperature for 30 min.23 The solvent was evaporated in vacuum and the remaining crude liquid was purified over a silica column (200–300 mesh; EA–n-hexane 1/2, 2/1, 6/1) to give b-[G2.0]-OH as a pale yellow oil (5.77 g, 60% yield).Synthesis of mPEG-2K-N3
Dried mPEG-2K (5.00 g, 2.5 mmol) was dissolved in 25 mL of anhydrous DCM. TEA (1.80 mL, 12.9 mmol) was added in the solution. The mixture was put in an ice-water bath with stirring. MsCl (1.00 mL, 12.9 mmol) diluted with 15 mL of anhydrous DCM was added dropwise. The reaction was kept at room temperature for 24 h. After concentration, the solution was precipitated in diethyl ether, filtrated and dried in vacuum. The dried product was dissolved in 60 mL of DMF, NaN3 (812.6 mg, 12.5 mmol) was added in the solution. The solution was stirred at 80 °C under nitrogen atmosphere for 24 h. After the DMF was evaporated, 20 mL of DCM was added. The solution was filtered to remove the excess NaN3, precipitated in 100 mL of diethyl ether twice and dried in vacuum. The resultant mPEG-2K-N3 was a white powder (4.53 g, 88.7% yield).Synthesis of b-[G1]-alkynyl
Propargyl bromide (5.95 g, 40 mmol) and TBAI (203.2 mg, 0.55 mmol) were added to b-[G1]-OH (660.8 mg, 5 mmol) in 30 mL of fresh distilled THF. When the mixture was cooled to 0 °C in an ice-water bath, NaH (800.0 mg, 20 mmol) was added slowly. The reaction was carried out at room temperature overnight. 80 mL of distilled water was added and the solvent was evaporated. The residue was extracted with EA (3 × 50 mL), washed with 50 mL of distilled water, dried with anhydrous Na2SO4 and filtered.24 The solution was condensed and purified through column chromatography (silica gel 200–300 mesh; PE–EA 8/1, 7/1). The product b-[G1]-alkynyl was a light yellow oil (731.9 mg, 86% yield).Synthesis of b-[G2]-alkynyl
Propargyl bromide (3.81 g, 32 mmol, 80 wt% in toluene) and TBAI (162.5 mg, 0.44 mmol) were added to b-[G2]-OH (1.28 g, 4 mmol) in 25 mL of fresh distilled THF. The mixture was cooled to 0 °C in an ice-water bath. NaH (640.0 mg, 16 mmol) was added slowly. The reaction was carried out at room temperature overnight. 80 mL of distilled water was added and the solvent was evaporated. The residue was extracted with EA (3 × 50 mL), washed with 50 mL of water, dried with anhydrous Na2SO4 and filtered. The solvent was evaporated and the product was purified through column chromatography (silica gel 200–300 mesh, PE–EA 8/1, 7/1). The resulting b-[G2]-alkynyl was a yellow oil (1.18 g, 82% yield).Synthesis of CIN-[G1]-alkynyl
Ion-exchange resin (510.6 mg) was added to b-[G1]-alkynyl (510.6 mg, 3 mmol) dissolved in 10 mL of methanol. The mixture was refluxed for 12 h. The ion-exchange resin was filtrated and the solvent was evaporated to give a-[G1]-alkynyl. The a-[G1]-alkynyl, CIN (982.8 mg, 6.6 mmol) and DMAP (185.1 mg, 1.5 mmol) were dissolved in 10 mL of distilled DCM. EDCI (1.27 g, 6.6 mmol) dissolved in 10 mL of dry DCM was added slowly into the solution in an ice-water bath. The mixture was stirred at room temperature for 24 h under nitrogen atmosphere. The mixture was diluted with DCM to 50 mL, washed with saturated solutions of NaHCO3 (3 × 50 mL) and NH4Cl (3 × 50 mL). The solution was dried with anhydrous Na2SO4, filtered and purified on a silica gel column (silica gel 200–300 mesh, DCM–methanol 10/1). The resultant CIN-[G1]-alkynyl was a yellow viscous solid (1.09 g, 93% yield).Synthesis of CIN-[G2]-alkynyl
Ion-exchange resin (716.9 mg) was added to b-[G2]-alkynyl (716.9 mg, 2 mmol) dissolved in 10 mL of methanol. The mixture was refluxed for 12 h. The ion-exchange resin was filtrated and the solvent was evaporated to give a-[G2]-alkynyl. The a-[G2]-alkynyl, CIN (1.31 g, 8.8 mmol) and DMAP (123.4 mg, 1.5 mmol) were dissolved in 10 mL of distilled DCM. EDCI (1.71 g, 8.8 mmol) dissolved in 10 mL of dry DCM was added slowly into the solution in an ice-water bath. The mixture was stirred at room temperature for 24 h under nitrogen atmosphere. The mixture was diluted with DCM to 50 mL, washed with saturated solutions of NaHCO3 (3 × 50 mL) and NH4Cl (3 × 50 mL). The solution was dried with anhydrous Na2SO4, filtered and purified on a silica gel column (silica gel 200–300 mesh, DCM–methanol 10/1). The resultant CIN-[G2]-alkynyl was a yellow viscous solid (2.83 g, 86% yield).Synthesis of mPEG-2K-[G1]-CIN
CIN-[G1]-alkynyl (117.1 mg, 0.3 mmol) and mPEG-2K-N3 (582.0 mg, 0.285 mmol) were dissolved in 3 mL of THF. DIPEA (15 μL, 0.09 mmol) was added. After the mixture became a homogeneous solution, an aqueous solution of sodium ascorbate (18.00 mg, 0.09 mmol in 1.8 mL of water) and CuSO4·5H2O (12.00 mg, 0.048 mmol in 1.2 mL of water) were added to the solution. The mixture was stirred vigorously. The reaction was monitored by thin layer chromatography (TLC) analysis (DCM–methanol 10/1). The mixture was diluted with 20 mL of distilled water and extracted with DCM (3 × 20 mL). The combined organic layer was dried with Na2SO4, concentrated and precipitated in diethyl ether. The precipitate was dialyzed (MWCO = 1000) against distilled water for 24 h. The white powder of mPEG-2K-[G1]-CIN was received after lyophilization (595.2 mg, 83% yield).Synthesis of mPEG-2K-[G2]-CIN
CIN-[G2]-alkynyl (159.8 mg, 0.2 mmol) and mPEG-2K-N3 (388.0 mg, 0.19 mmol) were dissolved in 2 mL of THF. DIPEA (10 μL, 0.06 mmol) was added after the mixture became a homogeneous solution, an aqueous solution of sodium ascorbate (12.00 mg, 0.06 mmol in 1.2 mL of water) and CuSO4·5H2O (8.00 mg, 0.032 mmol in 0.8 mL of water) were added to the solution. The mixture was stirred vigorously. The reaction was monitored via thin layer chromatography (TLC) analysis (DCM–methanol 10/1). The mixture was diluted with 20 mL of distilled water and extracted with DCM (3 × 20 mL). The combined organic layer was dried with Na2SO4, concentrated and precipitated in diethyl ether. The precipitate was dialyzed (MWCO = 1000) against distilled water for 24 h. The white powder of mPEG-2K-[G2]-CIN was received after lyophilization (425.4 mg, 76% yield).Critical micelle concentration (CMC) measurement
Nile red was used as a fluorescence probe to measure the critical micelle concentration (CMC) of the amphiphilic polymers in aqueous medium.23 The polymer solutions (1.5 mL) with concentrations ranging from 1 × 10−4 to 1 mg mL−1 were stirred with 20 μL of Nile red (1 mg mL−1) solution in THF for 24 h at room temperature. The absorbance at wavelength 662 nm was measured by fluorescence measurements with the excitation at 550 nm. The CMC calculated from the scatter plot of the fluorescence intensity corresponded to the concentration.Deprotonation of DOX·HCl
DOX·HCl (2 mg mL−1) was dissolved in deionized water. The pH value was slowly adjusted to 9.6 with addition of NaOH (1 M) aqueous solution in an ice-water bath. The mixture was centrifugated (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 r min−1 for 8 min) and washed with deionized water.25 The product was freeze-dried to receive doxorubicin. Each step in the procedure was performed in the dark.
000 r min−1 for 8 min) and washed with deionized water.25 The product was freeze-dried to receive doxorubicin. Each step in the procedure was performed in the dark.
Preparation of DOX loaded micelles
The solution of amphiphilic polymer (10 mg) and DOX (2.5 mg) in DMSO (1 mL) was added dropwise to distilled water (10 mL) under stirring. The mixture was transferred into dialysis tubing (MWCO = 1000) and dialyzed against deionized water at 4 °C for 12 h. The solution was centrifugated and freeze-dried to give DOX loaded micelles. The whole procedure was performed in the dark.Determination of drug loading content (DLC)
The content of DOX in the micelles was determined by UV measurement (maximum absorption wavelength at 480 nm) with the calibration curve of DOX–DMSO solution. Drug loading content (DLC) and drug encapsulation efficiency (DEE) were calculated according to the following formula:| DLC (wt%) = (weight of loaded drug/weight of drug loaded micelle) × 100% |
| DEE (%) = (weight of loaded drug/weight of drug in feeding) × 100% |
In vitro drug release
A certain amount of DOX loaded micelles was dispersed in 1 mL of phosphate buffered saline solution (pH = 7.4) and transferred into a dialysis tubing (MWCO = 1000). The dialysis tubing was immersed in 25 mL of PBS (pH = 7.4) and kept in a horizontal shaker at 37 °C with 170 rpm. 1 mL of the medium was removed at different time points and the same volume of fresh PBS was added. The released DOX was measured by a fluorescence detector with the excitation wavelength at 485 nm.Cell culture
NIH 3T3 fibroblasts and HepG2 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 IU mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified atmosphere with 5% CO2. The cells were harvested with 0.02% EDTA and 0.025% trypsin and rinsed.The cytotoxicity of the blank micelles
The cytotoxicity of the blank micelles was tested by Kit-8 assay (CCK-8, Dojindo, Japan) against NIH 3T3 fibroblasts. NIH 3T3 fibroblasts were seeded in 96-well plates at a density of 5 × 103 cells per well with 100 μL of DMEM. After 24 h incubation, the culture medium was removed and replaced with 100 μL of medium containing blank micelles. The cells were incubated for another 48 h. The culture medium was removed and the wells were rinsed with PBS (pH = 7.4). 100 μL of CCK-8 (volume fraction 10%) solution in DMEM was added to each well. The absorbance was measured after 2 h incubation.Cellular uptake
Confocal laser scanning microscopy (CLSM) was employed to examine the cellular uptake of DOX loaded micelles. 2 × 105 HepG2 cells in a logarithm phase in 200 μL of DMEM were seeded on 35 mm diameter glass dishes. After 24 h incubation, the culture medium was removed. DOX·HCl and DOX loaded micelles were dissolved in DMEM (DOX concentration of 10 μg mL−1), 200 μL of the solution was added in each dish. After incubation for 1, 2 and 6 h, the culture medium was removed and the dishes were rinsed with PBS (pH = 7.4).Flow cytometry measurements
HepG2 cells were seeded in 6-well plates at a density of 1 × 106 cells per well and incubated for 24 h. The cells were treated with drug loaded micelles at the same DOX concentration (10 μg mL−1) for 1 and 3 h, respectively. The culture medium was removed, the cells were washed with PBS three times and harvested with trypsinization. The cells were resuspended in PBS after centrifugation (1000 rpm, 5 min) and the fluorescence intensity was measured (excitation: 480 nm; emission: 590 nm).In vitro anticancer activity
HepG2 cells were seeded in 96-well plates at a density of 4 × 103 cells per well with 100 μL of DMEM. After 24 h incubation, the culture medium was removed and replaced with 100 μL of DMEM containing DOX·HCl and DOX loaded micelles with the same DOX concentration (10 μg mL−1). After incubation for 72 h, the culture medium was removed and the wells were rinsed with PBS (pH = 7.4). 100 μL of CCK-8 (volume fraction 10%) solution in DMEM was added to each well. The absorbance was measured after 2 h incubation.Results and discussion
The 1H NMR spectra of the intermediate and final products are presented in Fig. S1 in the ESI† and Fig. 1, respectively. In the spectra of A1 and A2, the signals at δ = 2.45 ppm and 4.22 ppm were assigned to the acetylene proton (i1 and i2) and the methylene protons (h1 and h2) adjacent to the alkyne group, respectively. The signals of the methyl protons (f1 and f2) in the acetal protection moiety were split and appeared at δ = 1.37 ppm and 1.44 ppm. It indicated the successful alkynylation of b-[G1]-OH and b-[G2]-OH as well as the preservation of an intact hydroxyl group with protection. The proton signals of propinyl were not changed and the signals of the methyl protons (f1 and f2) disappeared when comparing the spectra of A1 to B1 and A2 to B2 (Fig. S1†). Meanwhile, the chemical shifts of the proton signals in the cinnamate moieties were changed. The signals at δ = 6.48 ppm (j1 and j2) and 7.72 ppm (k1 and k2) were attributed to the protons of the double bond in the cinnamate moieties, the signal at δ = 7.52 ppm was assigned to the protons (m1 and m2) in the meta-position of the benzene ring of the cinnamate moieties, the proton signals (o1 + p1 and o2 + p2) of the ortho- and para-position appeared at δ = 7.39 ppm. This suggested that the predesigned molecules of CIN-[G1]-alkynyl and CIN-[G2]-alkynyl were successfully synthesized. | ||
| Fig. 1 The 1H NMR spectra of the mPEG-2K-[G1]-CIN-DOX (C1) and mPEG-2K-[G2]-CIN-DOX (C2) amphiphiles. | ||
In the mPEG-2K-[G1]-CIN and mPEG-2K-[G2]-CIN spectra (Fig. 1), new protons of PEG appeared. The signals from δ = 3.65 ppm to 3.47 ppm were attributed to the methylene protons (r1 and r2) of mPEG-2K, the methylene in the first repeat unit of mPEG adjoined to the azide group was especially identifiable at δ = 3.83 ppm (s1 and s2) and 4.50 ppm (t1 and t2), the terminal methoxy protons (q1 and q2) of mPEG appeared at δ = 3.38 ppm. Moreover, the protons (i′′1 and i′′2) in the triazole ring displayed a peak at δ = 7.73 ppm and the peak for the methylene protons (h′′1 and h′′2) within the triazole ring appeared at δ = 4.78 ppm, presenting a downfield shift to the peaks of the alkynyl protons (i′1 and i′2) and methylene protons (h′1 and h′2) adjacent to the alkyne. These results confirmed that a successful click reaction between the alkyne and azide was fulfilled and the triazole linkage was formed.
The click reaction was also detected by FTIR (Fig. S2 in the ESI†). The characteristic vibration of the azide group at around 2200 cm−1 wavenumber disappeared after the click reaction, which implied the success of the click reaction. The mPEG-2K-[G1]-CIN and mPEG-2K-[G2]-CIN were further confirmed by MS (Fig. S3 in the ESI†), the molecular weight variation in the MS spectra revealed the successful synthesis of the amphiphiles.
The thermal property of the two amphiphilic polymers were tested by DSC, both the cold crystallization and crystal melting processes were presented in Fig. S4 in the ESI.† As the glycerol dendrimer was amorphous, the immobilization of the dendritic segments on the terminal group of PEG via the click reaction reduced the crystallization capability of the PEG segments.26 When the cinnamate moieties in the dendritic segments increased from two in mPEG-2K-[G1]-CIN to four in mPEG-2K-[G2]-CIN, the melting temperature (Tm) of the PEG crystal in the polymers decreased from 51.5 °C to 48.9 °C. The ΔHm of the two amphiphiles was lower than that of mPEG-2K (166.2 J g−1), the ΔHms of the amphiphilic copolymers were 145.4 J g−1 for mPEG-2K-[G1]-CIN and 148.4 J g−1 for mPEG-2K-[G2]-CIN. This result seemed contradictory to the variation of Tm. However, it is reasonable while considering the glycerol dendrimer linkers. The flexibility of the dendritic glycerol linkers was very high, which enhanced the mobility of the PEG segments to decrease the Tm of the copolymer. However, the large stereo hindrance of the cinnamate moieties limited the movement of the PEG chains within certain domains, thus, resulting in the increase of the ΔHm of mPEG-2K-[G2]-CIN.
Critical micelle concentration (CMC) is an important parameter to polymeric micelles. The CMCs of both mPEG-2K-[G1]-CIN and mPEG-2K-[G2]-CIN micelles were tested using Nile red as fluorescence probe (Fig. S5 in the ESI†). The calculated CMCs of mPEG-2K-[G1]-CIN and mPEG-2K-[G2]-CIN micelles were 49 μg mL−1 and 28 μg mL−1, respectively (Table 1). The lower CMC of the mPEG-2K-[G2]-CIN polymeric micelles suggested that they were more stable than the mPEG-2K-[G1]-CIN micelles in aqueous medium. The results demonstrated that more cinnamate moieties in the lipophilic segments could stabilize the polymeric micelles due to the increase of hydrophobicity, and this conclusion is consistent with the regular rule of CMC in polymeric micelles.27
The sizes of both the mPEG-2K-[G1]-CIN and mPEG-2K-[G1]-CIN micelles were measured by DLS (Fig. S6 in the ESI†), the tested results were summarised in Table 1. The mean sizes of the two micelles were 81 and 42 nm, respectively. The mPEG-2K-[G1]-CIN micelles exhibited larger mean size. The TEM images of the micelles in Fig. 2 shows that the micelles were spherical nanoparticles and the sizes are consistent with the DLS results, which were about 80 nm for mPEG-2K-[G1]-CIN and 40 nm for the mPEG-2K-[G2]-CIN micelles. The size of the mPEG-2K-[G2]-CIN micelles was smaller than that of the mPEG-2K-[G1]-CIN micelles. This is attributed to more cinnamate moieties in mPEG-2K-[G2]-CIN micelles with stronger hydrophobic interactions as well as π–π interactions to compact the hydrophobic cores in the self-assembly.
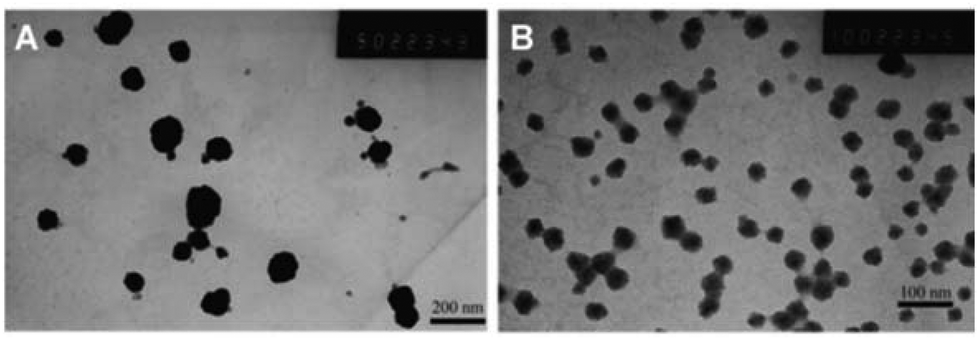 | ||
| Fig. 2 TEM photographs of the mPEG-2K-[G1]-CIN (A) and mPEG-2K-[G2]-CIN (B) micelles. | ||
The anticancer drug doxorubicin was used to study the drug loading behaviour of the micelles. The measured drug loading contents of the mPEG-2K-[G1]-CIN and mPEG-2K-[G2]-CIN micelles were 7.5 wt% and 15.7 wt%, respectively (Table 1). The corresponding encapsulation efficiencies were 55% and 64%. The mPEG-2K-[G2]-CIN micelles exhibited better drug loading efficacy.
The π–π interaction within the drug loaded micelles was characterized by UV-vis (Fig. 3A) and fluorescence spectra (Fig. 3B). The main absorbance of DOX is displayed at 482 nm. The DOX absorbance peak appeared at 506 nm for the micelles, the red shift, which indicates the π–π stacking interaction between DOX and the carriers, was observed within the DOX loaded micelles. In the fluorescence spectra of the DOX loaded micelles, drastic fluorescence quenching of DOX in the micelles suggested that a strong π–π stacking interaction was formed between DOX and the cinnamate moieties.28
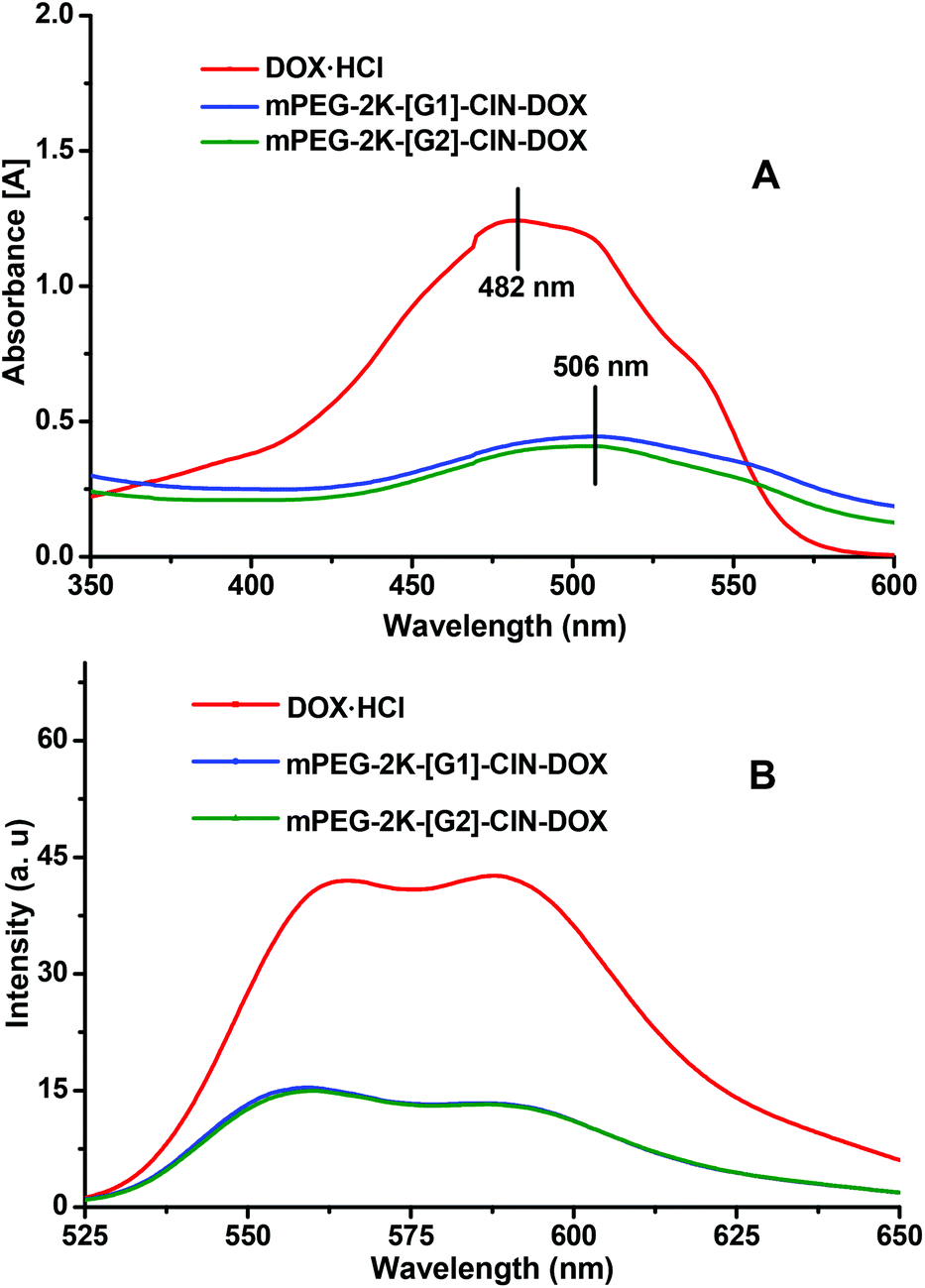 | ||
| Fig. 3 The UV-vis (A) and fluorescence (B) spectra of the DOX loaded micelles. | ||
The in vitro DOX release profiles of the drug loaded micelles are presented in Fig. 4. The cumulated release of DOX was 60% for mPEG-2K-[G1]-CIN-DOX and 70% for mPEG-2K-[G2]-CIN-DOX within 72 h. The DOX release rate of mPEG-2K-[G2]-CIN micelles was faster than that of mPEG-2K-[G1]-CIN-DOX. This is probably attributable to the relatively higher drug loading content, the higher concentration of DOX in mPEG-2K-[G2]-CIN micelles leads to the faster diffusion of DOX from the hydrophobic cores to the medium.
 | ||
| Fig. 4 Release profiles of DOX loaded mPEG-2K-[G1]-CIN-DOX and mPEG-2K-[G2]-CIN-DOX micelles in PBS (pH = 7.4) at 37 °C (n = 3). | ||
The toxicity of the blank micelles was evaluated. Fig. 5 shows the cell viability of NIH 3T3 fibroblasts incubated with the two blank micelles for 48 h. When the concentration of the blank micelles was 80 μg mL−1, the cell viability was nearly 100%, however, when the concentration was 200 μg mL−1, the cell viability was around 80%. There is nearly no difference in the cell viability between the two micelles at the same concentration, implying that the number of cinnamate moieties in the amphiphiles did not affect the cytotoxicity of the micelles.29 As 200 μg mL−1 is a very high concentration for future application in vitro and/or in vivo, it can be concluded that the micelles are suitable carrier candidates for drug delivery.
 | ||
| Fig. 5 The cytotoxicity of blank micelles against NIH 3T3 fibroblasts after 48 h incubation. | ||
Confocal laser scanning microscopy (CLSM) was used to examine the cellular uptake of drug loaded micelles (Fig. 6). DOX·HCl was used as control. As a water soluble drug, the internalization of DOX·HCl was fast, and a strong red florescence was observed both in the cytoplasm and nuclei of the HepG2 cells after 1 h incubation (C1 in Fig. 6). Red florescence was also observed in the cells treated with mPEG-2K-[G1]-CIN-DOX and mPEG-2K-[G2]-CIN-DOX micelles, however, nearly all the red florescence was located in the cytoplasm of the HepG2 cells (A1 and B1 in Fig. 6). The red florescence of DOX in the cells increased with the increase in incubation time.
 | ||
| Fig. 6 The laser confocal scanning microscopic images (CLSM) of the HepG2 cells incubated with DOX loaded micelles. A, B and C were mPEG-2K-[G1]-CIN-DOX, mPEG-2K-[G2]-CIN-DOX and DOX·HCl. 1, 2 and 3 represent the incubation times of 1, 3 and 6 h. The scale bar is 25 μm in all the images. | ||
Flow cytometry was used for the quantitative evaluation of the cellular internalization of the DOX loaded micelles (Fig. 7). The fluorescence intensity of both the DOX loaded micelles was strengthened when the incubation time was elongated from 1 h to 3 h. The mPEG-2K-[G1]-CIN-DOX and mPEG-2K-[G2]-CIN-DOX micelles showed similar cellular uptake, the fluorescence intensity of the two drug loaded micelles was comparative for both 1 and 3 h incubation.
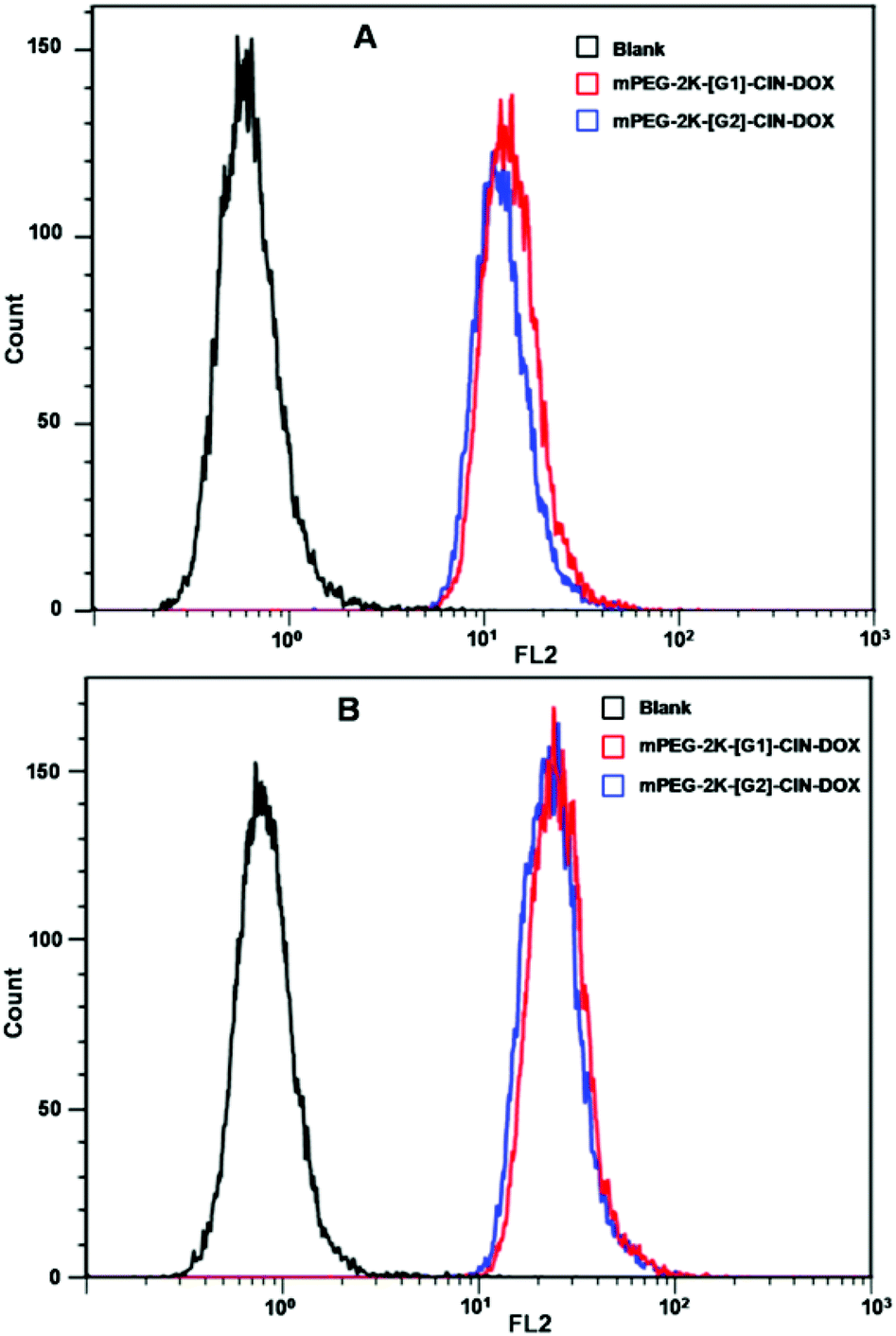 | ||
| Fig. 7 Fluorescence intensities of the flow cytometry results of drug loaded mPEG-2K-[G1]-CIN and mPEG-2K-[G2]-CIN micelles incubated with HepG2 cells, A: incubated for 1 h; B: incubated for 2 h. | ||
The drug loaded micelles and HepG2 cells were incubated to investigate the in vitro anticancer activity. As free DOX was hydrophobic and precipitated in the cell culture medium during the incubation, it could not be used as a control, thus, water soluble DOX·HCl was used as a control though it is not a perfect one. The in vitro anticancer activity of mPEG-2K-[G1]-CIN-DOX and mPEG-2K-[G2]-CIN-DOX micelles were tested, the half maximal inhibitory concentration (IC50) of the two drug loaded micelles were 0.2 and 0.15 μg mL−1, respectively (Fig. 8). The lower IC50 of the mPEG-2K-[G2]-CIN-DOX micelles indicated that they had better anticancer activity. This is probably due to the higher drug loading content and faster release rate, which maintains a relatively high DOX concentration in HepG2 cells to kill cells more efficiently. As the internalization of DOX·HCl via diffusion was much faster than that of drug loaded micelles via endocytosis, the IC50 of DOX·HCl was much lower than that of the DOX loaded micelles.
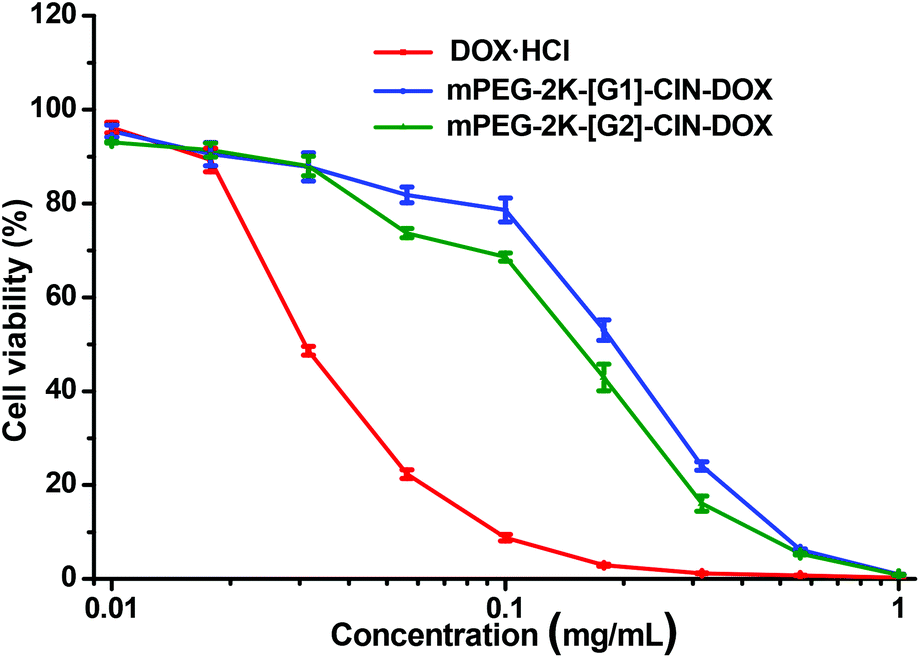 | ||
| Fig. 8 In vitro anticancer activity of the DOX loaded micelles against HepG2 cells for 72 h incubation. | ||
Conclusions
Amphiphilic polymers with π–π conjugated cinnamate moieties linked to glycerol dendrimers as lipophilic segments and PEG chains as hydrophilic segments were synthesized via click chemistry. The amphiphiles self-assembled into polymeric micelles in aqueous medium. The anticancer drug doxorubicin was encapsulated in the polymeric micelles. The effects of the generation of the glycerol dendrimer on the size, CMC, drug loading property, release profile and in vitro anticancer activity of the drug loaded micelles were investigated. The size and CMC of the mPEG-2K-[G2]-CIN micelles were lower than that of the mPEG-2K-[G1]-CIN micelles. The drug loading contents of the mPEG-2K-[G2]-CIN and mPEG-2K-[G1]-CIN micelles were 15.7 wt% and 7.5 wt%, respectively. The DOX released from the mPEG-2K-[G2]-CIN micelles was faster than that from the mPEG-2K-[G1]-CIN micelles. The cellular uptake of the two drug loaded micelles was comparative to HepG2 cells. The IC50 of the mPEG-2K-[G2]-CIN-DOX micelles was 0.15 μg mL−1, lower than that of the mPEG-2K-[G1]-CIN-DOX micelles. The DOX loaded mPEG-2K-[G2]-CIN micelles exhibited better anticancer activity in vitro.Acknowledgements
This research work was supported by the National Science Foundation for Excellent Young Scholars (no. 51222304), National Basic Research Program of China (National 973 program, no. 2011CB606206), National Science Foundation of China (NSFC, no. 31170921, 51133004) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1163).Notes and references
- M. Huo, A. Zou, C. Yao, Y. Zhang, J. Zhou, J. Wang, Q. Zhu, J. Li and Q. Zhang, Biomaterials, 2012, 33, 6393–6407 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS PubMed.
- L. L. Ma, P. Jie and S. S. Venkatraman, Adv. Funct. Mater., 2008, 18, 716–725 CrossRef CAS.
- Z. Zhu, C. Xie, Q. Liu, X. Zhen, X. Zheng, W. Wu, R. Li, Y. Ding, X. Jiang and B. Liu, Biomaterials, 2011, 32, 9525–9535 CrossRef CAS PubMed.
- S. Keereweer, I. M. Mol, J. D. F. Kerrebijn, P. B. A. A. Van Driel, B. Xie, R. J. Baatenburg de Jong, A. L. Vahrmeijer and C. W. G. M. Löwik, J. Surg. Oncol., 2012, 105, 714–718 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- R. B. Campbell, S. V. Balasubramanian and R. M. Straubinger, J. Pharm. Sci., 2001, 90, 1091–1105 CrossRef CAS PubMed.
- A. Sharma, E. Mayhew, L. Bolcsak, C. Cavanaugh, P. Harmon, A. Janoff and R. J. Bernacki, Int. J. Cancer, 1997, 71, 103–107 CrossRef CAS.
- X. Xu, H. Yuan, J. Chang, B. He and Z. Gu, Angew. Chem., Int. Ed., 2012, 51, 3130–3133 CrossRef CAS PubMed.
- T. Ogoshi, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2007, 129, 4878–4879 CrossRef CAS PubMed.
- C. Tu, L. Zhu, P. Li, Y. Chen, Y. Su, D. Yan, X. Zhu and G. Zhou, Chem. Commun., 2011, 47, 6063–6065 RSC.
- X. Deng, X. Xu, Y. Lai, B. He and Z. Gu, J. Biomed. Nanotechnol., 2013, 9, 1336–1344 CrossRef CAS PubMed.
- Y. Lai, Y. Lei, X. Xu, Y. Li, B. He and Z. Gu, J. Mater. Chem. B, 2013, 1, 4289–4296 RSC.
- Y. Lei, Y. Lai, Y. Li, S. Li, G. Cheng, D. Li, H. Li, B. He and Z. Gu, Int. J. Pharm., 2013, 453, 579–586 CrossRef CAS PubMed.
- D. Li, Y. Liang, Y. Lai, G. Wang, B. He and Z. Gu, Colloids Surf., B DOI:10.1016/j.colsurfb.2013.10.032.
- Y. Liang, Y. Lai, D. Li, B. He and Z. Gu, Mater. Lett., 2013, 97, 4–7 CrossRef CAS PubMed.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- A. L. Sisson, D. Steinhilber, T. Rossow, P. Welker, K. Licha and R. Haag, Angew. Chem., Int. Ed., 2009, 48, 7540–7545 CrossRef CAS PubMed.
- M. Wyszogrodzka and R. Haag, Chem.–Eur. J., 2008, 14, 9202–9214 CrossRef CAS PubMed.
- S. L. Elmer, S. Man and S. C. Zimmerman, Eur. J. Org. Chem., 2008, 3845–3851 CrossRef CAS PubMed.
- Y. Lai, Y. Long, Y. Lei, X. Deng, B. He, M. Sheng, M. Li and Z. Gu, J. Drug Targeting, 2012, 20, 246–254 CrossRef CAS PubMed.
- R. Liu, B. He, D. Li, Y. Lai, J. Z. Tang and Z. Gu, Polymer, 2012, 53, 1473–1482 CrossRef CAS PubMed.
- J. Gao, J. Ming, B. He, Y. Fan, Z. Gu and X. Zhang, Eur. J. Pharm. Sci., 2008, 34, 85–93 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- X. Zhang, L. Meng, Q. Lu, Z. Fei and P. J. Dyson, Biomaterials, 2009, 30, 6041–6047 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60267b |
| This journal is © The Royal Society of Chemistry 2014 |