Biomaterial arrays with defined adhesion ligand densities and matrix stiffness identify distinct phenotypes for tumorigenic and non-tumorigenic human mesenchymal cell types
Tyler D.
Hansen†
a,
Justin T.
Koepsel†
a,
Ngoc Nhi
Le
b,
Eric H.
Nguyen
a,
Stefan
Zorn
a,
Matthew
Parlato
a,
Samuel G.
Loveland
a,
Michael P.
Schwartz
*a and
William L.
Murphy
*abc
aDepartment of Biomedical Engineering, University of Wisconsin-Madison, Madison, WI, USA. E-mail: wlmurphy@wisc.edu; mpschwartz@wisc.edu
bMaterials Science Program, University of Wisconsin-Madison, Madison, WI, USA
cDepartment of Orthopedics and Rehabilitation, University of Wisconsin-Madison, Madison, WI, USA
First published on 22nd January 2014
Abstract
Here, we aimed to investigate migration of a model tumor cell line (HT-1080 fibrosarcoma cells, HT-1080s) using synthetic biomaterials to systematically vary peptide ligand density and substrate stiffness. A range of substrate elastic moduli were investigated by using poly(ethylene glycol) (PEG) hydrogel arrays (0.34–17 kPa) and self-assembled monolayer (SAM) arrays (∼0.1–1 GPa), while cell adhesion was tuned by varying the presentation of Arg-Gly-Asp (RGD)-containing peptides. HT-1080 motility was insensitive to cell adhesion ligand density on RGD-SAMs, as they migrated with similar speed and directionality for a wide range of RGD densities (0.2–5% mol fraction RGD). Similarly, HT-1080 migration speed was weakly dependent on adhesion on 0.34 kPa PEG surfaces. On 13 kPa surfaces, a sharp initial increase in cell speed was observed at low RGD concentration, with no further changes observed as RGD concentration was increased further. An increase in cell speed ∼two-fold for the 13 kPa relative to the 0.34 kPa PEG surface suggested an important role for substrate stiffness in mediating motility, which was confirmed for HT-1080s migrating on variable modulus PEG hydrogels with constant RGD concentration. Notably, despite ∼two-fold changes in cell speed over a wide range of moduli, HT-1080s adopted rounded morphologies on all surfaces investigated, which contrasted with well spread primary human mesenchymal stem cells (hMSCs). Taken together, our results demonstrate that HT-1080s are morphologically distinct from primary mesenchymal cells (hMSCs) and migrate with minimal dependence on cell adhesion for surfaces within a wide range of moduli, whereas motility is strongly influenced by matrix mechanical properties.
Introduction
Tumor cells have been characterized by “plasticity”, or the capacity to transition between mesenchymal (elongated) or amoeboid (rounded) migration mechanisms thought to be analogous to normal physiological function for motile cell types such as fibroblasts or immune cells.1–3 However, transformation to an aggressive tumorigenic phenotype alters many of the distinguishing features for cell motility, including cytoskeletal structure and cell adhesion properties.4–11 Direct comparisons between tumorigenic and non-tumorigenic cell types – thought to utilize analogous migration modes – have revealed important differences in function.12–16 For example, while HT-1080 fibrosarcoma cells (HT-1080s) were previously characterized by a mesenchymal mode that was qualitatively similar to motile fibroblasts in collagen,1,17 pronounced differences in cytoskeletal structure and cell adhesion properties have been identified through comparisons with human dermal fibroblasts (hDFs) cultured using identical 2D and 3D cell culture platforms.12–14 These studies highlight the importance of providing quantitative measures of cell function to classify tumor cell migration mechanisms, since features for identifying distinct modes of motility describe qualitative characteristics common to most motile cell types,18 and are not well-defined.19The extracellular matrix (ECM) plays an active role during tumor progression, and both biochemical and biophysical properties have been implicated in transition to an invasive phenotype.20–22 Previous studies on fibronectin-coated polyacrylamide substrates have identified a role for substrate stiffness in migration for glioma cells,23 neutrophils24 and vascular smooth muscle cells,25 while HT-1080 and DU-145 prostate carcinoma cell motility is dependent on a balance of biochemical and biophysical properties in 3D culture.26 Importantly, transformation by Rous sarcoma virus reduces adhesiveness on fibronectin-coated surfaces due to altered integrin function,4 while HT-1080s are quantitatively less adherent than primary fibroblasts,12 which demonstrates that transformed cell types are characterized by altered adhesion properties compared to non-tumorigenic cells. While adhesion properties have been proposed as defining features of tumor cell migration modes,1–3 most quantitative models of cell adhesion-dependent motility describe non-tumorigenic adherent cell types27–29 on tissue culture polystyrene (TCP) or glass substrates with moduli orders of magnitude higher (∼109 Pa)30,31 than most tissues (∼102–104 Pa).30,32,33 Due to the complexity of the extracellular matrix and limitations inherent to common 2D culture platforms,34 there is increasing motivation to take advantage of engineered culture platforms to systematically investigate biochemical and biophysical influences on tumor cell function.12,13,23,26,35–45
Here, we describe a strategy to investigate cell adhesion-dependent motility for HT-1080s using engineered biomaterial arrays to provide synthetic control over adhesion ligand presentation. We chose engineered 2D surfaces to relate defining properties proposed for HT-1080s based on qualitative 3D observations to quantitative migration models primarily developed in 2D culture. We investigated cell adhesion-dependent migration for HT-1080s on surfaces with mechanical properties comparable to low modulus cell culture platforms and a range of soft tissues30,32,33 using Arg-Gly-Asp (RGD)-functionalized poly(ethylene glycol) (PEG) hydrogels (moduli 0.34–17 kPa, Fig. 1 and 2).13,32,36,46 Self-assembled monolayer (SAM) arrays of alkanethiolates on gold47–51 were used to present an RGD-containing cell adhesion ligand52–55 on a substrate with mechanical properties (∼0.1–1 GPa)49 comparable to TCP or glass30,31 (Fig. 3). Finally to provide further insight into the migration mechanism, we compared morphologies for HT-1080s and human mesenchymal stem cells (hMSCs), a primary human mesenchymal cell type. Our results demonstrate that HT-1080s are characterized by distinct morphological features compared to hMSCs, and migrate with adhesion characteristics of a weakly-adherent cell type. Our results add to a growing body of evidence12,13,37 in contrast with the common view that HT-1080s migrate via a mesenchymal mechanism similar to normal mesenchymal cells.1,17
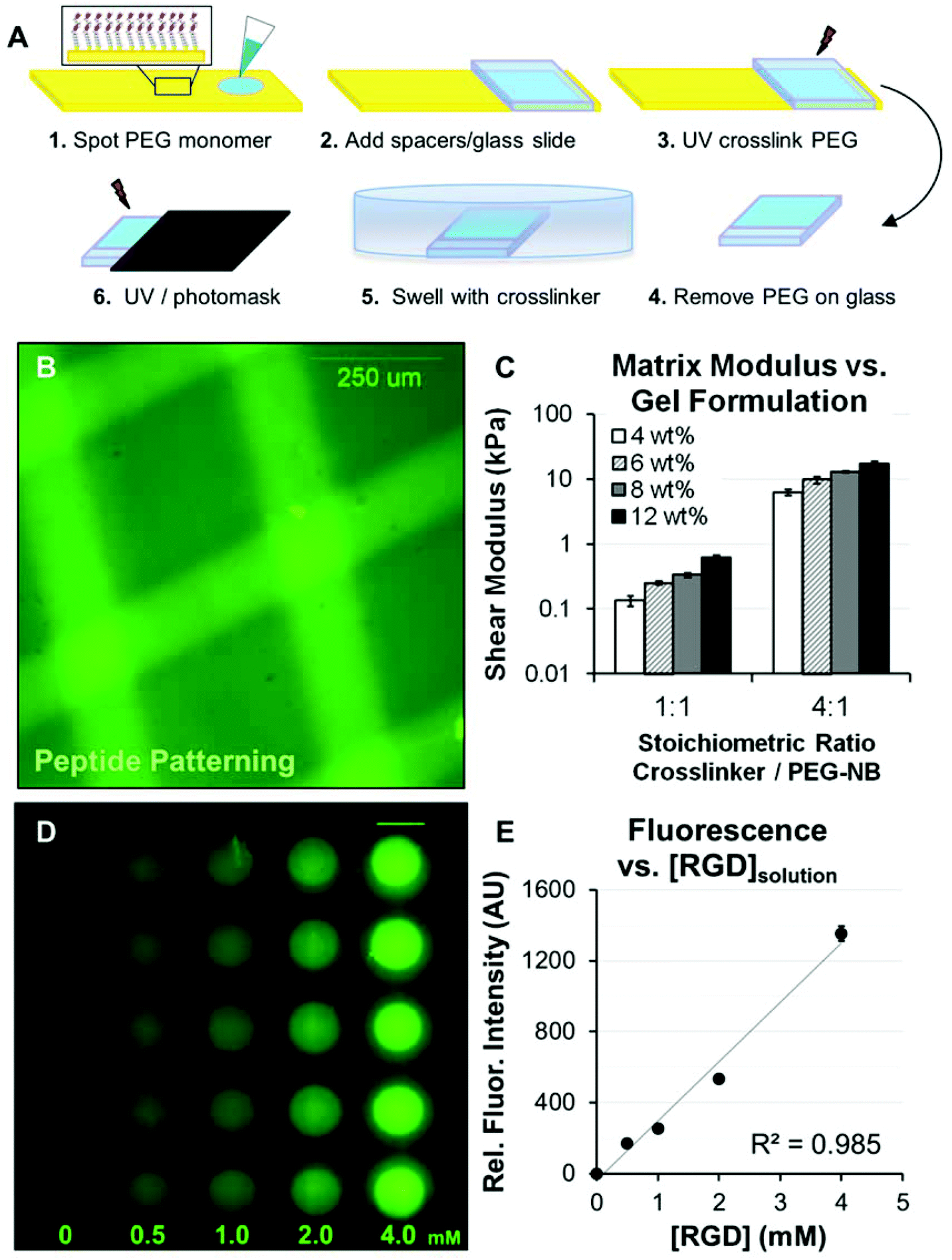 | ||
Fig. 1 Formation and patterning of poly(ethylene glycol) arrays (PEG arrays). (A) Schematic representation of the procedure for forming PEG arrays. PEG arrays were crosslinked with SH-PEG-SH3400 and functionalized with pendant Cys-Arg-Gly-Asp-Ser (CRGDS) peptides for adhesion. Scrambled, non-bioactive CRDGS peptide was included to maintain constant pendant group concentration (4 mM CRGDS + CRDGS) when investigating adhesion-dependent motility. Patterning: A PEG thin film was first formed with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 4:1 molar ratio SH-PEG-SH3400:PEG-NB and 0.5–4 mM CRGDS. The PEG hydrogel film was incubated in crosslinker solution at excess concentration overnight, followed by a second exposure to UV under a photomask to fully crosslink hydrogels in distinct regions (see also, Fig. 2) while maintaining the same RGD concentration. (B) A fluorescently tagged CRGDSK peptide was photopatterned into a PEG substrate to illustrate spatial control over polymer properties. (C) Elastic moduli for PEG hydrogels formed with different monomer concentrations, where reported values for wt% are based on the concentration of PEG-norbornene (8-arm, 20 K MW, PEG-NB) in the monomer solution (e.g., 4 wt% = 40 mg mL−1 PEG-NB). Shear modulus was also tuned by changing the molar ratio of SH-PEG-SH3400 to PEG-NB molecules (1:1 or 4:1, equivalent to 25% or 100% crosslinker thiol to total norbornene groups in the monomer solution). (D) Illustration of CRGDS incorporation into PEG hydrogels (8 wt%; 1:1 SH-PEG-SH:PEG-NB) for the range of concentrations investigated (reported for [CRGDS] in monomer solution). Relative CRGDS concentration was determined by labeling the N-terminus with Alexa Fluor 488 (see Methods). Scale bar = 2 mm. (E) Mean fluorescence intensity for PEG array spots functionalized with fluorescently tagged CRGDS. Error bars = standard deviation for (C) and (E). :1 or 4:1 molar ratio SH-PEG-SH3400:PEG-NB and 0.5–4 mM CRGDS. The PEG hydrogel film was incubated in crosslinker solution at excess concentration overnight, followed by a second exposure to UV under a photomask to fully crosslink hydrogels in distinct regions (see also, Fig. 2) while maintaining the same RGD concentration. (B) A fluorescently tagged CRGDSK peptide was photopatterned into a PEG substrate to illustrate spatial control over polymer properties. (C) Elastic moduli for PEG hydrogels formed with different monomer concentrations, where reported values for wt% are based on the concentration of PEG-norbornene (8-arm, 20 K MW, PEG-NB) in the monomer solution (e.g., 4 wt% = 40 mg mL−1 PEG-NB). Shear modulus was also tuned by changing the molar ratio of SH-PEG-SH3400 to PEG-NB molecules (1:1 or 4:1, equivalent to 25% or 100% crosslinker thiol to total norbornene groups in the monomer solution). (D) Illustration of CRGDS incorporation into PEG hydrogels (8 wt%; 1:1 SH-PEG-SH:PEG-NB) for the range of concentrations investigated (reported for [CRGDS] in monomer solution). Relative CRGDS concentration was determined by labeling the N-terminus with Alexa Fluor 488 (see Methods). Scale bar = 2 mm. (E) Mean fluorescence intensity for PEG array spots functionalized with fluorescently tagged CRGDS. Error bars = standard deviation for (C) and (E). | ||
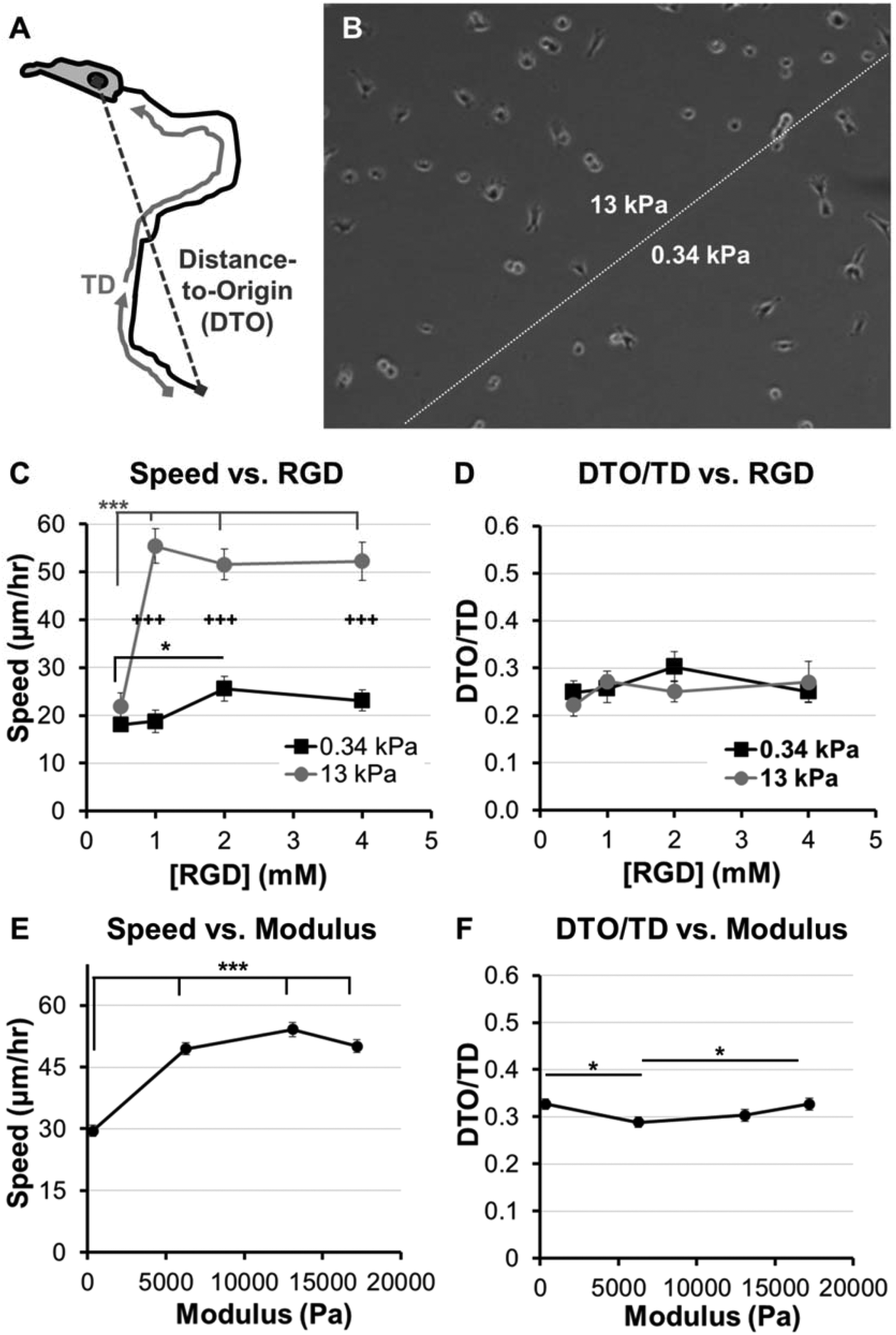 | ||
| Fig. 2 Adhesion and modulus-dependence for HT-1080s migrating on PEG-array surfaces. (A) Illustration of variables used to calculate Directionality (Directionality = Distance-to-Origin/Total Distance, DTO/TD). (B) Illustration of PEG thin film patterned to form two distinct modulus regions (see Methods, also Fig. 1A). (C) Cell speed and (D) Directionality (DTO/TD) for HT-1080s migrating on PEG thin films with varying RGD concentration (0.5–4 mM) and patterned to form regions with different moduli (8 wt% hydrogels; 1:1 or 4:1 molar ratio SH-PEG-SH:PEG-NB; elastic modulus = 0.34 or 13 kPa, respectively). (E) Cell speed and (F) Directionality (DTO/TD) for HT-1080s migrating on PEG thin films with varying moduli and constant 2 mM RGD concentration (8 wt% PEG-NB with 1:1 and 4:1 crosslinker; 4 and 12 wt% PEG-NB with 4:1 crosslinker; 0.34–17 kPa, see Fig. 1C). PEG-arrays with varying moduli and constant RGD concentration were formed as individual thin films. Error bars = standard error (N ≥ 30 for C–D, except 0.5 mM RGD high modulus condition, N = 11; N > 120 for E–F; * = p < 0.05; *** = p < 0.001; +++ = p < 0.001 for comparison between 0.34 and 13 kPa conditions). | ||
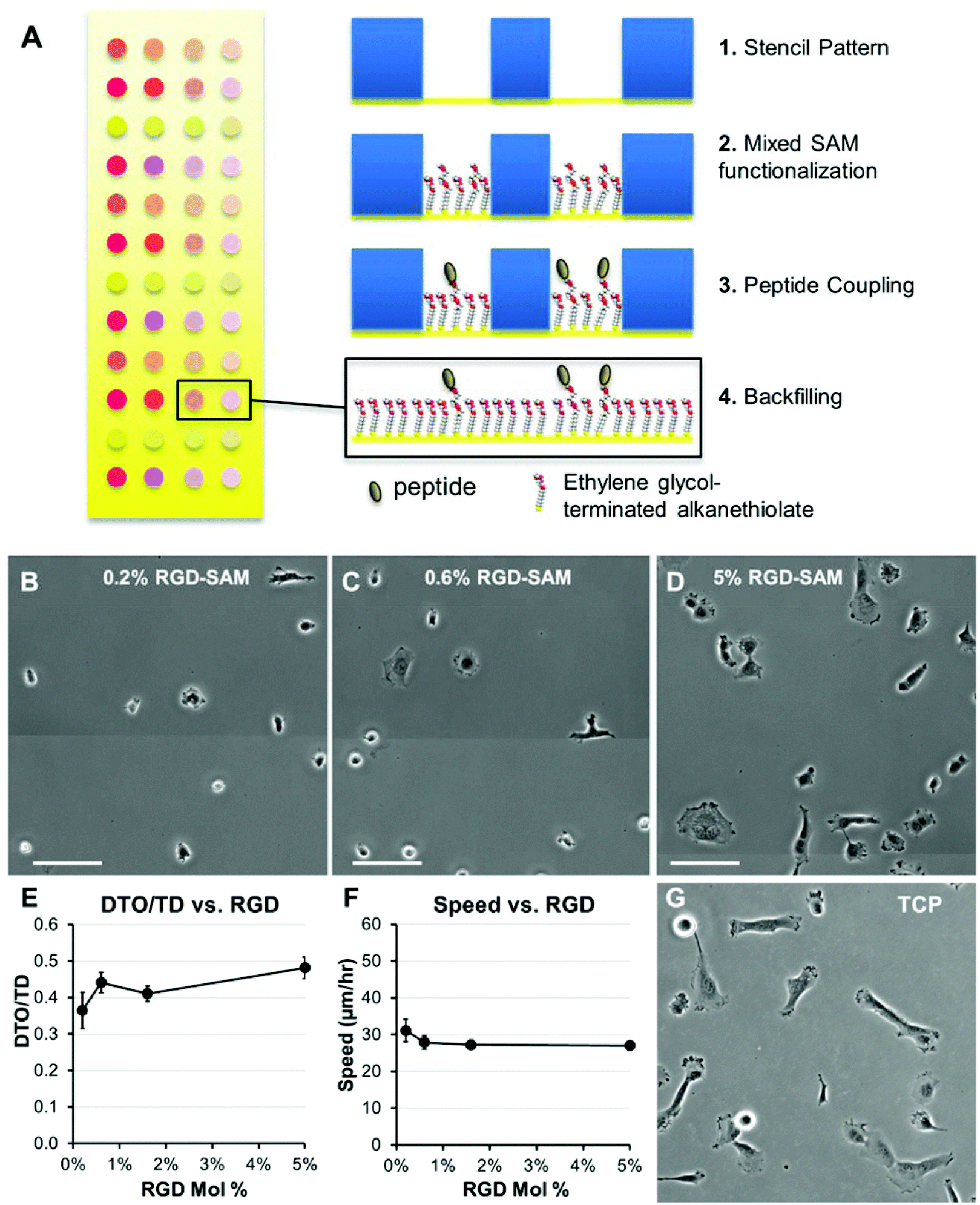 | ||
| Fig. 3 Migration and morphologies for HT-1080s on self-assembled monolayer (SAM) arrays. (A) Strategy for generating self-assembled monolayer (SAM) arrays. Adhesion was promoted by coupling GRGDSP peptide at a maximum density of 5% mol fraction, with lower densities representing successive 3-fold serial dilutions. Total peptide density was held constant by also incorporating non-bioactive GRGESP (5 mol% RGDSP + RGESP). HT-1080 morphologies on SAM-arrays presenting GRGDSP adhesion peptide (RGD-SAMs) at (B) 0.2 mol%, (C) 0.6 mol%, and (D) 5.0 mol% (Scale Bars = 50 μm). HT-1080s migrating on SAM-arrays with varying densities of CRGDSP: (E) Cell Speed and (F) Directionality (DTO/TD). Error bars = standard error (N ≥ 20 for all RGD densities; p > 0.05 for all comparisons). (G) HT-1080 morphologies on tissue culture polystyrene (TCP) were similar to 5 mol% RGD-SAMs. | ||
Results
We first investigated adhesion and stiffness influences on HT-1080 motility using poly(ethylene glycol) (PEG) hydrogels formed with elastic moduli that span reported values for many soft tissues (Fig. 1C).30,32,33 HT-1080s migrated with directionality (defined as the ratio of distance-to-origin to total distance, DTO/TD, Fig. 2A) that was minimally-dependent on RGD concentration (0.5–4 mM) for both low and high modulus PEG surfaces (0.34 kPa and 13 kPa, Fig. 2D), and when modulus was changed while holding RGD constant (Fig. 2F), which agrees with previous results in 2D and 3D culture.12,13,37 On 0.34 kPa PEG hydrogels, HT-1080 migration speed was weakly dependent on adhesion (Fig. 2C), reaching a maximum of 26 ± 3 μm h−1 for the 2 mM RGD condition. HT-1080s migrated on 13 kPa PEG hydrogels similarly to the 0.34 kPa formulation at the lowest RGD concentration investigated (0.5 mM RGD). However, cell speed for HT-1080s increased >two-fold to a maximum of 55 ± 3 μm h−1 when RGD concentration was increased from 0.5–1 mM RGD on 13 kPa PEG hydrogels, while further changes in RGD minimally influenced migration (Fig. 2C). The ∼two-fold increase in cell speed above 0.5 mM RGD for HT-1080s on 13 kPa surfaces is similar to previous results using ∼20–25 kPa PEG hydrogels,37 suggesting a threshold adhesion ligand density is required for effective motility, particularly on stiff substrates. Notably, HT-1080s migrated with >two-fold higher cell speed on 13 kPa compared to 0.34 kPa PEG surfaces for each of the 1–4 mM RGD conditions (Fig. 2C). HT-1080 migration speed also increased >1.5-fold on each of the 6–17 kPa formulations relative to 0.34 kPa PEG surface when RGD concentration was held constant at 2 mM (Fig. 2E). Thus, our results are consistent with a migration mode for HT-1080s that is weakly adhesion-dependent, whereas substrate stiffness plays a more pronounced role in motility.Quantitative models for adhesion-dependent motility were first derived for adherent cells migrating on tissue culture polystyrene (TCP) or glass,27–29 which are substrates with moduli orders of magnitude higher (∼109 Pa) than native human tissues30,32,33 or common biomimetic cell culture platforms (∼103–104 Pa).56 Therefore, we investigated HT-1080 motility using self-assembled monolayer (SAM) arrays of alkanethiolates on gold47–51 to present an RGD-containing peptide (RGD-SAMs) at defined densities52–55 (Fig. 3) on a substrate with mechanical properties (∼0.1–1 GPa)49 comparable to standard 2D culture platforms (e.g., TCP).30,31 HT-1080s adopted spread morphologies on 5% RGD-SAMs (Fig. 3D) that were similar to TCP (Fig. 3G), but became increasingly rounded as RGD density was decreased (Fig. 3B and C). HT-1080s migrated on RGD-SAMs with similar quantified directionality (DTO/TD) and speed for each of the RGD densities investigated (0.2–5% mol fraction, Fig. 3E and F). Therefore, HT-1080 motility was insensitive to adhesion on RGD-SAMs formed with this wide range of RGD densities. This result contrasts with the strong biphasic dependence on cell adhesion described previously for model adherent cell types.27–29 Interestingly, HT-1080s migrated on RGD-SAMs with cell speed (Fig. 3F) that was similar to the 0.34 kPa PEG hydrogel condition (Fig. 2C), and significantly lower than the maximum speed observed on higher modulus PEG hydrogels (Fig. 2C and E). Therefore, maximum cell speed for HT-1080s was observed on PEG hydrogels with intermediate stiffness relative to low modulus (0.34 kPa PEG) and high modulus (RGD-SAMs) substrates. While differences in chemical and structural properties limit conclusions that can be drawn for PEG compared to RGD-SAM surfaces, our results are consistent with a biphasic relationship between migration speed and substrate stiffness, such as described previously for neutrophils24 and vascular smooth muscle cells.25 Results for HT-1080s migrating on high modulus RGD-SAMs are consistent with PEG surfaces, pointing to a migration mechanism with minimal adhesion-dependence.
Tumor cells migrate with rounded morphologies that have been compared to amoeboid cells such as neutrophils, and elongated morphologies thought to be analogous to mesenchymal cells such as fibroblasts.1,2,17,57 On low and high modulus PEG hydrogels, HT-1080s adopted compact, rounded morphologies with similar projected cell areas on both surfaces (<600 μm2, Fig. 4A and B). In contrast, human mesenchymal stem cells (hMSCs, a model primary human mesenchymal cell type) were more spread on PEG surfaces (Fig. 4C), with projected cell area ∼2.5 to 3-fold higher than HT-1080s (Fig. 4A). HT-1080s were more spread on RGD-SAMs than on PEG (Fig. 5), which was reflected by projected cell area that increased from 830 ± 260 to 1030 ± 60 μm2 for 0.2% to 5% mol. fraction RGD (Fig. 5A). Projected cell area for hMSCs was ∼3.5-fold higher than HT-1080s on equivalent surfaces (Fig. 5A), increasing from 2980 ± 440 to 3870 ± 120 μm2 for 0.2% to 5% mol. fraction RGD-SAMs. HT-1080s were also characterized by distinct cytoskeletal structure and expression of vinculin-containing adhesions compared to hMSCs on RGD-SAMs (Fig. 5B–E). Vinculin was primarily delocalized for HT-1080s, with defined adhesions observed primarily for a subpopulation of cells on 5% RGD surfaces (Fig. 5B and C). HT-1080s expressed F-actin that was condensed on the edges of lamellipodia, cortically on the cell body for rounded cells, and as weakly organized filaments for some cells (Fig. 5B and C). In contrast, hMSCs were characterized by organized F-actin filaments and pronounced vinculin-containing adhesions (Fig. 5D and E), even on the lowest density RGD-SAMs (0.2% mol fraction), which is consistent with morphological features for a fibroblastic (mesenchymal) cell type.58–61 Our results here agree with previous comparisons of HT-1080s to human dermal fibroblasts12 and hMSCs to human umbilical vein endothelial cells54 on RGD-SAMs. Based on previous results on RGD-SAMs12,54 and direct comparisons between RGD-SAM and PEG surfaces here, we conclude that HT-1080s tend to adopt distinct morphologies compared to hMSCs and other adherent human primary cell types when cultured on high and low modulus 2D surfaces.
 | ||
| Fig. 4 Comparison of morphologies for HT-1080s and primary human mesenchymal stem cells (hMSCs) on PEG-arrays. (A) Average projected cell area for HT-1080s (black) and hMSCs (gray) on 0.34 and 13 kPa PEG hydrogels (2 mM RGD). 0.34 and 13 kPa hydrogels were formed with 8 wt% PEG-NB and 1:1 or 4:1 SH-PEG-SH:PEG-NB, respectively. Error bars = standard error (N > 60; *** = p < 0.001). Morphologies on 13 kPa PEG hydrogels for (B) HT-1080s and (C) hMSCs. | ||
 | ||
| Fig. 5 Comparison of morphologies for HT-1080s and hMSCs on RGD-SAM arrays. (A) Average projected cell area for HT-1080s (black) and hMSCs (gray) on RGD-SAM arrays. (B–D) Immunofluorescence images illustrating F-actin (Phalloidin, Red), Vinculin (Green), and Nuclei (DAPI, Blue) for (B, C) HT-1080s and (D, E) hMSCs on (B, D) 0.2% and (C, E) 5.0% RGD-SAM surfaces. | ||
Discussion
Mesenchymal motility describes a mechanism where cells generate traction forces through matrix attachments at the leading edge, while amoeboid cells squeeze or flow through the extracellular matrix with minimal cell adhesion requirements.1–3 Our results provide several pieces of evidence that HT-1080s migrate through a 2D mechanism that minimally depends on adhesion for most of the conditions investigated. HT-1080s migrated on 0.34 kPa PEG surfaces with quantified speed and directionality that were weakly influenced by RGD concentration (Fig. 2), which differs from pronounced biphasic motility described for adherent cell types such as fibroblasts or smooth muscle cells.27–29 While cell speed increased >two-fold from 0.5–1.0 mM RGD on 13 kPa PEG hydrogels, further increases in RGD concentration minimally influenced motility (Fig. 2). A similar trend was previously reported for HT-1080s on PEG hydrogels with higher modulus than the formulations used here,37 which suggests that efficient motility requires a minimal level of adhesion, but that migration is weakly-dependent on adhesion at higher adhesion ligand concentrations. Weak adhesion-dependence such as observed on low modulus PEG surfaces, and above a threshold RGD concentration on intermediate modulus PEG, are consistent with previous results for 2D and 3D HT-1080 motility investigated using PEG matrices.12,13,37Our results also demonstrated that matrix mechanical properties influenced HT-1080 motility when RGD concentration was held constant on PEG hydrogels. Specifically, cell migration speed for HT-1080s on PEG hydrogels increased almost two-fold for 6–17 kPa relative to 0.34 kPa surfaces, reaching a maximum at 13 kPa (Fig. 2). Previous studies on fibronectin (FN)-coated polyacrylamide substrates identified a role for substrate stiffness in mediating migration for glioma cells,23 neutrophils24 and vascular smooth muscle cells.25 Interestingly, substrate stiffness influenced neutrophil migration within a relatively narrower and lower range of moduli (∼3–13 kPa)24 than smooth muscle cells (∼1–45 kPa for higher adhesion surfaces; ∼45 to >300 kPa for low adhesion surfaces).25 The range of moduli that led to changes in cell speed for HT-1080s on PEG hydrogels here (0.34–17 kPa) was similar to the range reported for neutrophil migration previously (∼3–13 kPa),24 which is notable since neutrophils are a model amoeboid cell type characterized by a weakly-adherent phenotype.62,63 Glioma cell migration on FN-coated polyacrylamide was also dependent on modulus, and reported cell speeds on 0.8 kPa polyacrylamide and glass surfaces23 were similar to HT-1080s on 0.34 kPa PEG (Fig. 2) and RGD-SAM surfaces (Fig. 3), respectively. Therefore, substrate stiffness influenced HT-1080 motility similarly to amoeboid cells (e.g. neutrophils)24 and other aggressive tumorigenic cell types (e.g. glioma cells),23 but distinctly from adherent mesenchymal cells (e.g. smooth muscle cells, hMSCs).
Tumor cell migration modes have also been identified by differences in morphology and cytoskeletal organization.1,2 In particular, the mesenchymal (elongated) mode has been defined by F-actin dense protrusions, while amoeboid (rounded) tumor cells are characterized by cortical organization of the F-actin cytoskeleton.1,2 HT-1080s were previously characterized by cortical F-actin organization and quantitatively more rounded morphologies than primary hDFs in 2D and 3D culture.12 Here, HT-1080s adopted fan-shaped morphologies on 5% RGD-SAMs similarly to cells on TCP (Fig. 3 and 5), but became increasingly rounded when RGD density was decreased, which is consistent with previous results for 2D culture.12,64–67 In contrast, hMSCs adopted more spread morphologies on both PEG hydrogels and RGD-SAMs, leading to >2.5-fold higher projected cell area than HT-1080s for all surfaces investigated (Fig. 4 and 5). Rounded morphologies for HT-1080s on low and high modulus substrates are likely not due to artifacts of substrate stiffness or availability of cell adhesion ligands, as several primary human adherent cell types adopt spread morphologies on equivalent surfaces, including hMSCs, hDFs, and human umbilical vein endothelial cells (HUVECs).12,54,68,69 Thus, primary human adherent mesenchymal cell types attach and spread on PEG and RGD-SAM surfaces12,54,68,69 under conditions that lead to pronounced rounding for HT-1080s.
Methods
Surface modification
Glass coverslips (Fisher scientific) were functionalized with silane monolayers for subsequent use as substrates for PEG hydrogel formation. Firstly, they were activated on both sides for 5 min in oxygen plasma at 4 ppm gas pressure in a Diener plasma chamber (Diener electronic, Ebhausen, Germany). They were transferred in a homemade reaction chamber and successively purged with dry nitrogen gas. (3-Mercaptopropyl) trimethoxysilane (Sigma Aldrich) was dissolved in anhydrous Toluene (ACS) (v/v 0.5%) and transferred to the reaction chamber. The reaction proceeded overnight (8 h) under the exclusion of light. After the reaction, the coverslips were rinsed with toluene (ACS grade, Sigma Aldrich) and then sonicated in toluene, ethyl acetate (Chromasolv grade, Sigma Aldrich), ethanol (200 proof, Decon labs, King of Prussia, PA) and methanol (ACS grade, Sigma Aldrich). Coverslips and reaction vessel were rinsed with methanol, blow-dried and reassembled and purged with dry nitrogen gas. They were then cured at 110 °C for 2 h. After cooling down the slides were stored in a dry nitrogen atmosphere and under the exclusion of light until use. Non-adhesive SAMs for hydrogel array processing were formed on glass slides coated with a 50 Å Ti adhesion layer and a 1000 Å Au layer (Evaporated Metal Films, Inc., Ithaca, NY) were functionalized with a 11-tri(ethylene glycol)-undecane-1-thiol [HS–C11–(O–CH2–CH2)3–OH] (termed tri(ethylene glycol) alkanethiol) self-assembled monolayer (SAM) by incubation in a 0.1 mM ethanolic tri(ethylene glycol) alkanethiol solution overnight. They were then washed with ethanol and blown dried directly before use.SAM array production
SAM arrays were fabricated as described previously (Fig. 3A).52,54,68 Thin films of 100 Å Au <111>, 20 Å Ti on 1′′ × 3′′ × 0.040′′ glass were purchased from Platypus Technologies, LLC (Madison, WI, Cat. no. AU.0100.ALSI). Briefly, an elastomeric stencil containing arrays of 1.1 mm holes was placed on a bare gold surface to form an array of wells. Wells were then filled with 1 mM ethanolic alkanethiolate solutions containing mixtures of carboxylic acid-capped hexa(ethylene glycol) undecanethiol (HS–C11–(O–CH2–CH2)6–O–CH2–COOH, purchased from Prochimia, Sopot, Poland) and tri(ethylene glycol) alkanethiol and incubated for 10 minutes in a chamber containing a laboratory wipe soaked with ethanol to prevent evaporation. Alkanethiolate solutions were then aspirated and wells were rinsed with DIUF H2O. Carboxylate groups were then converted to active ester groups by adding a solution of 100 mM NHS and 250 mM EDC in DIUF H2O pH 5.5 to wells and incubated for 10 minutes. After an additional rinse with DIUF H2O, 300 mM solutions of peptide in PBS at pH 7.4 were added to each well and incubated for 1 h in a humidity controlled chamber to covalently couple peptides to each array spot. After a final rinse with DIUF H2O, regions surrounding array spots were backfilled with tri(ethylene glycol) alkanethiol to create a bioinert background surrounding the array spots. This was achieved by submerging the gold substrate and attached elastomeric stencil in an aqueous 0.1 mM HS–C11–EG3–OH solution (pH 2.0), removing the PDMS “stencil”, and incubating for 10 minutes. Following backfilling, the array was rinsed with 0.1 wt% SDS in DIUF H2O, DIUF H2O, and EtOH and then dried under a stream of N2. Arrays were stored in sterile DIUF H2O at 4 °C and used within 24 h.Peptide and polymer synthesis
A fibronectin derived peptide with the sequence Cys-Arg-Gly-Asp-Ser (CRGDS), a scrambled version CRDGS and a lysine containing version CRGDSK for fluorescent labeling were each synthesized using standard solid phase Fmoc-peptide synthesis, which was performed using a 36S automated peptide synthesizer (CS Bio, Menlo Park, CA) on rink amide resin using Fmoc protected amino acids (AnaSpec, Fremont, CA). Peptides purity >90% was verified using MALDI-TOF MS and HPLC, while concentrations in solution were determined using Elman's reagent (Pierce) to quantify thiol content by comparing against a standard curve generated with L-cysteine (Sigma-Aldrich). PEG-norbornene (8-arm PEG-NB 20k) was synthesized using a hydroxyl-functionalized multi-arm PEG core (JenKem Technology, USA) as previously described in detail.13,46 Directly after the synthesis the PEG norbornene macromers were purified by dialysis in 3500 kDa cut off membranes in DI water. Purity ≥88% was confirmed using H-NMR.PEG hydrogel formation
PEG hydrogels were formed with different moduli by changing the concentration of the 8-arm PEG-NB backbone and the stoichiometric ratio of SH-PEG-SH to PEG-NB (1:1 or 4:1 molar ratio SH-PEG-SH:8-arm PEG-NB, which is equivalent to 25% or 100% crosslinker SH to total norbornene groups, Fig. 1C). Adhesion properties were tuned by incorporating RGD (Arg-Gly-Asp)-containing peptides70,71 through a thiol-containing cysteine that was incorporated into the amino acid sequence during synthesis. Total pendant peptide concentration was held constant for adhesion-dependent migration studies on PEG surfaces by including non-bioactive CRDGS (4 mM CRGDS + CRDGS). We prepared polymer mixes in PBS buffer containing 8-arm PEG-NB 20k, PEG dithiol 3.4k and peptides to prepare defined surfaces described in the Results section, using 0.05 wt% Irgacure 2959 (wt/wt) as the photoinitiator (BASF Schweiz AG, Basel, Switzerland). 2 μL of the polymer mix were pipetted on top of an Au coated thiolated glass slide and the silanized coverslip was placed on top (Fig. 1A). The thin film between the two slides was polymerized by UV light (365 nm, OmniCure Series 1000, Lumen Dynamics Group Inc. Mississauga, Canada). The coverslip was lifted up and due to the non-adhesive coating on the gold slide and the adhesive coating on the coverslip the film remained complete and undamaged at the silanized coverslip. The formed hydrogel was thoroughly washed in PBS buffer and incubated overnight. On the next day, the hydrogel was sterilized by incubation in EtOH–H2O mix (v/v 70/30) for 30 min, successively repeated and thoroughly washed with sterile PBS buffer and used for cell experiments.
Formation of hydrogels with patterned modulus
We took advantage of the photoinitiated thiol–ene reaction to form PEG hydrogels at different moduli (0.34 and 13 kPa) while maintaining controlled RGD concentrations in the 0.5–4.0 mM range by patterning distinct regions with different crosslinking densities (Fig. 2B). Specifically, RGD-functionalized hydrogels were first formed with an off-stoichiometric ratio of thiol groups to norbornene arms (1:1 molar ratio of SH-PEG-SH:PEG-NB, or 25% ratio of crosslinker thiol to total norbornene groups), leaving unreacted norbornene arms that could subsequently be crosslinked by PEG-dithiol in a second post-encapsulation step. For the second step, excess PEG-dithiol was included in the buffer during swelling of the partially crosslinked hydrogel, after which the remaining PEG-NB arms were crosslinked using a photomask to provide defined regions with alternating low and high modulus and constant RGD concentration (Fig. 1 and 2). Then, a photomask with the appropriate pattern was applied and the hydrogel was exposed to UV light, forming additional cross-links with the appropriate pattern. Subsequently, the resulting hydrogel was thoroughly washed and sterilized as described above.
Fluorescent peptide incorporation
To illustrate photopatterning, we incorporated the pendant peptide CRGDSK using the same procedure (described for photopatterning modulus), after-which the available lysine group was fluorescently tagged for imaging as described below (Fig. 1B). To demonstrate controllable peptide incorporation into PEG hydrogels, hydrogel arrays were constructed using silanized glass slides and gold slides patterned with defined hydrophilic and hydrophobic regions. Briefly, thin films of Au/Ti on 1′′ × 3′′ × 0.040′′ glass were purchased from Evaporated Metal Films (Ithaca, NY, Cat. no. TA134) and submerged in 0.1 mM ethanolic HS–(CH2)11–O–(CH2)2–(CF2)5–CF3 solution (termed FluoraSAMs, Prochimia Surfaces, Sopot, Poland) overnight. Afterward, an elastomeric stencil containing arrays of 2.5 mm diameter holes was placed on the gold surface. The gold slides were treated for 1 minute in oxygen plasma at 4 ppm gas pressure in a Diener plasma chamber to selectively remove the FluoraSAM layer from the areas defined by the stencil holes. The elastomeric stencil was removed from the patterned surface and the slides were soaked in tri(ethylene glycol) alkane thiol solution overnight resulting in defined circular hydrophilic regions on a hydrophobic FluoraSAM-coated slide. The slides were then washed with ethanol and blow-dried directly before use.We formed arrays of hydrogels with 1:1 molar ratio of SH-PEG-SH to 8-arm PEG-NB molecules. The hydrogels were functionalized with 0–4.0 mM CRGDS as a pendant peptide. 0.75 μL droplets of precursor solution were pipetted into the hydrophilic regions of the gold slides and the hydrogels were cured between the patterned gold slides and silanized glass slides for 3 seconds under 365 nm UV light at a dose rate of 90 mW cm−2. Spot thickness was defined using 0.15 mm thick elastomer spacers. The hydrogel arrays were soaked in PBS for 1 hour to swell the hydrogel spots to equilibrium and remove non-crosslinked molecules. Relative CRGDS concentration was compared by labeling the N-terminus of incorporated peptide with Alexa Fluor 488. The arrays were treated overnight with 30 μM fluorescein-conjugated sulfodichlorophenol ester (Invitrogen, Grand Island, NY) in PBS, rinsed for 5 hours in PBS with multiple buffer exchanges prior to imaging. The fluorescently labeled arrays (Fig. 1D) were photographed using a Typhoon Trio Variable Mode Imager (GE Healthcare, Madison, WI; Courtesy of the UW-Madison Carbone Cancer Center Scientific Instrumentation Facility) and photopatterned fluorescent hydrogels (Fig. 1B) were imaged using a Nikon TI Eclipse Fluorescence microscope. Mean fluorescence intensity of the individual spots was performed using Nikon Elements image analysis software (Fig. 1E).
Cell culture, imaging and image analysis
Human mesenchymal stem cells (hMSCs, Lonza) were cultured in minimum essential medium, alpha (AMEM, Mediatech, Manassas, VA) containing 10% MSC qualified fetal bovine serum (Invitrogen, Carlsbad, CA) and 1% penicillin/streptomycin. HT-1080 fibrosarcoma cells (HT1080s, ATCC, Manassas, VA) were cultured in Eagle's minimal essential medium (EMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 100 μg mL−1 streptomycin/penicillin at 37 °C in a 5% CO2 atmosphere until 70% confluence. Cells were trypsinized, resuspended in basal media and seeded at densities of 1500 cells per cm2 for SAM arrays or 4000 cells per cm2 for PEG-arrays. For SAM arrays, unattached cells were removed by gently dipping slides into a 50 mL conical vial with warm culture media, and then samples were placed on the heated environmental chamber and imaged (T = 0 was 2 hours after seeding). Cells on PEG-arrays were allowed to attach overnight (14 h), placed in the environmental chamber and imaged. Cell migration was quantified for images collected at 15 min intervals for 6 h using a Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan). Cell migration was analyzed manually using NIS elements (Nikon, Tokyo, Japan). Cells that divided or interacted with other cells during analysis were excluded from analysis. For cells on PEG-arrays, morphologies were quantified using the “Draw Object” feature in NIS Elements to trace the outline of individual cells, from which projected cell area was automatically calculated using the “Automated Measurement” function. For cells on RGD-SAMs, stacked images of array spots were thresholded and then automated measurements of area and counts were tabulated using NIS Elements. For each spot, average projected cell area was calculated by dividing the thresholded actin staining (red channel) by the total number of nuclei in the same spot.Immunofluorescence (IF) imaging
IF staining followed the procedure provided with the focal adhesion staining kit (Millipore, FAK100). TRITC-conjugated phalloidin, DAPI and primary mouse anti-vinculin monoclonal antibody (Clone 7F9, 1:100 dilution) were included with the focal adhesion staining kit (Millipore, FAK100). The secondary antibody was Alexa-Fluor 488 goat anti-mouse IgG (Invitrogen, A-11001, 1:200 dilution).
Dynamic mechanical analysis of hydrogels
Dynamic mechanical analysis was performed using an Ares-LS2 rheometer (TA Instruments, New Castle, DE; Courtesy of the UW-Madison Soft Materials Laboratory) to evaluate the modulus of the hydrogels. Briefly, hydrogel samples were crosslinked in 8.0 mm diameter, 1.2 mm depth Teflon wells for 3 seconds using 365 nm UV light at a dose rate of 90 mW cm−2. The samples were swollen to equilibrium in PBS and cut to a final diameter of 8 mm using a hole punch. Samples were loaded onto 8 mm diameter cross-heads and loaded with 0.2 Newtons of normal force. If the samples were not sufficiently robust to withstand a 0.2 Newton normal force, the crossheads were set at a 1.0 mm gap distance instead. The cross-heads sheared the samples in an oscillatory manner at a constant frequency of 10 Hz. Shear strains ranged from 0.1% to 20%. All tests were carried out at room temperature. Shear stress, shear strain, and the moduli were calculated as follows: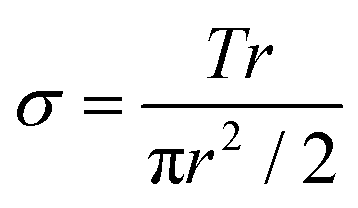 | (1) |
Eqn (1). Sample shear stress based on torque (T), sample radius (r), and the sample polar moment of inertia (πr2/2).
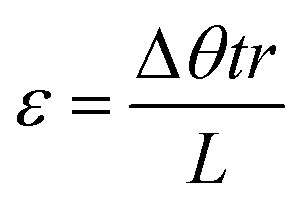 | (2) |
Eqn (2). Sample shear strain based on rotation rate (Δθ), time (t), sample radius (r), and sample height (L).
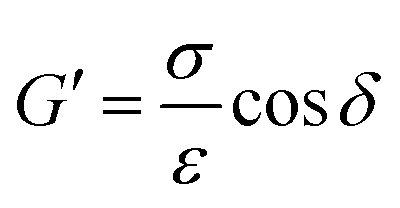 | (3) |
Eqn (3). Storage modulus (G′) of the sample was calculated by taking the sample stress (σ) divided by the sample strain (ε) and multiplying by the cosine of the phase angle (δ). As the testing was carried out at multiple strains, a strain-averaged value was computed. Loss modulus (G′′) was also computed but the sine of the phase angle (δ) was taken instead.
Conclusions
Our combined results demonstrate that HT-1080s are characterized by a phenotype that is distinct from primary human mesenchymal cell types and migrate with weak adhesion-dependence on both low and high modulus substrates, while matrix modulus had a more pronounced influence on cell speed. Previous studies demonstrated that HT-1080s migrate in poly(ethylene glycol) (PEG) hydrogels with minimal dependence on adhesion,12–14 and differ in quantified migration, cytoskeletal structure, and adhesion properties relative to primary human dermal fibroblasts (hDFs) in 2D and 3D culture.12 Here, HT-1080s migrated through a mechanism that minimally depends on adhesion on substrates with a wide range of moduli (∼103–109 Pa), which is consistent with previous studies using 2D and 3D culture platforms.12,13,37 HT-1080s adopted rounded morphologies on each of the matrices investigated here, while cortical F-actin organization was observed even when cultured on very stiff RGD-SAM surfaces, which contrasts with spreading and well-defined F-actin filaments observed for primary human mesenchymal stem cells (hMSCs, a model mesenchymal cell type). Our results provide new evidence that contrasts with the common view that HT-1080s migrate via a mesenchymal mechanism similar to normal primary adherent cells.1,17 While fibrosarcomas are tumors of mesenchymal origin,72 an N-Ras mutation for HT-1080s constitutively activates signaling pathways that regulate global cell function (e.g., proliferation and migration), which disrupts the F-actin cytoskeleton, increases proliferation, and induces anchorage independent growth.66,67,73–75 Therefore, while HT-1080s are characterized by properties that are distinct from primary human mesenchymal cell types in 2D and 3D culture,12,13 several observations here and previously12,13,37 suggest a migration mechanism that is influenced by phenotypic changes induced by transformation, including a weakly-adherent phenotype, rounded morphologies, and cortical F-actin organization.66,67,73–75Acknowledgements
Funding was provided by the National Institutes of Health (NIH R01 HL093282-01A1, NIH R21 EB016381-01, NIH 1UH2 TR000506-01) and a donation in the memory of Gerald E. Whitbeck (generously provided by his family and Follett Corp). NNL thanks the National Science Foundation for a Graduate Research Fellowship (Grant DGE-0718123), the University of Wisconsin Graduate Engineering Research Scholars, and the Gates Millennium Scholars Program.References
- P. Friedl and K. Wolf, Nat. Rev. Cancer, 2003, 3, 362–374 CrossRef CAS PubMed.
- V. Sanz-Moreno and C. J. Marshall, Curr. Opin. Cell Biol., 2010, 22, 690–696 CrossRef CAS PubMed.
- C. D. Madsen and E. Sahai, Dev. Cell, 2010, 19, 13–26 CrossRef CAS PubMed.
- L. C. Plantefaber and R. O. Hynes, Cell, 1989, 56, 281–290 CrossRef CAS.
- T. Volk, B. Geiger and A. Raz, Cancer Res., 1984, 44, 811–824 CAS.
- M. Schliwa, T. Nakamura, K. R. Porter and U. Euteneuer, J. Cell Biol., 1984, 99, 1045–1059 CrossRef CAS.
- A. Raz and B. Geiger, Cancer Res., 1982, 42, 5183–5190 CAS.
- T. David-Pfeuty and S. J. Singer, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6687–6691 CrossRef CAS.
- M. Cottlerfox, W. Ryd, B. Hagmar and C. H. Fox, Int. J. Cancer, 1980, 26, 689–694 CrossRef CAS.
- J. Bubenik, P. Perlmann, E. M. Fenyo, T. Jandlova, E. Suhajova and M. Malkovsky, Int. J. Cancer, 1979, 23, 392–396 CrossRef CAS.
- D. R. Coman, Cancer Res., 1944, 4, 625–629 Search PubMed.
- M. P. Schwartz, R. E. Rogers, S. P. Singh, J. Y. Lee, S. G. Loveland, J. T. Koepsel, E. S. Witze, S. I. Montanez-Sauri, K. E. Sung, E. Y. Tokuda, Y. Sharma, L. M. Everhart, E. H. Nguyen, M. H. Zaman, D. J. Beebe, N. G. Ahn, W. L. Murphy and K. S. Anseth, PLoS One, 2013, 8, e81689 Search PubMed.
- M. P. Schwartz, B. D. Fairbanks, R. E. Rogers, R. Rangarajan, M. H. Zaman and K. S. Anseth, Integr. Biol., 2010, 2, 32–40 RSC.
- R. J. Petrie, N. Gavara, R. S. Chadwick and K. M. Yamada, J. Cell Biol., 2012, 197, 439–455 CAS.
- E. Van Goethem, R. Poincloux, F. Gauffre, I. Maridonneau-Parini and V. Le Cabec, J. Immunol., 2010, 184, 1049–1061 CrossRef CAS PubMed.
- F. Sabeh, R. Shimizu-Hirota and S. J. Weiss, J. Cell Biol., 2009, 185, 11–19 CrossRef CAS PubMed.
- K. Wolf, I. Mazo, H. Leung, K. Engelke, U. H. von Andrian, E. I. Deryugina, A. Y. Strongin, E. B. Brocker and P. Friedl, J. Cell Biol., 2003, 160, 267–277 CrossRef CAS PubMed.
- D. A. Lauffenburger and A. F. Horwitz, Cell, 1996, 84, 359–369 CrossRef CAS.
- A. Huttenlocher and A. R. Horwitz, Cold Spring Harbor Perspectives in Biology, 2011, p. 3 Search PubMed.
- P. P. Provenzano and P. J. Keely, J. Cell Sci., 2011, 124, 1195–1205 CrossRef CAS PubMed.
- C. M. Nelson and M. J. Bissell, Semin. Cancer Biol., 2005, 15, 342–352 CrossRef PubMed.
- M. J. Bissell, H. G. Hall and G. Parry, J. Theor. Biol., 1982, 99, 31–68 CrossRef CAS.
- T. A. Ulrich, E. M. D. Pardo and S. Kumar, Cancer Res., 2009, 69, 4167–4174 CrossRef CAS PubMed.
- K. M. Stroka and H. Aranda-Espinoza, Cell Motil. Cytoskeleton, 2009, 66, 328–341 CrossRef CAS PubMed.
- S. R. Peyton and A. J. Putnam, J. Cell. Physiol., 2005, 204, 198–209 CrossRef CAS PubMed.
- M. H. Zaman, L. M. Trapani, A. Siemeski, D. MacKellar, H. Y. Gong, R. D. Kamm, A. Wells, D. A. Lauffenburger and P. Matsudaira, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10889–10894 CrossRef CAS PubMed.
- S. P. Palecek, J. C. Loftus, M. H. Ginsberg, D. A. Lauffenburger and A. F. Horwitz, Nature, 1997, 385, 537–540 CrossRef CAS PubMed.
- P. A. Dimilla, J. A. Stone, J. A. Quinn, S. M. Albelda and D. A. Lauffenburger, J. Cell Biol., 1993, 122, 729–737 CrossRef CAS.
- P. A. Dimilla, K. Barbee and D. A. Lauffenburger, Biophys. J., 1991, 60, 15–37 CrossRef CAS.
- T. R. Cox and J. T. Erler, Dis. Models Mech., 2011, 4, 165–178 CrossRef CAS PubMed.
- K. Miyake, N. Satomi and S. Sasaki, Appl. Phys. Lett., 2006, 89 Search PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- I. Levental, P. C. Georges and P. A. Janmey, Soft Matter, 2007, 3 Search PubMed.
- E. A. Dubiel, Y. Martin and P. Vermette, Chem. Rev., 2011, 111, 2900–2936 CrossRef CAS PubMed.
- M. H. Zaman, Nat. Rev. Cancer, 2013, 13, 596–603 CrossRef CAS PubMed.
- E. S. Witze, M. K. Connacher, S. Houel, M. P. Schwartz, M. K. Morphew, L. Reid, D. B. Sacks, K. S. Anseth and N. G. Ahn, Dev. Cell, 2013, 26, 645–657 CrossRef CAS PubMed.
- J. C. Hoffmann and J. L. West, Integr. Biol., 2013, 5, 817–827 RSC.
- D. Loessner, J. A. Flegg, H. M. Byrne, J. A. Clements and D. W. Hutmacher, Integr. Biol., 2013, 5, 597–605 RSC.
- K. E. Sung, N. Yang, C. Pehlke, P. J. Keely, K. W. Eliceiri, A. Friedl and D. J. Beebe, Integr. Biol., 2011, 3, 439–450 RSC.
- M. Hakanson, M. Textor and M. Charnley, Integr. Biol., 2011, 3, 31–38 RSC.
- D. Loessner, K. S. Stok, M. P. Lutolf, D. W. Hutmacher, J. A. Clements and S. C. Rizzi, Biomaterials, 2010, 31, 8494–8506 CrossRef CAS PubMed.
- D. W. Hutmacher, D. Loessner, S. Rizzi, D. L. Kaplan, D. J. Mooney and J. A. Clements, Trends Biotechnol., 2010, 28, 125–133 CrossRef CAS PubMed.
- S. I. Fraley, Y. F. Feng, R. Krishnamurthy, D. H. Kim, A. Celedon, G. D. Longmore and D. Wirtz, Nat. Cell Biol., 2010, 12, 598–U169 CrossRef CAS PubMed.
- E. L. Baker, J. Lu, D. Yu, R. T. Bonnecaze and M. H. Zaman, Biophys. J., 2010, 99, 2048–2057 CrossRef CAS PubMed.
- C. Fischbach, R. Chen, T. Matsumoto, T. Schmelzle, J. S. Brugge, P. J. Polverini and D. J. Mooney, Nat. Methods, 2007, 4, 855–860 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- S. H. Shabbir, J. L. Eisenberg and M. Mrksich, Angew. Chem., Int. Ed., 2010, 49, 7706–7709 CrossRef CAS PubMed.
- M. Mrksich, Acta Biomater., 2009, 5, 832–841 CrossRef CAS PubMed.
- F. W. DelRio, C. Jaye, D. A. Fischer and R. F. Cook, App. Phys. Lett., 2009, 94, 131909–131903 CrossRef PubMed.
- M. Mrksich, Chem. Soc. Rev., 2000, 29, 267–273 RSC.
- E. Sabatani, I. Rubinstein, R. Maoz and J. Sagiv, J. Electroanal. Chem., 1987, 219, 365–371 CrossRef CAS.
- J. T. Koepsel, E. H. Nguyen and W. L. Murphy, Integr. Biol., 2012, 4, 914–924 RSC.
- J. T. Koepsel and W. L. Murphy, ChemBioChem, 2012, 13, 1717–1724 CrossRef CAS PubMed.
- J. T. Koepsel, S. G. Loveland, M. P. Schwartz, S. Zorn, D. G. Belair, N. N. Le and W. L. Murphy, Integr. Biol., 2012, 4, 1508–1521 RSC.
- G. A. Hudalla and W. L. Murphy, Soft Matter, 2011, 7, 9561–9571 RSC.
- A. Asthana and W. S. Kisaalita, Drug Discovery Today, 2013, 18, 533–540 CrossRef CAS PubMed.
- E. Sahai and C. J. Marshall, Nat. Cell Biol., 2003, 5, 711–719 CrossRef CAS PubMed.
- K. Katoh, Y. Kano, M. Amano, K. Kaibuchi and K. Fujiwara, Am. J. Physiol., Cell Physiol., 2001, 280, C1669–C1679 CAS.
- K. A. Beningo, M. Dembo, I. Kaverina, J. V. Small and Y. L. Wang, J. Cell Biol., 2001, 153, 881–887 CrossRef CAS.
- G. Totsukawa, Y. Yamakita, S. Yamashiro, D. J. Hartshorne, Y. Sasaki and F. Matsumura, J. Cell Biol., 2000, 150, 797–806 CrossRef CAS.
- L. Lewis, J. M. Verna, D. Levinstone, S. Sher, L. Marek and E. Bell, J. Cell Sci., 1982, 53, 21–36 CAS.
- T. Lammermann and M. Sixt, Curr. Opin. Cell Biol., 2009, 21, 636–644 CrossRef PubMed.
- A. F. Brown, J. Cell Sci., 1982, 58, 455–467 CAS.
- V. Niggli, D. Schlicht and S. Affentranger, Biochem. Biophys. Res. Commun., 2009, 386, 688–692 CrossRef CAS PubMed.
- V. Niggli, M. Schmid and A. Nievergelt, Biochem. Biophys. Res. Commun., 2006, 343, 602–608 CrossRef CAS PubMed.
- S. Gupta, S. Stuffrein, R. Plattner, M. Tencati, C. Gray, Y. E. Whang and E. J. Stanbridge, Mol. Cell. Biol., 2001, 21, 5846–5856 CrossRef CAS.
- S. Gupta, R. Plattner, C. J. Der and E. J. Stanbridge, Mol. Cell. Biol., 2000, 20, 9294–9306 CrossRef CAS.
- J. T. Koepsel, P. T. Brown, S. G. Loveland, W. J. Li and W. L. Murphy, J. Mater. Chem., 2012, 22, 19474–19481 RSC.
- J. T. Koepsel and W. L. Murphy, Langmuir, 2009, 25, 12825–12834 CrossRef CAS PubMed.
- E. Ruoslahti, Annu. Rev. Cell Dev. Biol., 1996, 12, 697–715 CrossRef CAS PubMed.
- M. D. Pierschbacher and E. Ruoslahti, Nature, 1984, 309, 30–33 CrossRef CAS.
- A. I. Baba and C. Câtoi, in Comparative Oncology, The Publishing House of the Romanian Academy, Bucharest, 2007, Available from: http://www.ncbi.nlm.nih.gov/books/NBK9549/ Search PubMed.
- R. Brown, C. J. Marshall, S. G. Pennie and A. Hall, EMBO J., 1984, 3, 1321–1326 CAS.
- A. Hall, C. J. Marshall, N. K. Spurr and R. A. Weiss, Nature, 1983, 303, 396–400 CrossRef CAS.
- S. Rasheed, N. Wa, E. M. Toth, P. Arnstein and M. B. Gardner, Cancer, 1974, 33, 1027–1033 CrossRef CAS.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |