DOI:
10.1039/C3BM60283D
(Paper)
Biomater. Sci., 2014,
2, 735-744
Human versus porcine tissue sourcing for an injectable myocardial matrix hydrogel
Received
12th November 2013
, Accepted 15th January 2014
First published on 21st January 2014
Abstract
Heart failure (HF) after myocardial infarction (MI) is a leading cause of death in the western world with a critical need for new therapies. A previously developed injectable hydrogel derived from porcine myocardial matrix (PMM) has had successful results in both small and large animal MI models. In this study, we sought to evaluate the impact of tissue source on this biomaterial, specifically comparing porcine and human myocardium sources. We first developed an analogous hydrogel derived from human myocardial matrix (HMM). The biochemical and physical properties of the PMM and HMM hydrogels were then characterized, including residual dsDNA, protein content, sulfated glycosaminoglycan (sGAG) content, complex viscosity, storage and loss moduli, and nano-scale topography. Biochemical activity was investigated with in vitro studies for the proliferation of vascular cells and differentiation of human cardiomyocyte progenitor cells (hCMPCs). Next, in vivo gelation and material spread were confirmed for both PMM and HMM after intramyocardial injection. After extensive comparison, the matrices were found to be similar, yet did show some differences. Because of the rarity of collecting healthy human hearts, the increased difficulty in processing the human tissue, shifts in extracellular matrix (ECM) composition due to aging, and significant patient-to-patient variability, these studies suggest that the HMM is not a viable option as a scalable product for the clinic; however, the HMM has potential as a tool for in vitro cell culture.
Introduction
Cardiovascular disease is the number one killer in the western world impacting millions of lives each year.1 Heart failure (HF) after myocardial infarction (MI) has limited therapeutic options, including pharmacotherapy, device-based hemodynamic support and orthotopic heart transplantation, and thus new therapies are critically needed. A minimally invasive approach using an injectable therapy, such as a biomaterial alone to stimulate endogenous repair, is a desirable approach and has had success in small and large animal MI models.2,3 Previously, a tissue specific porcine myocardial matrix (PMM) hydrogel, derived from decellularized porcine left ventricular myocardium, was developed as a potential injectable therapy for treating MI.4 This porcine biomaterial has been extensively characterized and tested in small and large animal MI-models with positive results.5–8 In the large animal MI model, the material was delivered via a cardiac injection catheter, and increased cardiac muscle, reduced infarct fibrosis, and improved both global and regional cardiac function, suggesting it has significant translational potential.8
Decellularization techniques have been applied to produce naturally derived biomaterials, which mimic the native tissue environment including both structural and biochemical cues.9,10 In the last decade, decellularized xenogeneic and allogeneic derived biomaterials have been implanted into millions of patients with acceptable tissue responses and positive clinical outcomes.11 The biomaterials have typically been from planar tissue sources such as small intestine submucosa, pericardium, dermis, and bladder, and used as surgical patches or wound healing scaffolds.9,11 The natural components of the extracellular matrix (ECM) are known to be important for cellular migration, attachment, proliferation, viability, differentiation, and maturation and it has been noted that each tissue has a unique ECM composition.12 In fact, there is building evidence that suggests the tissue-specific nature of the ECM is important for driving progenitor and stem cell differentiation and maturation, and promoting regeneration.8,13–17 It was previously shown that the PMM promotes maturation of human embryonic stem cell derived cardiomyocytes and cardiac differentiation of rat cardiac progenitor cells.13,14 While this suggests that species specificity may not be critical, it is possible that using a biomaterial sourced from decellularized human tissue could be more desirable and create a better mimic of the native human myocardial ECM. A human derived biomaterial could also bypass some regulatory hurdles since they avoid certain ethical concerns, immunogenic challenges, and issues with xenogeneic disease transfer. In this study, we developed an injectable human myocardial matrix (HMM) hydrogel, derived from decellularized human cadaveric donor hearts, to evaluate whether there are important differences in tissue sourcing when generating a cardiac specific hydrogel for treating MI.
Results and discussion
Fabrication of human myocardial matrix hydrogel
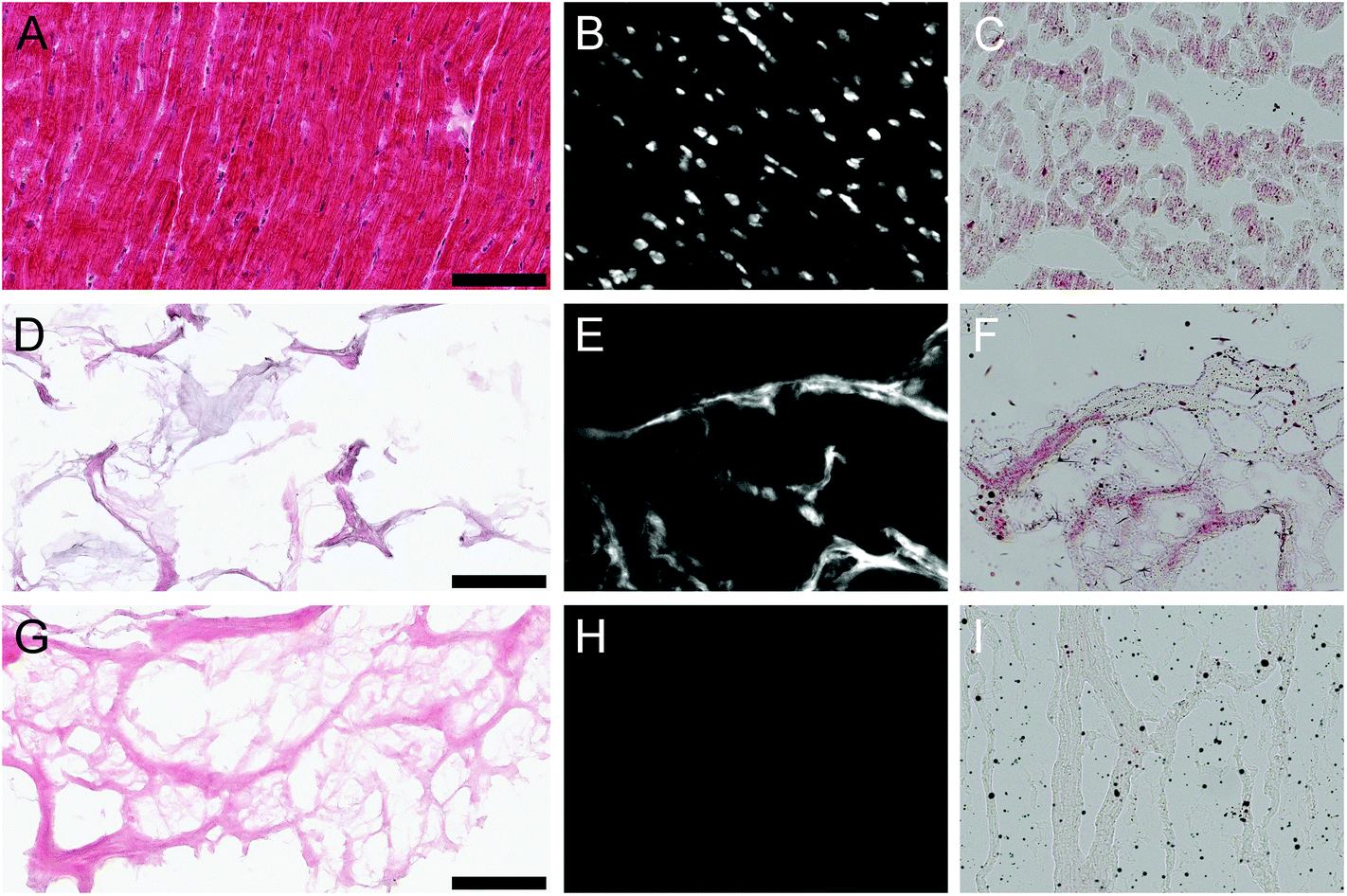
The processing protocol to generate the PMM hydrogel contains 4 main steps: decellularization in SDS, lyophilization, milling, and digestion. Application of this protocol for decellularization of cadaveric human myocardial tissue (Fig. 1A–C) was insufficient for complete decellularization (Fig. 1D) and subsequent processing into a hydrogel. Significant DNA content and lipid content remained (Fig. 1E and F) leading to a lack of in vitro gelation via self-assembly at physiological conditions. Thus, the decellularization process had to be optimized for human tissue including longer decellularization in SDS, and additional DNA and lipid removal steps. This modified decellularization protocol lead to the successful removal of DNA and lipid content as confirmed initially with histology (Fig. 1G–I). Even after optimizing the protocol for removal of cellular content, including DNA and residual lipids, patient-to-patient variability was observed between the materials. Over fifty percent of the decellularized human hearts did not self-assemble into hydrogels at physiological conditions after digestion, but instead formed a white precipitate after adjusting the pH to 7.4. Thus, this prevented the hearts from being used further, either as an injectable myocardial matrix hydrogel or a liquid substrate coating for cell culture. Similar issues with gelation have not been observed with the PMM and could potentially be correlated to age or species difference for the HMM. The optimized protocol was applied to seven human hearts with patient ages that ranged from 41 to 69 years with an average age of 52 ± 10 years.
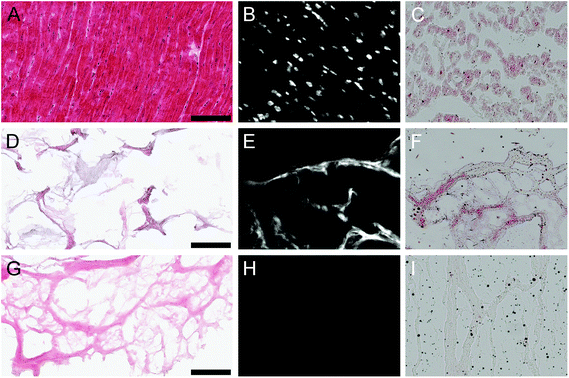 |
| | Fig. 1 Histological analysis of human heart decellularization. Human myocardial tissue fresh (A–C), unsuccessfully decellularized with previously developed porcine myocardial matrix (PMM) protocol (D–F), and fully decellularized with the optimized protocol for human myocardial tissue (G–I). Shown are Hematoxylin and Eosin staining (A, D, G), Hoechst staining for dsDNA (B, E, H), and Oil Red O staining for lipids (C, F, I). The top row shows clear distinguishable nuclei and significant lipid staining in the fresh human myocardial tissue. The middle row indicates incomplete decellularization with positive Hematoxylin in purple, Hoechst, and Oil Red O staining in red. The bottom row shows the optimized human protocol with successful removal of dsDNA and lipid content. Scale bars apply to all images and are 100 μm. | |
Donor age is a significant distinction between the two sources considering tissue was harvested from young adult pigs for PMM, compared to the older human patient donors for HMM. With age there is increased adipose tissue deposition, fibrosis, ECM cross-linking and corresponding stiffening of cardiac tissue.18–21 These factors could have contributed to the increased challenges observed in decellularization of adult human myocardium. Complete decellularization of a tissue is important for host acceptance and triggering an appropriate remodeling outcome. The double stranded DNA (dsDNA) content is considered to be a potential indicator of remaining cellular debris and degree of decellularization.22,23 Complications with removal of DNA from human myocardium have previously been reported.24 Preceding decellularization protocols for human myocardium have also needed additional DNA-removal steps beyond the use of decellularization detergents alone.24,25 Godier-Furnémont et al. utilized surgical explants of human myocardium as a basis for creating a patch in which the application of DNase and RNase enzymes were also used.25 Oberwallner et al. used a variety of techniques for decellularizing sheet forms of human myocardium biopsies and removed residual DNA by incubation with fetal bovine serum (FBS).24 Neither of these protocols investigated the lipid content or removal, potentially due to their use of the ECM scaffold in patch or sheet form. Unlike the porcine hearts, the human hearts contained significant amounts of lipid, which prevented in vitro gelation at physiological conditions after digestion. We therefore added an isopropyl alcohol rinse to remove the lipids.
Previous decellularization techniques of human myocardium used small biopsies from living patients,24,25 but here we chose to decellularize the entire left ventricle from cadaveric donors who did not present with cardiac disease, but whose hearts were rejected for transplant. This was to achieve larger quantities for application and avoid creating site-specific deficits to healthy patients. The damage inflicted to the myocardium due to tissue biopsies, although small, can never fully regenerate and heal. Additionally, the patients undergoing open-heart surgery who are candidates for heart biopsies typically have medical conditions involving diseased myocardium. Thus, biopsies are not a clinically relevant approach for sourcing human myocardial tissue to develop an injectable myocardial matrix therapy as diseased myocardium would not contain the same ratio of ECM components.
Biochemical characterization
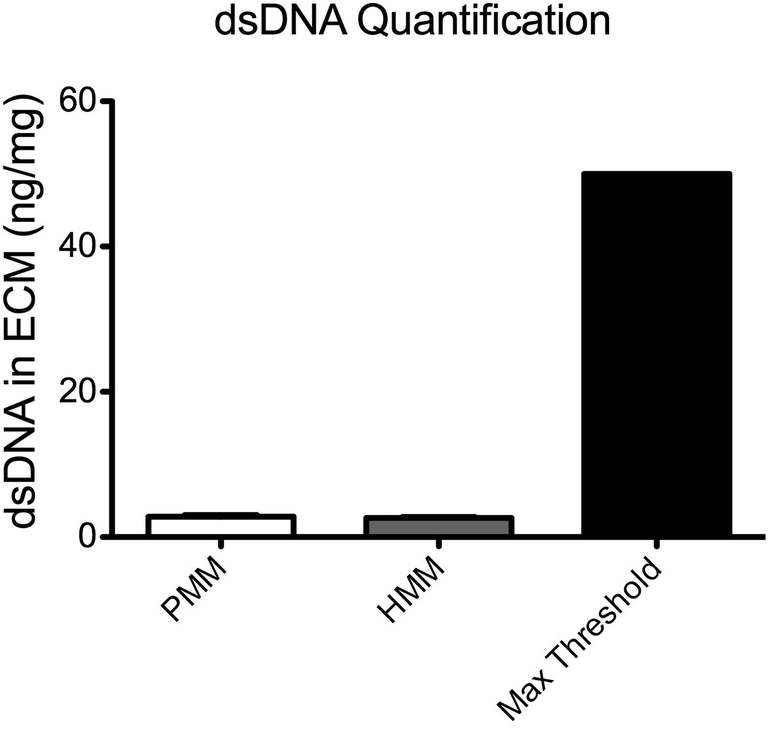
To quantitatively characterize the degree of decellularization, the DNA content from both PMM and HMM was isolated and quantified with a PicoGreen assay (Fig. 2). The PMM had 2.80 ± 0.232 ng of DNA per mg of dry ECM and the HMM had 2.64 ± 0.133 ng DNA per mg of dry ECM, with no significant difference between the two materials. These values are well below a published recommended threshold of 50 ng mg−1 dry ECM for decellularized biomaterials.26 Nucleic acid concentrations were also assessed by absorbance using a NanoDrop machine and no measurable signal was detected. It has been previously shown that removal of DNA content is important in decellularized biomaterials for minimizing potentially negative immune responses once implanted.23,26 Thus, both biomaterials were successfully decellularized to minimize cellular content within the scaffold.
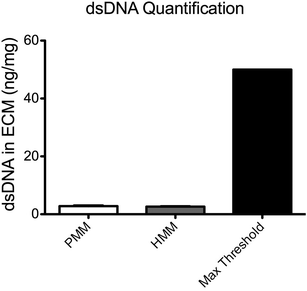 |
| | Fig. 2 Quantification of DNA in the myocardial matrix materials. Plotted is the quantified dsDNA isolated from PMM or HMM per mg of dry ECM. DNA content is very low in both materials and there is no significant difference in the dsDNA content between PMM and HMM. | |
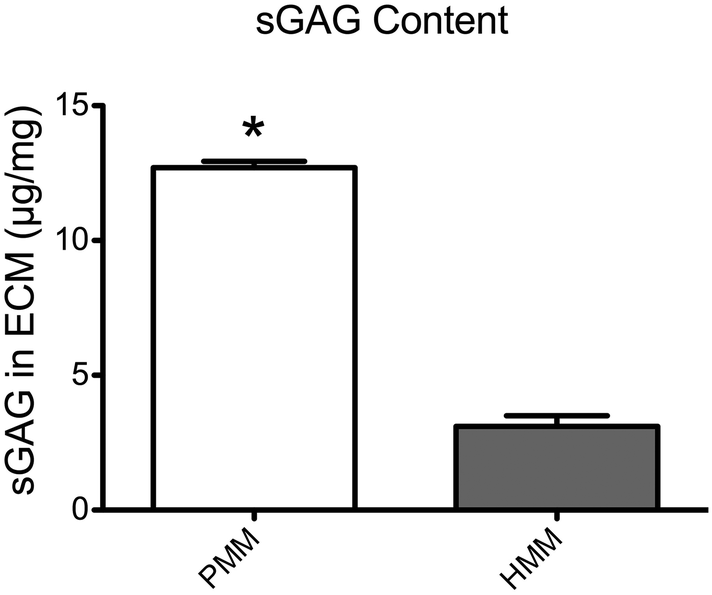
Next, the sulfated glycosaminoglycan content of the ECM materials was investigated. The sGAG content in these ECM scaffolds is potentially important for their regenerative capacity due to the impact on cellular functions and ability to sequester native growth factors.27,28 Utilizing a DMMB assay, the sGAG content of both matrices was quantified and showed a significantly higher level in the PMM compared to HMM (Fig. 3). This decreased content of sGAG in the human based material potentially correlates with the increased processing that was required for sufficient decellularization. Also, the shift in ECM composition in aging hearts due to increased fibrosis or structural proteins, such as collagen, could be another contributing factor for decreased sGAG content in the HMM.20,21
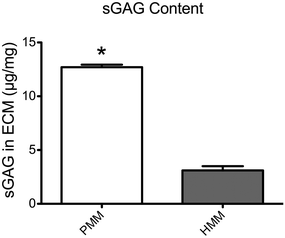 |
| | Fig. 3 Quantification of sulfated glycosaminoglycan (sGAG) content from PMM and HMM as assessed with a DMMB assay. The PMM contains significantly more sGAG than in HMM. *p < 0.0001. | |
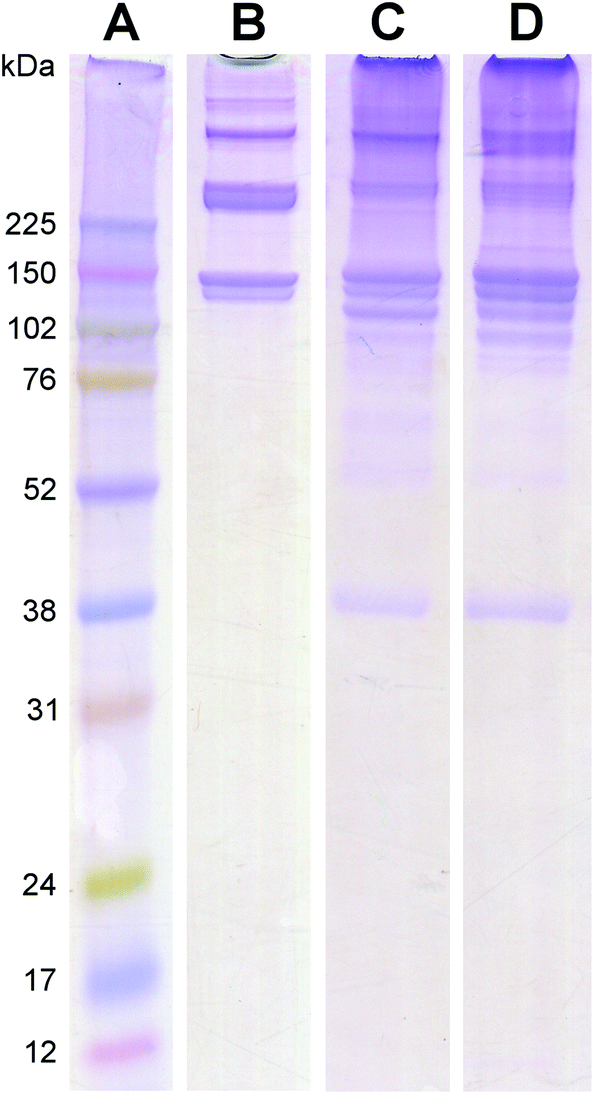
The protein and peptide contents for both the PMM and HMM were characterized with mass spectrometry analysis (Table 1) and visually assessed with PAGE (Fig. 4). PAGE showed similar bands to the collagen control as well as numerous other bands over a wide range of molecular weights, indicating the complexity of the materials. The banding patterns of the PMM and HMM were very similar, although there were some slight differences, particularly in the collagen bands. Shifts in ECM composition with increased collagen content and crosslinking have been previously observed in aging myocardium, and in addition to species variation, could account for the differences seen here between HMM and PMM.18,20,21 The diverse protein composition of the PMM and HMM were confirmed with the results from mass spectrometry. The myocardium is known to contain a variety of the structural ECM proteins due to the substantial dynamic requirements of the tissue under regular cyclic contraction.21 A few additional ECM proteins were identified in HMM compared to PMM (Table 1). However, mass spectrometry is not an all-inclusive technique because of challenges with analyzing a complex ECM sample and with the limitations of current databases. Due to species differences, the materials were analyzed under different peptide databases. There are, however, numerous similarities between the two materials.
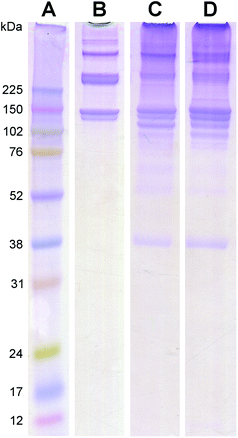 |
| | Fig. 4 PAGE showing molecular weight bands created by the standard ladder (A), rat tail collagen as a control (B), PMM (C), and HMM (D). While PMM and HMM have similar bands and are predominantly collagen, there are some minor differences. | |
Table 1 ECM components identified from mass spectrometry analysis
| |
Porcine myocardial matrixa |
Human myocardial matrix |
|
Data previously reported.13
|
| Collagen I |
X |
X |
| Collagen II |
X |
|
| Collagen III |
X |
X |
| Collagen IV |
X |
X |
| Collagen V |
X |
|
| Collagen VI |
X |
|
| Collagen XII |
|
X |
| Elastin |
X |
X |
| Fibrillin-1 |
X |
X |
| Fibrinogen |
X |
X |
| Fibronectin |
X |
X |
| Fibulin-2 |
|
X |
| Fibulin-3 |
X |
|
| Fibulin-5 |
X |
X |
| Heparan sulfate proteoglycan |
|
X |
| Laminin |
X |
X |
| Lumican |
X |
|
| Periostin |
|
X |
Physical properties
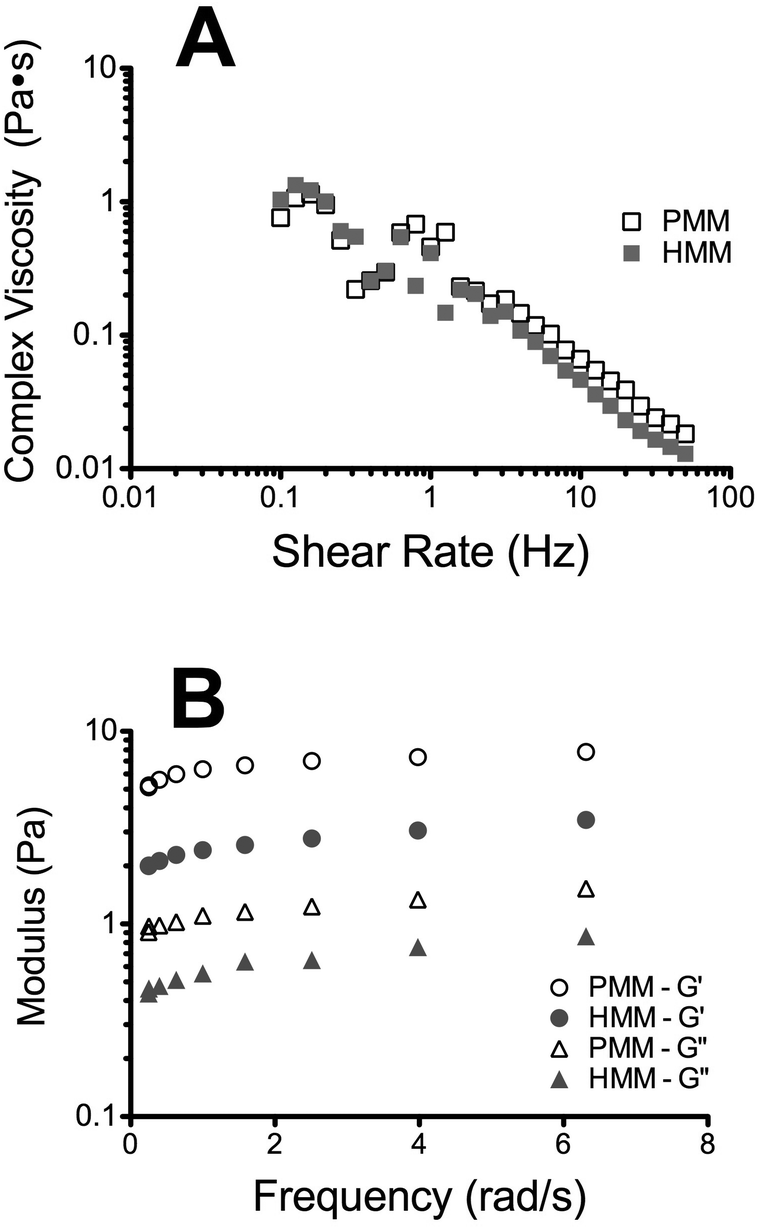
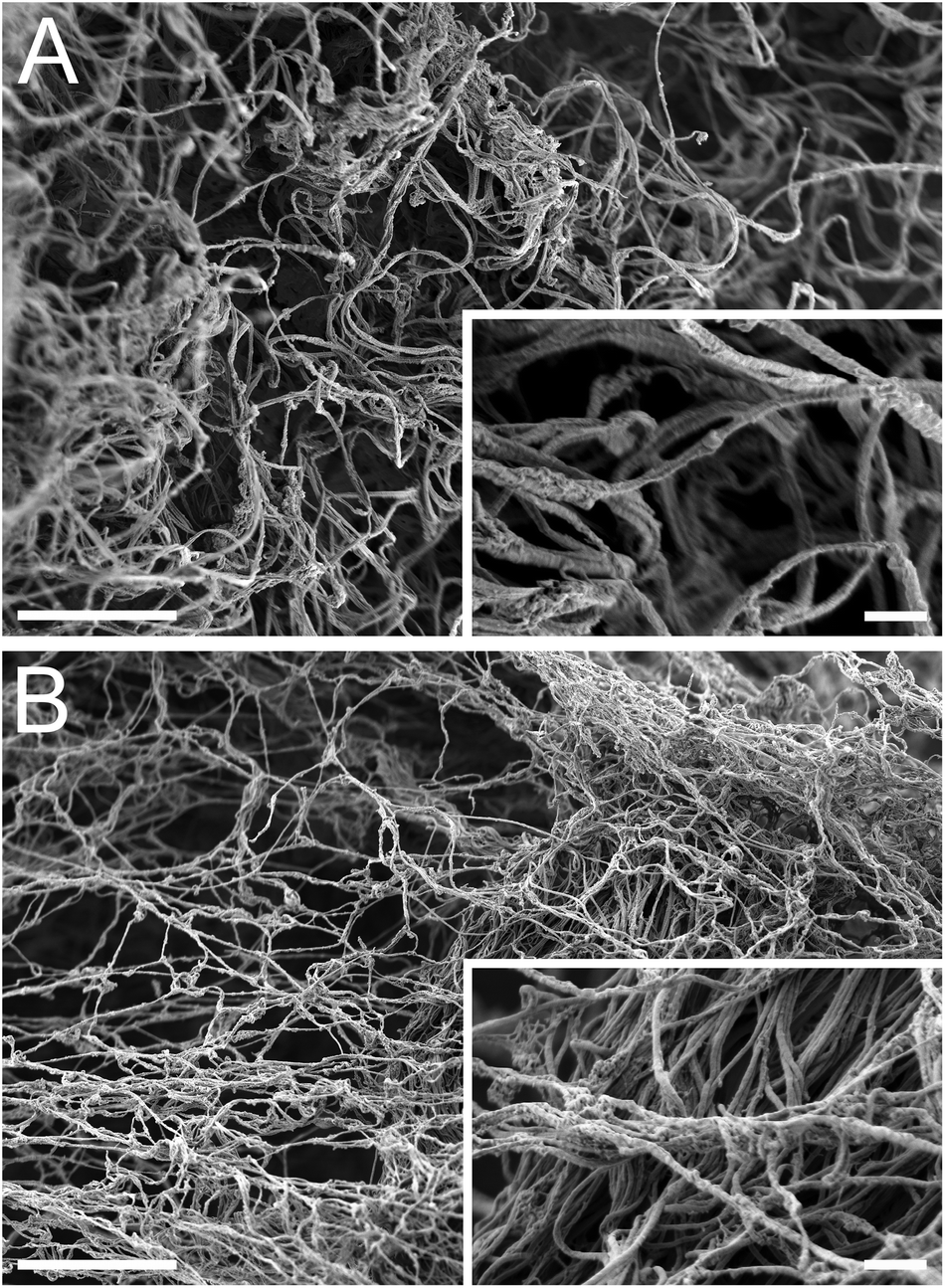
Complex viscosity of the liquid form of the matrices was measured with parallel plate rheometry (Fig. 5A). Both matrices had similar linear profiles when plotted on a log–log scale, indicating that they are shear thinning. This is an important characteristic for translation of an injectable biomaterial when a catheter approach is being utilized. A highly viscous or a shear thickening material could potentially impede or hinder delivery through a catheter. In vitro gelation and self-assembly of both the PMM and HMM materials was then confirmed and their mechanical properties and nano-scale topography were investigated. Both the HMM and PMM form weak hydrogels that hold their form. Measured with a parallel plate rheometer, the storage and loss moduli (Fig. 5B) were both significantly higher for the PMM compared to HMM (p < 0.0001). At 1 Hz, the storage modulus of PMM was 6.08 ± 0.38 Pa and the loss modulus was 1.08 ± 0.01 Pa. For the HMM, the storage modulus was 2.53 ± 0.17 Pa and the loss modulus was 0.57 ± 0.04 Pa. This is not surprising, due to the challenges we faced in processing the HMM and the increased issues due to human lipid content within the material, which impedes gelation. From scanning electron microscopy (SEM) it was observed that both matrices self-assembled into very similar complex networks of linear fibers, producing a nano-scale topography (Fig. 6). This topography is on a scale smaller than a cell and thus can be sensed and manipulated by infiltrating cells upon injection and gelation in host tissue.
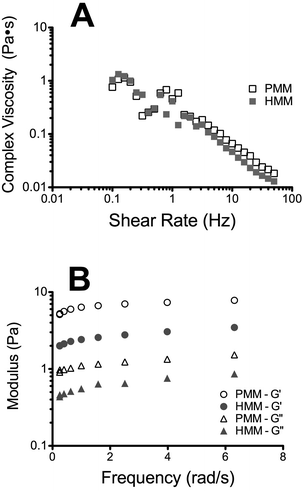 |
| | Fig. 5 Characterization of the mechanical properties using a parallel plate rheometer. Complex viscosity of the liquid form was assessed at 25 °C over a range of shear rates (A). Both materials are plotted on a log–log scale and are shear thinning in the liquid form. Storage modulus (G′) and loss modulus (G′′) were measured after incubation for 24 hours at 37 °C and example curves over a range of frequencies are plotted (B). | |
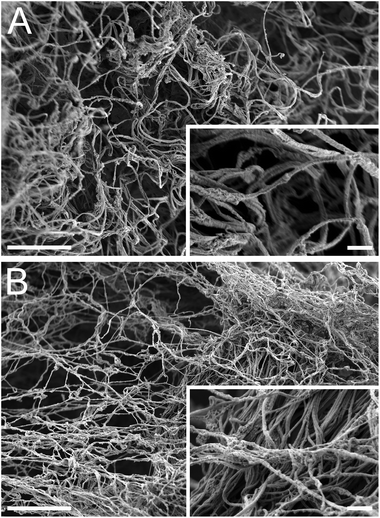 |
| | Fig. 6 Scanning electron microscopy images of both PMM (A) and HMM (B) gels after incubation at 37 °C for 24 hours. The materials produced a complex mesh of nano-scale fibers that were formed via self-assembly. Scale bars for the large images are 4 μm and the smaller inset images are 400 nm. | |
Bioactivity
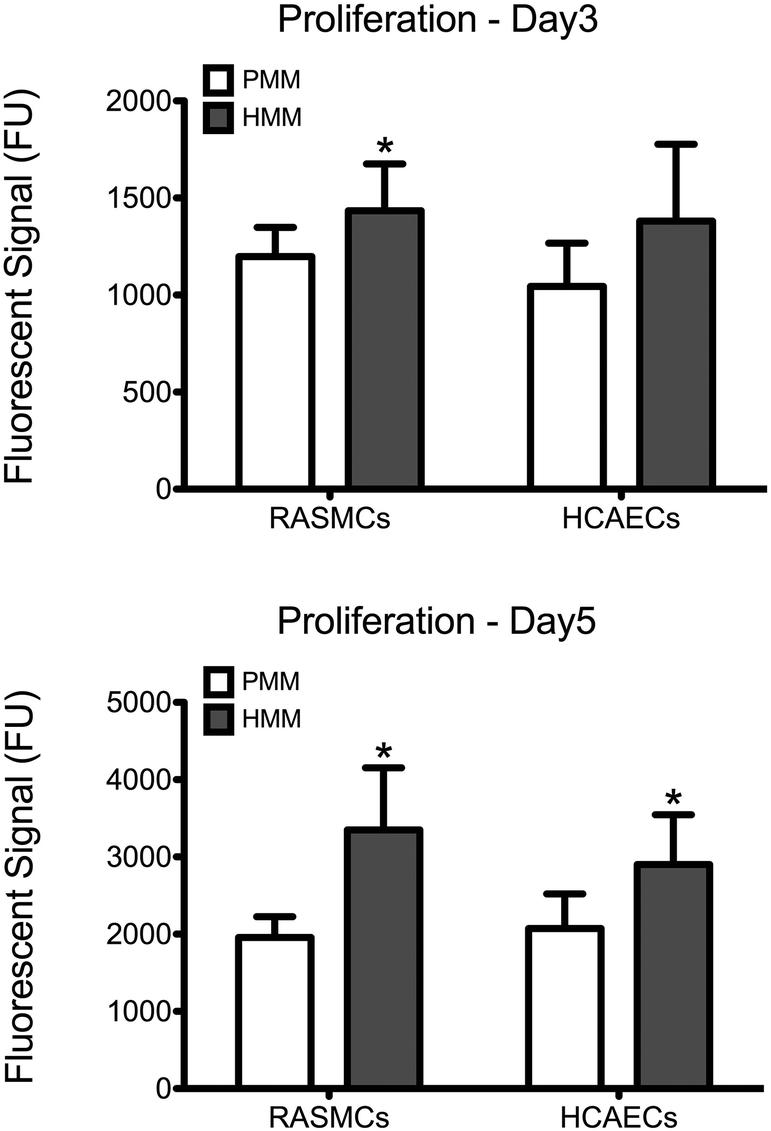
After material characterization, the bioactivity of both matrices was tested in vitro. Cell culture coatings by adsorption of HMM or PMM were utilized in the following studies. This method of generating ECM cell culture coatings has previously been characterized and shown to produce unique and complex coatings.13 Either rat aortic smooth muscle cells (RASMCs) or human coronary artery endothelial cells (HCAECs) were cultured for 3 or 5 days on PMM or HMM and relative cellular quantities were measured (Fig. 7). It has been previously shown that the PMM hydrogel supports infiltration of neovasculature and significant numbers of proliferating cells in rat models.4,7 However, in vitro, HMM consistently had increased cellular proliferation over the PMM coatings. For both day 3 and day 5, HMM lead to significant increases in proliferation for RASMCs over PMM coated surfaces. On day 3 the HCAECs had no significant changes in proliferation, but by day 5 the HMM lead to a significant increase again. The differences in proliferation of these vascular cell types is likely not due to species variation since both coatings are xenogeneic for the RASMCs and the HMM is allogeneic for only the HCAECs. However, this trend could be related to the age difference between the material tissue sources. It was previously shown that the degradation products from porcine small intestinal submucosa (SIS) ECM from older tissue sources lead to greater proliferation of perivascular stem cells.29
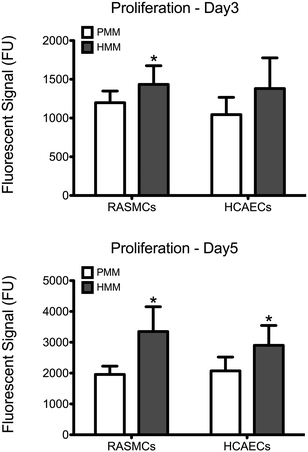 |
| | Fig. 7 Proliferation of rat aortic smooth muscle cells (RASMCs) and human coronary artery endothelial cells (HCAECs) on cell culture coatings of PMM or HMM. Cells were cultured in growth medium for 3 or 5 days and relative proliferation was quantified with a PicoGreen assay. *p < 0.05. | |
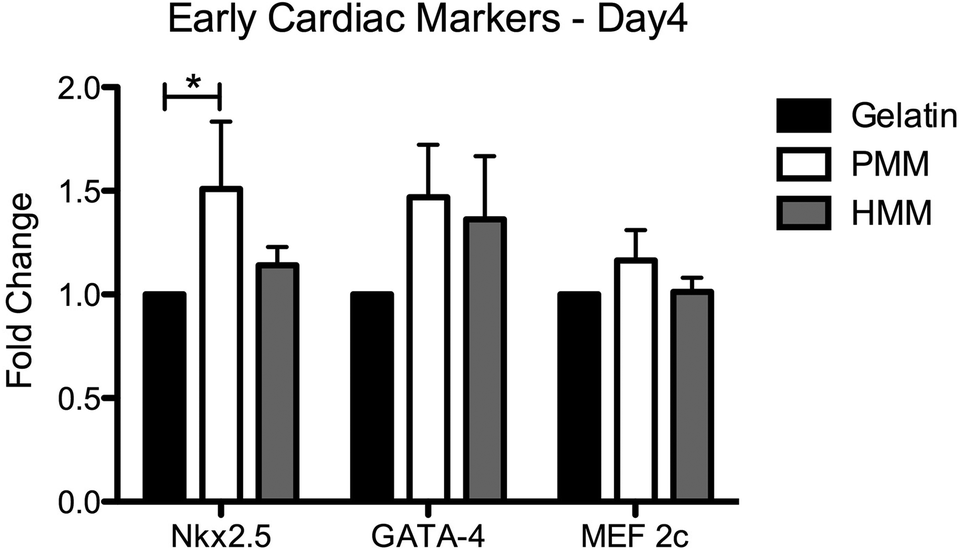
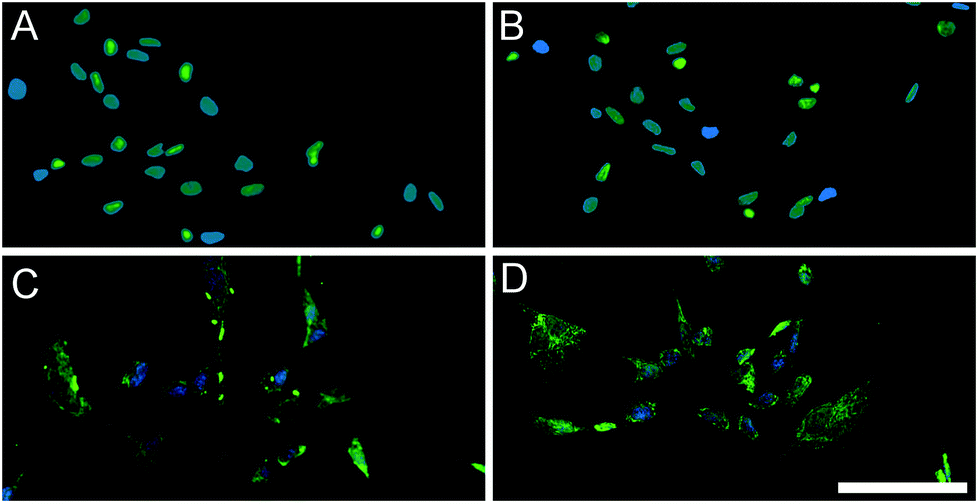
We also assessed the effect of both matrices on the expression of early cardiac transcription factors with human fetal cardiomyocyte progenitor cells (hCMPCs). After four days of culture on coatings made from gelatin, PMM, or HMM, there was a significant increase in the early cardiac marker Nkx2.5 for the PMM coatings compared to standard culture conditions (Fig. 8). There was a trend toward an increase for GATA-4 for both myocardial matrices, although this was not statistically significant. In addition, there were no significant differences between the PMM and HMM coatings with regards to gene expression levels and no morphological differences as observed from bright field images (data not shown). The similar levels of gene expression on PMM and HMM for the early cardiac marker, Nkx2.5, and expression of a cardiac sarcomeric protein, troponin T (TnT), were visually confirmed with IHC staining (Fig. 9). Here we have shown that the biochemical cues from the myocardial matrices enhance early stages of differentiation for hCMPCs, which correlates well with previous studies.14 French et al. cultured rat c-kit+ cardiac progenitor cells on PMM coatings compared to collagen. By day 2 in culture, they had significant increases in the cardiomyocyte lineage markers Nkx2.5, troponin, and alpha-myosin heavy chain with trends for increases in GATA-4.14 It has also been shown that the PMM hydrogel supports the infiltration of c-kit+ progenitors,7 which is hypothesized to play a role in facilitating cardiac regeneration.8 Overall, there were slight changes in the bioactivity of the two matrices. Only PMM was capable of significantly increasing expression of Nkx2.5, while cells cultured on HMM had increased proliferation over PMM. These results suggest that PMM may be more effective at promoting differentiation of cardiac progenitors. It is however unclear how these results relate to the in vivo setting where both proliferation and differentiation are crucial for myocardial regeneration.
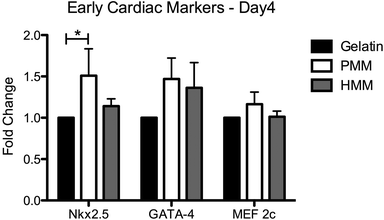 |
| | Fig. 8 RT-PCR for early cardiac markers for human cardiomyocyte progenitor cells (hCMPCs) after culture for four days. Cells were cultured on 2D coatings made from gelatin, PMM, or HMM in proliferation media. *p < 0.05. | |
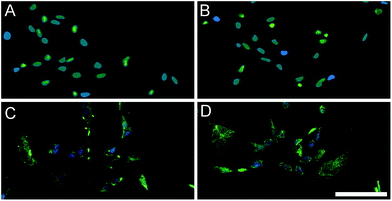 |
| | Fig. 9 hCMPCs after 4 days of culture on PMM (A, C) or HMM (B, D) coatings. Nuclei were stained with Hoechst and shown in blue. An early cardiac marker, Nkx2.5, was stained in green (A, B) and cardiac troponin T was also labeled in green (C, D). The scale bar is 100 μm for all images. | |
In vivo gelation
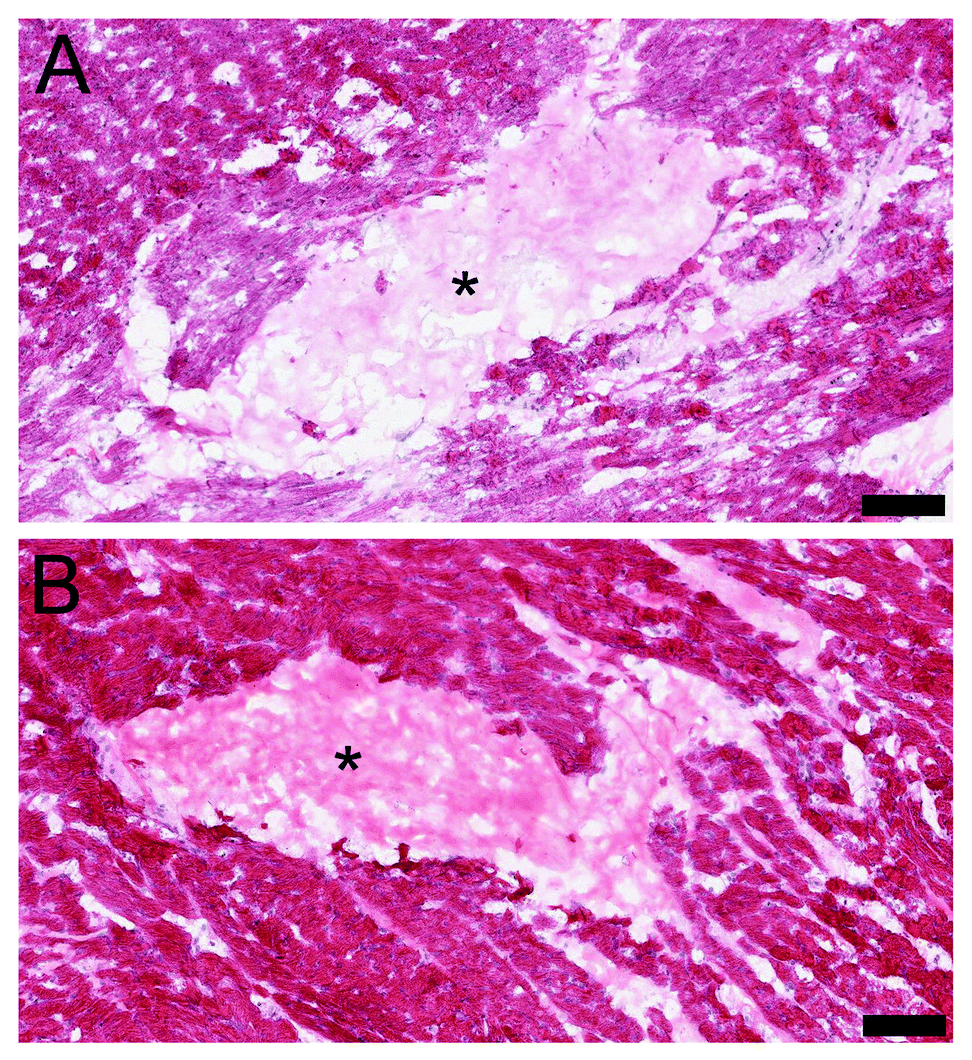
Following the in vitro results, the in vivo gelation was tested by direct injection into the healthy rat myocardium. Both materials showed successful gelation from the bolus injection within the myocardium (Fig. 10). It was noted that a similar porous macrostructure of both of the myocardial matrices was observed upon injection, which is potentially important for cellular infiltration.4 Cellular infiltration, as shown in the previous studies with PMM, is essential for the positive regenerative outcomes that can be stimulated by these injectable biomaterials.7,8 After reaching body temperature the matrices self-assemble into a hydrogel form containing a nano-scale ultrastructure. The porous formation is a crucial aspect of these biomaterials, since overly dense materials can block cellular influx and lead to a negative tissue response such as fibrous encapsulation.30 In a porcine MI model, the influx of cells into the PMM followed by degradation of the biomaterial caused a significant increase in cardiac muscle near the endocardium and a significant decrease in collagen content in the infarcted region.7
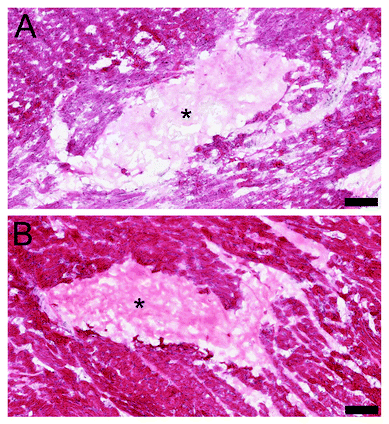 |
| | Fig. 10
In vivo gelation of PMM (A) and HMM (B) in the endocardium of the left ventricle of a Sprague Dawley rat. The injection bolus is indicated with (*) and is surrounded by healthy myocardium. Tissue sections are stained with Hematoxylin and Eosin (H&E). Image scale bars are 100 μm. | |
Assessment of HMM and PMM for translation
Although some differences between the materials were observed, they contained numerous similarities, such as dsDNA content, ECM protein composition and complexity, shear thinning viscosity profiles, nano-scale fibrous structures from self-assembly, and gelation in vivo. Differences between PMM and HMM materials include the methods required for decellularization, sGAG content, collagen banding pattern, and storage and loss moduli. In vitro bioactivity analysis indicated that HMM did not cause a significant increase in the expression of Nkx2.5 for hCMPCs like the PMM, but did lead to an increase in proliferation of two vascular cell types over PMM. There are, however, many setbacks that diminish the potential allogeneic benefits of HMM as a viable clinical therapy. For example, complications arose in producing the HMM material due to significant patient-to-patient variability. Differences between the matrices and complications associated with HMM were potentially correlated to patient age since cadaveric tissue donors were significantly older than the porcine sources. It has also been previously shown that with aging there is an increase in ECM crosslinking and shifts in ECM composition, especially due to increased fibrosis and adipose tissue deposition in aging human hearts.20,21 Tissue age for ECM materials has formerly been reported to cause variation in material properties.29 These changes in ECM due to aging can lead to a decreased regenerative capacity of the corresponding decellularized biomaterials as well.31 Also, the human hearts collected from this study are relatively rare considering they were rejected as candidates for transplantation and yet were healthy, involving no serious cardiovascular conditions. From a translational and clinical perspective, the variability within the production of the HMM and shifts in ECM composition and bioactivity due to aging indicates that PMM is a more promising choice for the basis of a new therapy. However, HMM still has potential for applications as a cell culture substrate for in vitro studies, such as those examining stem cell differentiation and different cardiomyopathies.
Conclusion
Here we have shown the development of an injectable human myocardial matrix hydrogel that was extensively compared to a previously studied porcine myocardial matrix hydrogel as a potential injectable therapy for treatment of ischemic cardiomyopathy. Such studies are important due to the increasing evidence of tissue specific cues within ECM constructs for promoting in vitro differentiation and in vivo regeneration. Increased difficulty in processing and significant patient-to-patient variability of the HMM suggests HMM is not a viable option for translation as an injectable biomaterial therapy; however, the HMM still has potential applications including use as a means for studying diseased human ECM, or as a cell culture coating for differentiation protocols.
Experimental
Porcine myocardial matrix
The porcine myocardial matrix was previously developed and detailed methods are described elsewhere.4 In brief, fresh porcine myocardial tissue from the left ventricle was isolated and cut into small pieces, rinsed, and then spun in sodium dodecyl sulfate (SDS) solution for 3–5 days with daily solution changes. Once fully decellularized the tissue was rinsed, frozen, lyophilized, and milled into a fine powder. The porcine myocardial matrix powder was then digested with pepsin for 48 hours and brought to physiological conditions for salt and pH. The material was then frozen and lyophilized and kept at −80 °C for long-term storage. Before use the material was resuspended with water back to the original concentration of the liquid form of 6 mg mL−1.
Human myocardial matrix
Human hearts were obtained from organ donors whose hearts could not be used for transplantation under an institutionally approved protocol. The previously published protocol for decellularizing porcine myocardial tissue4 was not sufficient for decellularizing human myocardial tissue and needed additional modifications. The human heart decellularization protocol starts with the isolation of the entire left ventricle by the removal of epicardial adipose tissue, papillary muscles, chordae tendineae, and valves. The tissue was then cut into small cubes with side lengths of 1–3 mm each. The tissue was placed into 1 L beakers with 25–35 g of tissue per beaker and spun at 125 rpm in ultrapure water for 30 minutes. The tissue was then rinsed and spun in 1% (wt/vol) SDS (Fischer Scientific, Fair Lawn, NJ) phosphate buffered saline (PBS), and 0.5% penicillin streptomycin (PS) of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1 (Gibco, Life Technologies, Grand Island, NY) for 2 hours. The solution was then replaced daily for 6–8 days until the tissue was white in color. The tissue was then rinsed by spinning in water for 2 hours and placed into isopropyl alcohol (IPA) (Fisher Scientific, Fair Lawn, NJ) for 12–24 hours for lipid removal. Next, the tissue was rinsed twice in water for 15 minutes each and divided into 50 mL conicals with a DNase/RNase solution. The DNase/RNase solution consists of 40 U mL−1 DNase (Sigma-Aldrich, St. Louis, MO) and 1 U mL−1 RNase A (Qiagen, Hilden, Germany) in 40 mM tri-hydrochloric acid (HCl), 6 mM magnesium chloride, 1 mM calcium chloride, and 10 mM sodium chloride at pH 7.4. The conicals with tissue in the DNase/RNase solution were incubated at 37 °C on a shaker plate for 24 hours. Following incubation the tissue was rinsed with water and placed back into the SDS in PBS with PS and spun for 24 hours at 125 rpm. The tissue was then briefly shaken in 0.001% Triton X-100 solution (Sigma-Aldrich, St. Louis, MO), then spun in the same solution for 30–45 minutes and finally spun in water again for 24 hours. Then the tissue was shaken in water, frozen, lyophilized and milled using a Wiley® Mini-Mill into a fine powder through a #40 sieve. Two decellularized human hearts were combined for all analysis and testing. The human material was then treated with the same methods as the porcine myocardial matrix powder for all further processing.
000 U mL−1 (Gibco, Life Technologies, Grand Island, NY) for 2 hours. The solution was then replaced daily for 6–8 days until the tissue was white in color. The tissue was then rinsed by spinning in water for 2 hours and placed into isopropyl alcohol (IPA) (Fisher Scientific, Fair Lawn, NJ) for 12–24 hours for lipid removal. Next, the tissue was rinsed twice in water for 15 minutes each and divided into 50 mL conicals with a DNase/RNase solution. The DNase/RNase solution consists of 40 U mL−1 DNase (Sigma-Aldrich, St. Louis, MO) and 1 U mL−1 RNase A (Qiagen, Hilden, Germany) in 40 mM tri-hydrochloric acid (HCl), 6 mM magnesium chloride, 1 mM calcium chloride, and 10 mM sodium chloride at pH 7.4. The conicals with tissue in the DNase/RNase solution were incubated at 37 °C on a shaker plate for 24 hours. Following incubation the tissue was rinsed with water and placed back into the SDS in PBS with PS and spun for 24 hours at 125 rpm. The tissue was then briefly shaken in 0.001% Triton X-100 solution (Sigma-Aldrich, St. Louis, MO), then spun in the same solution for 30–45 minutes and finally spun in water again for 24 hours. Then the tissue was shaken in water, frozen, lyophilized and milled using a Wiley® Mini-Mill into a fine powder through a #40 sieve. Two decellularized human hearts were combined for all analysis and testing. The human material was then treated with the same methods as the porcine myocardial matrix powder for all further processing.
Material characterization
Histological analysis.
Tissue samples were fresh frozen before and after decellularization for histological analysis. Then 10 μm cryosections were stained with Hematoxylin and Eosin (H&E), Hoechst, and Oil Red O. Slides were imaged with Leica Aperio ScanScope® CS2, Carl Zeiss Observer D1, and Zeiss Imager A1 respectively.
Material composition.
DNA was isolated from the HMM and PMM using a NucleoSpin® Tissue kit (Macherey-Nagel, Duren, Germany). Nucleic acid content was quantified with a Thermo Scientific NanoDrop 2000c spectrophotometer. The dsDNA content was then quantified (n = 3) with a Quant-iT™ PicoGreen® dsDNA Assay Kit (Invitrogen, Eugene, OR). The sulfated glycosaminoglycan (sGAG) content was quantified (n = 3) with a DMMB assay.32 The DMMB protocol was implemented as previously described on the liquid form of the matrices. The DMMB working solution was made fresh each time without dilution and Chondroitin Sulfate (Sigma-Aldrich, St. Louis, MO) was used as the standard. Protein fragment size was characterized using a NuPAGE 12% Bis-Tris Gel (Novex, Life Technologies, Carlsbad, CA). Protein composition was characterized using mass spectrometry. The samples were initially prepared as previously reported.33 The recovered peptides from the FASP method were then extracted using Aspire RP30 desalting columns (Thermo Scientific) before injection into the mass spectrometer. Trypsin-digested peptides were analyzed by liquid chromatography (LC)-MS/MS with nanospray ionization. All nanospray ionization experiments were performed using a QSTAR-Elite hybrid mass spectrometer (ABSCIEX) interfaced to a nanoscale reversed-phase high-pressure liquid chromatography (Tempo) using a 10 cm 180 ID glass capillary packed with 5 μm C18 Zorbax™ beads (Agilent). The buffer compositions were as follows. Buffer A was composed of 98% H2O, 2% ACN, 0.2% formic acid, and 0.005% TFA; buffer B was composed of 100% ACN, 0.2% formic acid, and 0.005% TFA. Peptides were eluted from the C-18 column into the mass spectrometer using a linear gradient of 5–60% Buffer B over 60 min at 400 μL min−1. LC-MS/MS data were acquired in a data-dependent fashion by selecting the 6 most intense peaks with charge state of 2 to 4 that exceeds 20 counts, with exclusion of former target ions set to “120 seconds” and the mass tolerance for exclusion set to 100 ppm. Time-of-flight MS were acquired at m/z 400 to 1800 Da for 0.5 seconds with 12 time bins to sum. MS/MS data were acquired from m/z 50 to 2000 Da by using “enhance all” function and 24 time bins to sum, dynamic background subtract, automatic collision energy, and automatic MS/MS accumulation with the fragment intensity multiplier set to 6 and maximum accumulation set to 2 s before returning to the survey scan. Peptide identifications were made using Mascot® (Matrix Sciences) and paragon algorithm executed in Protein Pilot 3.0 (ABSCIEX).
Viscosity and in vitro gelation characterization.
The complex viscosities of the resuspended liquid form of both matrices (n = 3) were measured with TA Instruments ARG2 Rheometer using a 20 mm parallel plate geometry as previously described.6 Gelation in vitro was confirmed by incubation of the liquid form at 37 °C for 24 hours. Gels of 500 μL in volume were assessed (n = 3) for storage (G′) and loss moduli (G′′) as previously described.6 The storage moduli and loss moduli were assessed over a range of frequencies. For statistical analysis the moduli values at 1 Hz were compared.
Gels were imaged for nano-scale fiber formation with scanning electron microscopy (SEM) similar to previously described methods.6 Samples of 100 μL volume were gelled in a 96-well plate for 24 hours at 37 °C. The gels were then fixed in a solution of 4% paraformaldehyde and 4% glutaraldehyde for 24 hours. Then the gels were dehydrated with a series of gradated ethanol rinses. Fixed and dehydrated hydrogels were loaded into Teflon sample holders and processed in an automated critical point drier (Leica EM CPD300, Leica, Vienna), which was set to perform 40 exchange cycles of CO2 at medium speed and 40% stirring. The fill and heating steps were performed at slow speed, while the venting step was performed at medium speed. Mounted samples were then sputter coated (Leica SCD500, Leica, Vienna) with approximately 7 nm of platinum while being rotated. The samples were then imaged on a FE-SEM (Sigma VP, Zeiss Ltd, Cambridge, UK) at 0.6 kV using the in-lens SE1 detector.
In vitro cell culture
ECM cell culture coatings.
Cell culture coatings from PMM or HMM were made fresh each time by re-suspending the ECM in 0.1 M acetic acid to a concentration of 1 mg mL−1. The solution was then incubated on the tissue culture plastic for 1 hour at 37 °C. After incubation the solution was removed and the surface was rinsed 2–3 times with PBS before cells were plated as previously reported.13
Proliferation of vascular cells.
Two relevant vascular cell types were used for this study: rat aortic smooth muscle cells (RASMCs) and human coronary artery endothelial cells (HCAECs). The RASMCs were isolated as previously described4,34 and cultured on collagen coated plates during expansion in growth media of Dulbecco's Modified Eagle's Medium (DMEM) (Corning Cellgro, Manassas, VA) with 10% Fetal Bovine Serum (FBS) (Sigma-Aldrich, St. Louis, MO) and 1% Penicillin Streptomycin (PS) (Gibco, Life Technologies, Grand Island, NY). The HCAECs were purchased (Cell Applications Inc, San Diego, CA) and cultured in MesoEndo Endothelial cell media (Cell Applications). The wells of a polystyrene 96-well microtiter plate were coated with either PMM or HMM (n = 8) as described above. On day 0, a total of 5 × 102 RASMCs or HCAECs were seeded into each well. Then, 24 hours later the media was changed and wells were rinsed twice with PBS. Every two days, the media was exchanged for fresh media and cells were rinsed twice with PBS. Following 3 and 5 day time point intervals, relative cell proliferation was measured by quantifying dsDNA content in each well with Quant-iT™ PicoGreen® dsDNA Assay Kit (Invitrogen, Eugene, OR).
Human cardiomyocyte progenitor cells.
Human fetal cardiomyocyte progenitor cells (hCMPCs) were isolated from tissue harvested under standard informed consent procedures and prior approval of the ethics committee of the University Medical Center Utrecht. All cell studies were done in compliance with the University of California, San Diego Institutional Review Board. hCMPCs were isolated from human fetal hearts by selection with Sca-1+ magnetic bead sorting and cultured on 0.1% gelatin in growth media during expansion and passaged as previously described.35,36 Growth media for these studies consisted of 25% EGM-2 (3% EGM-2 single quotes (Cambrex, cat. no. CC-4176) in EBM-2 (Cambrex, cat. no. CC-3156)) and 75% M199 (BioWhittaker, cat. no. BE12-119F), 10% FBS (Hyclone, cat. no. CH30160.30), 1× MEM non-essential amino acids (BioWhittaker, cat. no. BE13-114E) and 1% penicillin/streptomycin (Sigma, cat. no. P4458). For gene expression analysis and immunohistochemistry, the hCMPCs were plated in 6-well plates with 70000 cells (P7-11) per well on either gelatin, PMM, or HMM coatings in growth media. Cells on day 2 were either analyzed or passaged and re-plated on new coatings, and then analyzed at day 4. All experiments were done in triplicate using cells from four separate donors that were studied in parallel (n = 4).
RT-PCR on hCMPCs.
The hCMPCs RNA was isolated by NucleoSpin RNAII column (Macherey-Nagal, cat no. 740955.250). The RNA concentration and purity was then measured using a Thermo Scientific NanoDrop 2000c spectrophotometer. The cDNA was synthesized using iScript cDNA synthesis Kit (cat. no. 170-8891, Bio-Rad) and quantitative RT-PCR amplification was detected in a MyIQ single-color real-time polymerase chain reaction system using iQ SYBR Green Supermix (170-8884, Bio-Rad). Primers used included the following: GATA-4 (fw – GTTTTTTCCCCTTTGATTTTTGATC, rv – AACGACGGCAACAACGATAAT), hNkx2.5 (fw – CCCCTGGATTTTGCATTCAC, rv – CGTGCGCAAGAACAAACG), MEF2c (fw – TCGGGTCTTCCTTCATCAG, rv – GTTCATCCATAATCCTCGTAATC), and the house keeping gene GAPDH (fw – CTCTGACTTCAACAGCGACA, rv – TCTCTCTCTTCCTCTTGTGC).
IHC on hCMPCs.
Cells were fixed with 4% PFA for 15 minutes, permeabilized with 0.1% Trion X-100, 1% BSA in PBS for 10 minutes and blocked with 10% goat serum in PBS for 1 hour. The following primary antibodies were then separately incubated overnight on the samples at 4 °C in 1% goat serum: Nkx2.5 (Santa Cruz, sc-14033 at 1:100) and TnT (Santa Cruz, sc-20643 at 1:100). The cells were then incubated for 1 hour at room temperature with a goat anti-rabbit secondary (Alexa488, A11008) in 0.01% Triton X-100, 0.1% BSA in PBS and counterstained with Hoechst.
In vivo gelation
All experiments in this study were performed in accordance with the guidelines established by the committee on Animal Research at the University of California, San Diego, and the American Association for Accreditation of Laboratory Animal Care. In vivo gelation was tested in female Sprague Dawley (SD) rats. Injections were performed as previously described.4,37 Both the human and porcine myocardial matrices resuspended in liquid form at 6 mg mL−1 concentration were separately injected three times each with a volume of 50 μL into the left ventricular free wall. After 20 minutes the animals were sacrificed, then the hearts were harvested and rapidly frozen in OCT. Short axis sections of 10 μm were taken and stained with H&E to confirm material spread and gelation within the wall of the heart.
Statistical analysis
All graphs with error bars are plotted as mean ± standard deviation (SD) and studies were done in triplicate unless otherwise indicated. Significance was determined with an unpaired student's t-test or a one-way ANOVA using a Tukey post-hoc test with a p < 0.05.
Acknowledgements
The authors would like to thank Dr Ghassemian and Matt Joens for their technical expertise in mass spectrometry and scanning electron microscopy, respectively. The authors would also like to thank Lifesharing for their assistance in obtaining the human cardiac tissue. Funding for this study was provided by the NIH Heart, Lung, Blood Institute 5R01HL113468 (KLC) and the Leducq Foundation Transatlantic Career Development Award (RG). TDJ received funding during this project from the NSF as a graduate student fellow, the NHLBI as a training grant recipient, and as a Powell Fellow from the Powell Foundation. KLC is co-founder, board member, and holds equity interest in Ventrix, Inc.
Notes and references
- V. L. Roger, A. S. Go, D. M. Lloyd-Jones, E. J. Benjamin, J. D. Berry, W. B. Borden, D. M. Bravata, S. Dai, E. S. Ford, C. S. Fox, H. J. Fullerton, C. Gillespie, S. M. Hailpern, J. A. Heit, V. J. Howard, B. M. Kissela, S. J. Kittner, D. T. Lackland, J. H. Lichtman, L. D. Lisabeth, D. M. Makuc, G. M. Marcus, A. Marelli, D. B. Matchar, C. S. Moy, D. Mozaffarian, M. E. Mussolino, G. Nichol, N. P. Paynter, E. Z. Soliman, P. D. Sorlie, N. Sotoodehnia, T. N. Turan, S. S. Virani, N. D. Wong, D. Woo and M. B. Turner, Circulation, 2012, 125, e2–e220 CrossRef PubMed.
- A. A. Rane and K. L. Christman, J. Am. Coll. Cardiol., 2011, 58, 2615–2629 CrossRef CAS PubMed.
- T. D. Johnson and K. L. Christman, Expert Opin. Drug Deliv., 2012, 1–14 CAS.
-
J. M. Singelyn, J. A. DeQuach, S. B. Seif-Naraghi, R. B. Littlefield, P. J. Schup-Magoffin and K. L. Christman, Biomaterials, Elsevier Ltd, 2009, vol. 30, pp. 5409–5416 Search PubMed.
- J. M. Singelyn and K. L. Christman, Macromol. Biosci., 2011, 11, 731–738 CrossRef CAS PubMed.
- T. D. Johnson, S. Y. Lin and K. L. Christman, Nanotechnology, 2011, 22, 494015 CrossRef PubMed.
- J. M. Singelyn, P. Sundaramurthy, T. D. Johnson, P. J. Schup-Magoffin, D. P. Hu, D. M. Faulk, J. Wang, K. M. Mayle, K. Bartels, M. Salvatore, A. M. Kinsey, A. N. Demaria, N. Dib and K. L. Christman, J. Am. Coll. Cardiol., 2012, 59, 751–763 CrossRef CAS PubMed.
- S. B. Seif-Naraghi, J. M. Singelyn, M. A. Salvatore, K. G. Osborn, J. J. Wang, U. Sampat, O. L. Kwan, G. M. Strachan, J. Wong, P. J. Schup-Magoffin, R. L. Braden, K. Bartels, J. A. DeQuach, M. Preul, A. M. Kinsey, A. N. Demaria, N. Dib and K. L. Christman, Sci. Transl. Med., 2013, 5, 173ra125 CrossRef PubMed.
- P. M. Crapo, T. W. Gilbert and S. F. Badylak, Biomaterials, 2011, 32, 3233–3243 CrossRef CAS PubMed.
- T. Gilbert and T. Sellaro, Biomaterials, 2006, 27, 3675–3683 CAS.
- S. F. Badylak and T. W. Gilbert, Semin. Immunol., 2008, 20, 109–116 CrossRef CAS PubMed.
- S. Uriel, E. Labay, M. Francis-Sedlak, M. L. Moya, R. R. Weichselbaum, N. Ervin, Z. Cankova and E. M. Brey, Tissue Eng., Part C, 2009, 15, 309–321 CrossRef CAS PubMed.
- J. A. DeQuach, V. Mezzano, A. Miglani, S. Lange, G. M. Keller, F. Sheikh and K. L. Christman, PLoS One, 2010, 5, e13039 Search PubMed.
- K. M. French, A. V. Boopathy, J. A. DeQuach, L. Chingozha, H. Lu, K. L. Christman and M. E. Davis, Acta Biomater., 2012, 8, 4357–4364 CrossRef CAS PubMed.
- J. Cortiella, J. Niles, A. Cantu, A. Brettler, A. Pham, G. Vargas, S. Winston, J. Wang, S. Walls and J. E. Nichols, Tissue Eng., Part A, 2010, 16, 2565–2580 CrossRef CAS PubMed.
- T. L. Sellaro, A. K. Ravindra, D. B. Stolz and S. F. Badylak, Tissue Eng., 2007, 13, 2301–2310 CrossRef CAS PubMed.
- N. C. Cheng, B. T. Estes, H. A. Awad and F. Guilak, Tissue Eng., Part A, 2009, 15, 231–241 CrossRef CAS PubMed.
- M. L. Burgess, J. C. McCrea and H. L. Hedrick, Mech. Ageing Dev., 2001, 122, 1739–1756 CrossRef CAS.
- C. Leeuwenburgh, P. Wagner, J. O. Holloszy, R. S. Sohal and J. W. Heinecke, Arch. Biochem. Biophys., 1997, 346, 74–80 CrossRef CAS PubMed.
-
N. G. F. Anna Biernacka, Aging and Disease, JKL International LLC, 2011, vol. 2, p. 158 Search PubMed.
- R. R. de Souza, Biogerontology, 2002, 3, 325–335 CrossRef CAS.
- T. J. Keane, R. Londono, N. J. Turner and S. F. Badylak, Biomaterials, 2012, 33, 1771–1781 CrossRef CAS PubMed.
- M. H. Zheng, J. Chen, Y. Kirilak, C. Willers, J. Xu and D. Wood, J. Biomed. Mater. Res., 2005, 73B, 61–67 CrossRef CAS PubMed.
- B. Oberwallner, A. Brodarac, Y.-H. Choi, T. Saric, P. Anić, L. Morawietz and C. Stamm, J. Biomed. Mater. Res., 2013 Search PubMed.
- A. F. G. Godier-Furnémont, T. P. Martens, M. S. Koeckert, L. Wan, J. Parks, K. Arai, G. Zhang, B. Hudson, S. Homma and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7974–7979 CrossRef PubMed.
- J. E. Reing, B. N. Brown, K. A. Daly, J. M. Freund, T. W. Gilbert, S. X. Hsiong, A. Huber, K. E. Kullas, S. Tottey, M. T. Wolf and S. F. Badylak, Biomaterials, 2010, 31, 8626–8633 CrossRef CAS PubMed.
- S. B. Seif-Naraghi, D. Horn, P. J. Schup-Magoffin and K. L. Christman, Acta Biomater., 2012, 8, 3695–3703 CrossRef CAS PubMed.
- R. Raman, V. Sasisekharan and R. Sasisekharan, Chem. Biol., 2005, 12, 267–277 CrossRef CAS PubMed.
- S. Tottey, S. A. Johnson, P. M. Crapo, J. E. Reing, L. Zhang, H. Jiang, C. J. Medberry, B. Reines and S. F. Badylak, Biomaterials, 2011, 32, 128–136 CrossRef CAS PubMed.
- J. E. Valentin, J. S. Badylak, G. P. McCabe and S. F. Badylak, J. Bone Joint Surg. Am., 2006, 88, 2673–2686 CrossRef PubMed.
- B. M. Sicari, S. A. Johnson, B. F. Siu, P. M. Crapo, K. A. Daly, H. Jiang, C. J. Medberry, S. Tottey, N. J. Turner and S. F. Badylak, Biomaterials, 2012, 33, 5524–5533 CrossRef CAS PubMed.
- I. Barbosa, S. Garcia, V. Barbier-Chassefière, J.-P. Caruelle, I. Martelly and D. Papy-García, Glycobiology, 2003, 13, 647–653 CrossRef CAS PubMed.
- J. R. Wisniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359–362 CrossRef CAS PubMed.
- J. D. San Antonio, M. J. Karnovsky, M. E. Ottlinger, R. Schillig and L. A. Pukac, Arterioscler. Thromb., 1993, 13, 748–757 CrossRef CAS.
- R. Gaetani, P. A. Doevendans, C. H. G. Metz, J. Alblas, E. Messina, A. Giacomello and J. P. G. Sluijter, Biomaterials, 2012, 33, 1782–1790 CrossRef CAS PubMed.
- A. M. Smits, P. van Vliet, C. H. Metz, T. Korfage, J. P. Sluijter, P. A. Doevendans and M.-J. Goumans, Nat. Protoc., 2009, 4, 232–243 CrossRef CAS PubMed.
- K. L. Christman, H. H. Fok, R. E. Sievers, Q. Fang and R. J. Lee, Tissue Eng., 2004, 10, 403–409 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.