 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterisation and evaluation of the impact of microfabricated pockets on the performance of limbal epithelial stem cells in biodegradable PLGA membranes for corneal regeneration
Ilida
Ortega
*a,
Robert
McKean
b,
Anthony J.
Ryan
c,
Sheila
MacNeil
*a and
Frederik
Claeyssens
*a
aBiomaterials and Tissue Engineering Group, Department of Materials Science and Engineering, Kroto Research Institute, University of Sheffield, Broad Lane, S3 7HQ, Sheffield, UK. E-mail: I.Ortega@sheffield.ac.uk; S.MacNeil@sheffield.ac.uk; F.Claeyssens@sheffield.ac.uk
bThe Electrospinning Company Ltd, Rutherford Appleton Laboratory, Harwell Oxford, OX11 0QX, UK
cDepartment of Chemistry, University of Sheffield, S3 7HF, UK
First published on 16th January 2014
Abstract
Scarring of the cornea affects thousands of people every year, significantly reducing the quality of life and potentially leading to corneal blindness. Although cultured limbal epithelial cells have been used to regenerate scarred corneas for more than 15 years, the culture strategies do not deliver cells under the physiological conditions they experience in vivo. One of the main characteristics of stem cells is their ability to self-renew to maintain a tissue for a lifetime. Stem cells’ unique characteristics are thought to be at least partially due to their location within enclosed protective microenvironments or niches. For corneal stem cells these are located in intricate microenvironments or niches situated within areas of the limbal region known as the Palisades of Vogt. These are located in the limbus which is the area between the cornea and sclera. In this study we introduced micropockets into biodegradable microfabricated membranes and explored the potential contribution of these structures to limbal cell migration and their ability to deliver cells to a 3D cornea model. Membranes with micropockets were characterized using SEM, OCT, light microscopy and nanoindentation. Results indicate that the micropockets enhance the migration of cells from limbal explants and cells transfer readily from the membranes to the ex vivo cornea model.
Introduction
Corneal disease is the fourth most important cause of blindness worldwide (Word Health Organisation, WHO), and one of the main causes of the loss of corneal transparency is epithelial limbal stem cell deficiency, which can be caused by illnesses such as Aniridia or Steven Johnson's syndrome as well as by external factors, e.g. chemical burns or trauma.Limbal stem cells (LSC) are located in the limbus which is the narrow circular area between the cornea and sclera; they are specifically located in intricate microenvironments or niches situated within areas known as the Palisades of Vogt.1,2 The limbal stem cell niche or limbal crypt is a remarkable example of a stem cell niche and its role in corneal epithelial regeneration is well described. When the cells of the limbus are destroyed, adjacent cells from the conjunctiva move over the cornea, resulting in scar tissue with reduced vision or even blindness.2,3 Current treatments for corneal disease are (i) the use of corneal grafts and (ii) the use of stem cell carriers. The surgery of corneal grafting4–8 is well established and overall very successful. However patients who lack any residual limbal epithelial cell (LEC) population cannot benefit from donor corneas in the absence of LEC to repopulate them. Essentially these patients need further treatment to restore the LEC population. While donor cornea transplantation is well established and commonly available, there are currently relatively few specialist centres worldwide where LEC are cultured and transplanted to the patient. LECs are cultured from the contra-lateral eye if unaffected or from donor eyes when both eyes are affected (when immunosuppression must be used). In some studies oral mucosa has been used as an alternative epithelial cell source when both eyes are affected.9
For the last 15 years the most common methodology for LEC expansion and transfer to the cornea has been to expand cells on pieces of donor human amniotic membrane. This construct is then grafted onto the denuded cornea where the amniotic membrane acts as a sacrificial substrate, breaking down over several weeks or months, leaving the LEC in place.10–12 For those specialist centres around the world who have pioneered this technology the success rate of LEC grafting on amniotic membrane is initially very high: 85% for patients treated with LEC and amniotic membrane within the first year of treatment. After 3 and 5 years the success rate drops to 72% and 45% respectively.13,14
Different materials have been used for designing cell delivery devices for corneal applications: collagen15 and polyethylene glycol-based membranes16,17 have been described. Our group has recently developed a biodegradable polylactide-co-glycolide (PLGA) electrospun membrane for the culture of LEC and the support of cell outgrowth from limbal explants and demonstrated that cells grow well on this membrane and also transfer from it as it breaks down.18,19 Moreover, we recently reported research towards the fabrication of PLGA membranes containing artificial limbal stem cell micropockets20 using a combination of microstereolithography and electrospinning.
Several groups are now developing stem cell microenvironments (niches) for different tissues using a broad range of techniques, e.g. Lutolf and coworkers described the use of combinatorial methods for the development of artificial microarrays to direct single stem cell fate.21 Truckenmüller and coworkers22 reported the use of thermal imprinting methods for the development of protective micropockets. Our group has recently reported the use of microstereolithography for the creation of a polyethylene glycol artificial limbus containing epithelial microenvironments.17 Other authors such as Khademhosseini and Langer have also reported the use of microfabrication techniques for the development of polyethylene glycol-based stem cell wells23–25 and the use of other fabrication techniques such as 2-photon polymerization to produce artificial stem cell niches has been also described.26
Electrospinning is a very versatile manufacturing process in which the choice and design of electrospinning collectors play a major role in the final electrospun product. Many authors have reported different architectures and shapes achieved using patterned collectors.27–29 Recently, our group published the use of microstructured molds as underlying structures for patterned electrospinning applications.20
The aim of this study is to characterise and evaluate the contribution of micropockets to the performance of LEC growing on these membranes to see whether their inclusion confers any benefits on these cells and their ability to regenerate a new corneal epithelium. We combined microstereolithography and electrospinning to produce membranes containing microfabricated pockets. We characterised these pockets in some detail and looked at the response of the cells, both cultured cells and cells growing out from explants, to membranes containing these pockets. In addition, we developed a methodology for the batch production of reproducible, structured electrospun membranes which can be readily scaled-up for clinical applications. Limbal explants were included in this study as the ability of the membranes to be combined in theatre with small limbal explants and used in a one stage procedure for corneal regeneration is something that could simplify the whole process of corneal regeneration for surgeons and hence make this technique accessible to many more patients worldwide.
Materials and methods
Fabrication of collectors for electrospinning
The electrospun ring membranes were fabricated as previously reported.20 The constructs were fabricated by a combination of microstereolithography and electrospinning techniques. Polyethylene diacrylate (PEGDA) templates were custom-designed and cured using an in-house microstereolithography set-up equipped with a blue laser (MBL-III 473 nm; 150 mW, a full description in ref. 16). The PEGDA templates were created to be between 1.2 and 1.6 cm in diameter and 1 mm in height; the structures were equipped with microfabricated pockets in horseshoe shape of sizes of 300–500 μm. The PEGDA rings were directly cured on electroplated aluminium sheets that were then used as electrospinning collectors.Scale-up scaffold fabrication process at the Electrospinning Company Ltd
In order to scale-up the fabrication process and generate reproducible structured electrospun membranes, arrays of 36 PEGDA microfabricated templates were supplied to the Electrospinning Company, directly attached to the aluminium plates (300 mm × 150 mm). The plates were in turn fixed to a linear translation stage (Pro115-300, Aerotech Ltd, Reading, UK) directly underneath the spinning head (270 mm from needles to the collector) and grounded. The translation stage parameters (travel distance = 150 mm, translation speed = 500 mm min−1, dwell time = 2 s) were tuned to ensure that the membrane produced was of uniform thickness (50 microns) across the entire area of the plate. PLGA (50/50 DL-lactide (52 mol%):glycolide (48 mol%), 44 kg mol−1, Purac, the Netherlands) was dissolved in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) (Sigma Aldrich, Dorset, UK) by gently stirring at room temperature to produce solutions of suitable viscosity for electrospinning (20 wt%). The polymer solution was loaded into a 10 ml plastic syringe (Becton Dickinson, Oxford, UK) and delivered at a constant feed rate of 3200 μl h−1, using a programmable Harvard PHD4400 syringe pump (Harvard Apparatus, Kent, UK) via PTFE tubing (1/16′′ O.D.) and a 5 port manifold (Kinesis, St Neots, UK) to 4 blunt tipped stainless steel needles with internal diameters of 0.8 mm (Stainless Tube and Needle Company, Tamworth, UK). The needles were in turn connected to a positive high voltage unit (Glassman High Voltage Inc., High Bridge, NJ, USA) and solutions were electrospun with an applied voltage of 15.5 kV. The temperature and relative humidity of the electrospinning environment were maintained throughout the process at 25 °C and 25%, respectively. After 6.5 ml of solution had been processed, the coated aluminium plates were dried under vacuum at 20 °C for 72 hours to remove any residual solvent. Once dry, the scaffolds were cut into 22 mm diameter discs with the microstructured ring positioned in the centre and gently peeled away from the PEGDA template. The scaffold discs were then placed into 12-well non-tissue culture treated plates (BD Falcon, Beckton Dickinson, Oxford, UK), held down with custom-made retaining rings, sealed in polypropylene bags and terminally sterilised with gamma irradiation at a dose of 32 kGy (Synergy Health Plc, Swindon, UK). See the schematic for the production of electrospun rings in Fig. 1.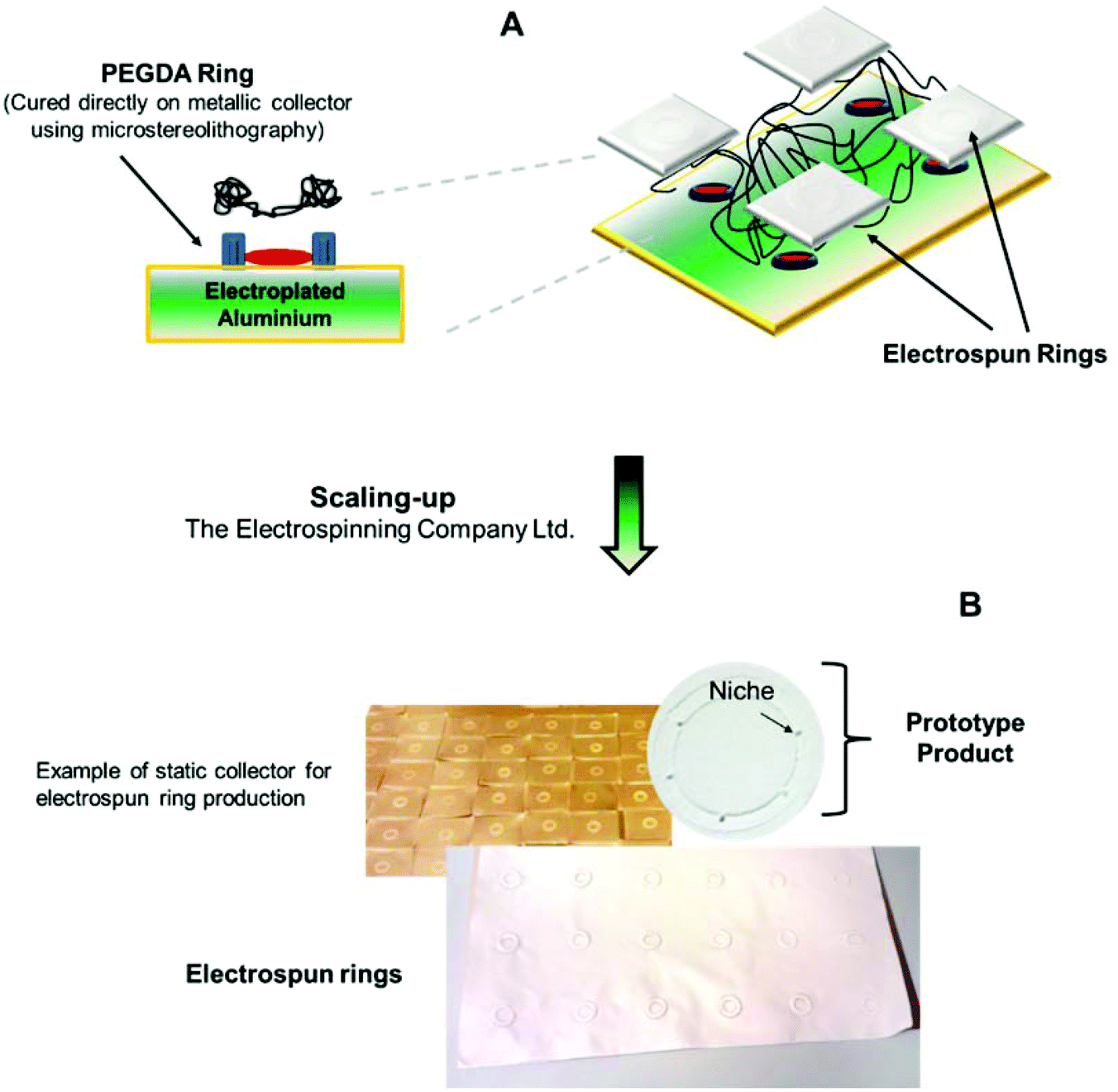 | ||
| Fig. 1 Schematic of electrospun ring fabrication. Panel (A) shows the combination of microstereolithography and electrospinning techniques for the microfabrication of biodegradable PLGA rings equipped with micropockets. Panel (B) shows the scaling-up procedures investigated at the Electrospinning Company Ltd showing an image of a prototype product produced in their facilities. | ||
Cell isolation and rabbit limbal explant dissection
Rabbit limbal fibroblasts (RLF) and rabbit limbal epithelial cells (RLE) were isolated from rabbit eyes (obtained from Alison Weather, Hook Farm, UK). For the isolation of primary rabbit limbal epithelial cells the limbal region was separated from the rest of the cornea and then cut into segments under a dissection microscope.For the isolation of rabbit limbal explants those segments were disinfected in iodine for 1 min and cut into small pieces (100–500 μm) with a scalpel. For cell isolation the segments were immersed in 2.5 U ml−1 Dispase II solution for one hour at 37 °C. Epithelial cells were then scraped, collected in media and then spun down at 1000 rpm for 5 minutes; the cells were then seeded into a T 25 flask containing irradiated 3T3s. The rabbit limbal epithelial cells were cultured in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMEM + Glutamax: Ham's F12, 10% fetal bovine serum, 1 U ml−1 penicillin, 100 mg ml−1 streptomycin, 2.5 μg ml−1 amphotericin, 10 ng ml−1 of EGF and 5 μg ml−1 of insulin. RLE cells were used at passage 1. Rabbit limbal fibroblasts (RLF) were isolated from stromal tissue remaining after isolation of RLE and they were cultured in DMEM containing 10% fetal bovine serum, 1 U ml−1 penicillin, 100 mg ml−1 penicillin–streptomycin, 2 mM L-glutamine and 0.625 mg ml−1 amphotericin. RLF were used between passages 4 and 7.
:1 DMEM + Glutamax: Ham's F12, 10% fetal bovine serum, 1 U ml−1 penicillin, 100 mg ml−1 streptomycin, 2.5 μg ml−1 amphotericin, 10 ng ml−1 of EGF and 5 μg ml−1 of insulin. RLE cells were used at passage 1. Rabbit limbal fibroblasts (RLF) were isolated from stromal tissue remaining after isolation of RLE and they were cultured in DMEM containing 10% fetal bovine serum, 1 U ml−1 penicillin, 100 mg ml−1 penicillin–streptomycin, 2 mM L-glutamine and 0.625 mg ml−1 amphotericin. RLF were used between passages 4 and 7.
Ex vivo 3D cornea model
For setting up the 3D corneal models, rabbit eyes were first disinfected using 3% Videne antiseptic solution (Ecolab) and then immersed into 0.14% ammonium hydroxide (Sigma Aldrich) for 5 minutes followed by washing with PBS. The epithelium in both the central cornea and the limbal region was then removed by scraping using a sclerotome knife. The corneas were mechanically supported by a combination of 0.5% agar (Sigma Aldrich) and 5 mg ml−1 collagen from a rat tail (Fluka). The corneas were cultured in the epithelial culture medium described above. Negative controls consisting of deliberately denuded corneas were maintained in culture for the same periods of time. The negative controls confirmed the lack of formation of a new epithelium in the absence of any added cells.For the transfer of cultured cells, PLGA rings of 1.2 and 1.6 mm diameter were mechanically supported by 6-well plate cell crowns and a total of 20000 cells were then seeded into the area of the pockets as described above for the viability assay. The scaffolds were kept in culture for 24 hours and then placed on the organ model. For the experiments with rabbit limbal explants, the pieces of limbus were directly placed on the rings and placed on the organ model immediately afterwards. The explants were placed directly on the niches using a dissecting microscope; the scaffolds were previously coated with fibrin glue (a 1:1 mixture of fibrinogen at a concentration of 18.75 mg ml−1 and thrombin at a concentration of 2.5 U ml−1). The membranes with either cultured cells or tissue explants were then placed on the deliberately denuded corneas using different conditions: cells facing upwards/cells facing downwards and air–liquid interface/submerged. The organ culture models were kept in culture for 4 weeks and then the corneas were fixed using 3.7% formaldehyde and processed for conventional histology to produce 6 μm paraffin sections (Microtome Leica RM 2145) and then stained with haematoxylin and eosin (H&E).
Characterisation of ring membrane complexity and mechanical properties
The complexity of the electrospun membrane was studied in detail by OCT, SEM and phase contrast imaging. The different parts of the construct were imaged and the correlation between OCT and SEM images was examined. Phase contrast microscopy was carried out using an inverted Olympus CK40 microscope and SEM was performed using a Philips X-L 20 microscope. The OCT system used in this study was equipped with a laser source (Santec HSL-2000) operated at 10 kHz rate with 10 mW output power and a central wavelength of 1300 nm.Fibre alignment was measured inside the niches using ImageJ software. A central fibre of the niche was assigned as a reference and angular differences were measured with respect to the reference fibre. Data were collected into groups of 5 ranging in angular differences from 5° up to 75°; 5 samples were analysed and a total of 75 fibres were measured.
The mechanical properties of the outer ring and the niche were studied using a nanoindenter (Hysitron Triboscope TS70) attached to a Bruker Dimension 3100 Atomic Force Microscope (AFM) which is used for imaging the sample. Load/unload curves were obtained using a starting load of 50 μN and a conospherical tip of 100 μm radius of curvature. The conospherical tip was positioned directly inside the niches and in random areas outside the micropocket. The reduced modulus (Er) was calculated using equations based on the Oliver–Pharr method.30
Cell viability and cell morphology on PLGA microfabricated membranes and niches
Cell viability on the electrospun rings was evaluated using the 3-(dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Rabbit limbal fibroblasts were seeded on PLGA rings, and on PLGA plain membranes and glass coverslips as positive controls. RLF were seeded specifically in the areas of the micropockets under a dissection microscope (Wild Heerbrugg M 3Z) and using an Eppendorf Micropipette (0.5–10 μl range) dispensing volumes of 3–6 μl in each niche (20000 cells). For plain membranes and glass coverslip controls, cells were seeded in the same way (areas of seeding are indicated with red arrows in Fig. 4). The cells were kept for 6 days in an incubator at 37 °C and 5% CO2. After that, the media was removed and RLF were washed with PBS. MTT (0.5 mg ml−1) was added for 40 minutes (37 °C). Acidified isopropanol (1 μl HCl in 1 ml isopropanol) was then added for dissolving the formazan crystals resulting from MTT reduction. The samples were measured using a BIO-TEK ELx 800 microplate reader at the wavelength of 540 nm and referenced at the wavelength of 630 nm.
For fluorescence imaging, rabbit limbal fibroblasts were seeded on the membranes at a concentration of 1 × 105 cells per ring and epithelial cells were seeded at a concentration of 5 × 104 cells per ring. RLF were stained with phalloidin–TRITC or phalloidin–FITC (to label actin filaments) and epithelial cells were also labeled with anti-vinculin staining. Cells were fixed in 10% formalin in PBS for 30 min at room temperature. Phalloidin–FITC was then added 1:500 into PBS for 30 min. RLE were stained using monoclonal anti-vinculin produced in mouse (Sigma Aldrich). After fixation with 3.7% formaldehyde the samples were permeabilized with Triton X-100 (0.1%) for 30 minutes and then washed with PBS. The cells were blocked with 10% goat serum (Sigma Aldrich) for 1 hour and then incubated with primary antibody diluted (1:150 in 1% goat serum) for another hour. The membranes were then washed with PBS and incubated with biotinylated secondary anti-mouse antibody (1:1000 in 1% goat serum, Vector Labs) for 1 hour at room temperature and further washed with PBS. Finally, the epithelial cells were incubated with tertiary antibody FITC–streptavidin (1:100 in 1% goat serum, Vector Labs) for 30 min at room temperature and then treated with the nuclear staining DAPI and phalloidin–TRITC. Cells were imaged inside and outside the microfeatures using a confocal scanning microscope (Carl Zeiss LSM510-META, Germany) and ImageXpress (Axon Instrument, USA).
Outgrowth of cells from limbal explants on PLGA membranes
Explant outgrowth was analysed using confocal microscopy and SEM. For SEM imaging, cells were washed with PBS and then fixed in 2.5% glutaraldehyde for 1 hour followed by dehydration with increasing concentration gradients of ethanol (from 35% to 100%). The samples were dried using hexamethyldisilazane (HMDS)–ethanol 1:1 for 1 h and finally treated with 100% HMDS for 5 min. The electrospun rings were sputter-coated with gold (emscope SC 500 coater) and analysed using a Philips X-L 20 microscope. Explant outgrowth was also studied using phalloidin–TRITC/DAPI and BrdU (bromodeoxyuridine) staining. Explants were left to attach overnight with a minimum amount of media; after 24 hours the samples were treated with BrdU solution (3.2 μM) for 48 hours. Samples were then carefully washed with BrdU-free cell culture media and kept in culture between 2 and 3 weeks.
The extent of cell outgrowth was also quantified using the ex vivo model. A single explant was located in the micropocket using a dissection microscope; the scaffold containing the explant was placed on the wounded organ culture model and kept in culture for 4 weeks. The samples were fixed for histology. H&E images were taken across the whole rabbit cornea and the extent of outgrowth from a single explant was quantified using a combination of H&E images.
Immunostaining
PLGA electrospun rings seeded with limbal epithelial cells were fixed and immunolabelled after 24 hours and after 2 weeks in culture. Limbal explants were fixed after 2 and 3 weeks in culture. In both cases the samples were fixed with 3.7% formalin, permeabilized with 0.5% Triton-X for 20 min and blocked with 10% goat serum for 1 hour. Samples were incubated with mouse monoclonal antibody cytokeratin 3 (CK3, Merck Millipore) and P63 (Merck Millipore) in 1% goat serum overnight at 4 °C. After PBS washes, the rings were treated with biotinylated secondary anti-mouse antibody (1:1000 in 1% goat serum, Vector Labs) for 1 hour at room temperature and tertiary antibody FITC–streptavidin (1:100 in 1% goat serum, Vector Labs) for 30 min at room temperature; samples were finally treated with the nuclear staining DAPI. For BrdU immunolabelling the samples were fixed with 3.7% formalin, permeabilized with 0.5% Triton-X, washed with PBS and incubated in 2 M HCl for 1 h at 37 °C.
The samples were then neutralised with 0.1 M borate buffer (pH = 8.5) for 20 min and rinsed with PBS followed by blocking using 2.5% BSA (1 h). Samples were incubated with monoclonal mouse anti-BrdU overnight at 4 °C. After PBS washes, the rings were treated with biotinylated secondary anti-mouse antibody and tertiary antibody FITC–streptavidin as described before for CK3 and P63 staining.
Immunohistochemistry procedures were performed in the histology sections obtained from the organ culture models. The sections were dewaxed in xylene and rehydrated in 100% ethanol, 70% ethanol and distilled water. The sections were then delineated with a Dako pen and treated with 0.05% trypsin (Aldrich) for 20 minutes (37 °C). After washing with PBS the samples were blocked with 10% goat's serum for 1 hour and treated with CK3 and P63 as described in the above paragraph.
Results
Examination of the 3D architecture of the membranes containing microfabricated pockets
The stereolithography-enabled electrospinning process used for creating the 3D corneal rings allows the control and creation of structures with parts having different fibre densities and alignment. The most reproducible membrane structures were achieved by placing and fabricating the PEGDA template directly onto the electroplated aluminium sheet and translating the flat plate collector during the electrospinning process to generate a uniform thickness material. Alternative collector configurations, such as rotating mandrels, were found to give less satisfactory results due to the need for adhesives/conductive tape to hold the PEGDA template in place, resulting in poor consistency even for samples from the same batch (results not shown).As the underlying structure of membrane fibres can play an important role in cell morphology, it was important to look at the structure of the membranes containing micropockets in detail. Fig. 2 shows the individual parts of those membranes. Part (a) corresponds to the microfabricated pockets which in this case are horseshoe shaped. Fibre alignment was studied within the pockets: 61% of the fibres showed high alignment (±1–5°) with the fibre direction fixed as a reference (47°). Part (b) corresponds to the outer ring which is a high density mesh of randomly oriented fibres. Part (c) is the centre of the hybrid membrane which again shows a high density of fibres with random alignment and part (d) corresponds to the area connecting the outer ring to the central membrane. As reported previously, the height of the PEGDA collectors influences the internal dimensions of the different parts of the scaffold.20 In particular, the height of the collectors has a direct impact on (i) the thickness of the central membrane of the constructs, and (ii) the relative area of parts (d) and (c). In this work we used collectors of 1 mm height as highlighted in section 2.1.
 | ||
| Fig. 2 Panel (A) is a schematic of a corneal electrospun ring highlighting the different parts of the construct. Panels (a–d) are SEM micrographs of the four areas highlighted in the schematic of the ring. Panel (a) corresponds to a micropocket showing a certain degree of fibre alignment, panel (b) is a lateral view of the ring scaffold containing a micropocket, panel (c) shows random oriented fibres from the central part of the hybrid membrane and panel (d) shows aligned fibres from the area joining the outer ring and the central area. Plot (B) is a histogram showing the degree of fibre alignment within the micropocket which was measured quantifying the angular variance between fibres plotting frequency vs. angular difference (in degrees). Panel (B′) is a phase contrast figure of a micropocket showing a high degree of fibre alignment. | ||
The differences in fibre density in the four areas of the ring-membranes were studied using OCT. The results were compared with SEM imaging (shown in Fig. 3). The samples were scanned in different areas. First of all, the areas of the niches were chosen (Fig. 3a) and different scans were performed in parallel directions towards the centre of the construct (directions marked with the yellow lines (b, c, d) in Fig. 3a).
 | ||
| Fig. 3 Panel (A) shows the correlation between SEM and OCT images for different areas of the electrospun membranes showing different fibre densities. Panel (a) is a SEM micrograph of an electrospun micropocket showing the scanning direction (red arrow) and different areas of scanning (dotted yellow lines). Panels (b–d) are the OCT scans corresponding to the 3 different yellow dotted regions highlighted in panel (a). Panel (e) is an OCT scan for a lateral view of the sample and panel (f) is its corresponding SEM (the scanning direction is highlighted in red). Plot (B) shows the load and unload curves for two different areas of the ring scaffold: inside the micropocket (grey dotted line) and in the periphery of the micropocket (outside pocket; black line). | ||
The consecutive scans b, c and d corresponded to the area just before the niche (b) and the beginning of the niche (c) and across the widest part of the niche (d). A second scan perpendicular to the niche was performed (the direction of the scan shown with a red arrow on (f)). The second scan allows one to see the differences in the density of the membrane in the area connecting the ring and the outer membrane to the central area of the membrane. The differences in densities are clear when comparing OCT scans (Fig. 3e) with SEM figures (Fig. 3f).
Mechanical properties were studied in areas of different fibre densities; nanoindentation was performed inside and outside the niche areas (see Fig. 3B). In areas of high fibre density (where the fibres are randomly distributed) the load/unload curves obtained using a 100 μm tip presented a typical shape for a stiff material with an average value of reduced modulus (Er) of Er = 0.027 ± 0.008 GPa (n = 3). The load/unload curves for the micropocket areas showed a high degree of elasticity. In contrast, the material within the niches offered little to no resistance to the indenter.
Performance of limbal cells on PLGA plain membranes and membranes with micropockets
A previous paper20 reported cell viability studies performed in both micropocket containing membranes and plain PLGA membranes and demonstrated that cells were viable in both types of membranes and that they proliferated in both at comparable rates. These results are confirmed in Fig. 4. This panel shows MTT staining absorbance values after 6 days of RLF on plain PLGA membranes (white), on membranes containing microstructured rings (grey) and on a control glass coverslip for comparison (black). There were no significant differences in viability observed between cells cultured on plain membranes and those cultured on membranes with micropockets (p > 0.05, Student's t-test, n = 6); however significant differences were seen between cells cultured on the PLGA scaffolds and glass coverslip controls (p < 0.05, Student's t-test, n = 6). These results are in agreement with data previously shown by our group.20 A closer analysis of the MTT-treated membranes showed differences in the distribution of cells in the membranes after 6 days in culture. Cells located in the pockets seemed to migrate towards the centre of the membranes following the direction laid down by the underlying electrospun fibres the niche and the central membrane (Fig. 4h). This was not observed for the cells placed on the plain membranes (Fig. 4g). In Fig. 4f there is clearly a teardrop shape in cells leaving the scaffolds which can be seen more readily at higher power in Fig. 4h.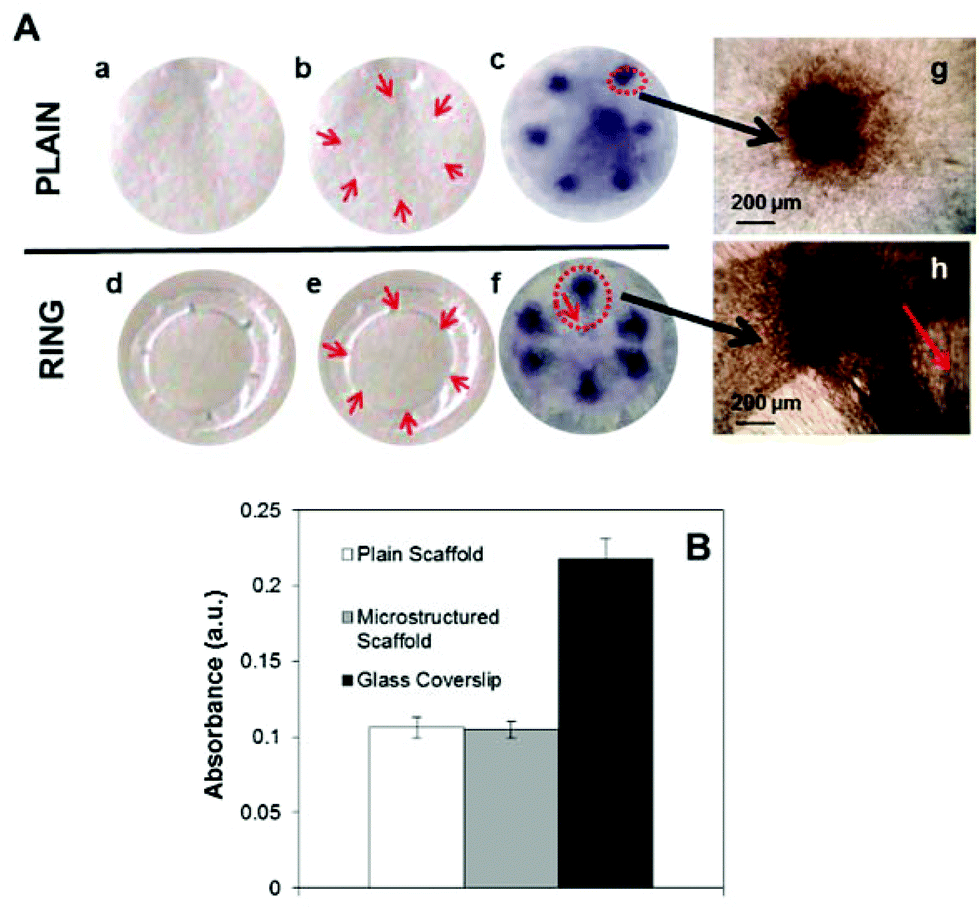 | ||
| Fig. 4 Panels (a) and (d) in panel (A) show a plain PLGA scaffold and a microfabricated ring respectively. Panels (b) and (e) highlight with red arrows the areas of the scaffolds where cells were seeded. Panel (f) in panel (A) shows MTT staining of cells placed in 6 areas corresponding to the location of the micropockets present in the ring. The same protocol was used to place the cells in the plain membranes shown in (c). Panels (g) and (h) show high magnification micrographs of the MTT stained cells after 6 days in the plain membrane (g) and the cells placed over the microfabricated pockets (h). The red arrow indicates the direction of alignment of the fibres. Panel (B) shows MTT staining of RLF after 6 days in culture on plain PLGA membranes (white), membranes containing microstructured rings (grey) and a control glass coverslip (black). There were no significant differences in viability between cells cultured in plain and microfabricated scaffolds. | ||
Cell morphology and characterisation of rabbit limbal cells on electrospun ring membranes
Cell morphology was also studied inside and outside the micropockets. Both rabbit limbal fibroblasts and epithelial cells presented different morphology in different areas. The morphology was dictated by the underlying structure.Panels (a–c) in panel (A) in Fig. 5 show RLF stained with phalloidin–FITC extended across the niche structure and following the parallel oriented fibres. Fig. 5d–f show RLF stained with phalloidin–FITC in a random fibre area of the membrane. The fibroblasts inside the micropockets show a more elongated morphology. The same effect was observed for epithelial cells. Panel (g) in panel (A) of Fig. 5 corresponds to a confocal image of epithelial cells taken inside the micropocket; Fig. 5h corresponds to an epithelial cell in the central area of the membrane. In both cases the cells were stained for vinculin (green, showing focal adhesion points) and for phalloidin–TRITC (red). RLF and RLE were co-cultured in the microfabricated rings and imaged again inside and outside the micropockets. Panels (m) and (n) in panel (B) of Fig. 5 show co-cultures of RLF and RLE. The fibroblasts were pretreated with CellTracker Green prior to seeding and the epithelial cells were stained for CK3 and they are shown in red. Elongated fibroblasts and epithelial cells can be observed in Fig. 5m which was taken inside a micropocket. Fig. 5n was taken in the periphery of the micropocket where the underlying scaffold presented randomly distributed fibres.
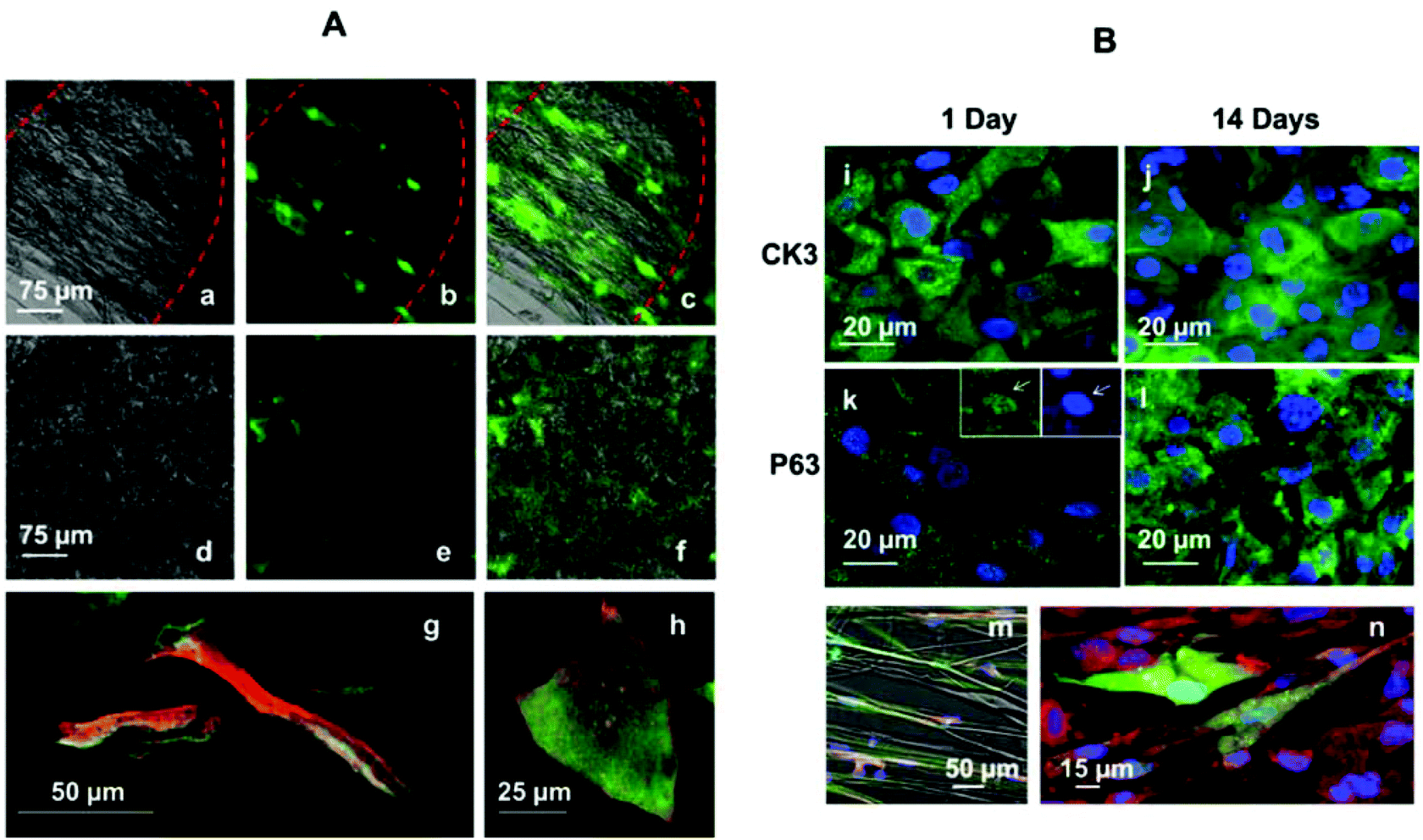 | ||
| Fig. 5 Panel (A) shows RLF and RLE imaged inside and outside the electrospun microfeatures. Panel (B) shows cells on PLGA rings stained for CK3 (differentiation marker) and P63 (stem cell marker) as well as the ability of the membranes to support co-cultures of RLF and RLE. Panels (a–c) are fluorescent and optical images of RLF stained with phalloidin–FITC after 6 days in culture inside a microfabricated pocket: panel (a) is an optical image of the pocket, panel (b) is a fluorescence image of the aligned cells inside the pocket and panel (c) is a merged panel of (a) and (b). Panels (d–f) are fluorescence and optical images of RLF stained with phalloidin–FITC after 6 days in a plain membrane with random fibres: panel (d) is an optical image of the random fibres in the membrane, panel (e) is a fluorescence image of cells on the membrane and panel (c) is a merged panel of (d) and (e). Panel (g) is a confocal image showing rabbit limbal epithelial cells inside a microfabricated pocket and panel (h) is a rabbit limbal epithelial cell in the central part of the hybrid membrane (where the fibres are randomly organised). Panels (i) and (j) correspond to CK3 positive cells at 1 and 14 days of culture. Panels (k) and (l) show cells positive for P63 at 1 and 14 days of culture. In panel (k) a high magnification micrograph is shown highlighting the presence of staining in the nuclei. Panels (m) and (n) correspond to co-cultures of RLE and RLF inside and outside the niche. RLF were preloaded with CellTracker Green, and RLE were immunolabelled with CK3 (red). | ||
Rabbit limbal epithelial cells were seeded on PLGA rings at passage 1 and immunolabelled with CK3 and P63 at different time points. CK3 is a cytokeratin expressed in corneal epithelium together with CK12, and P63 is a stem cell and transient amplifying cell marker. No differences in the expression of either CK3 or P63 were observed between cells inside and outside of the micropockets (images not shown). P63 was positive in the nuclei but also in the cytoplasm as reported before by our group19 (Fig. 5k and 5l) and Ck3 was observed in the cytoplasm (Fig. 5i and 5j). The markers were expressed at both of the two time points studied. After 14 days the cells increased in number, forming an 80–90% confluent monolayer and demonstrating the ability of limbal epithelial cells to proliferate in these 3D membranes.
Cell outgrowth from rabbit limbal explants located on electrospun pockets
Rabbit limbal explants were located in PLGA rings previously treated with fibrin glue; the pieces of tissue were placed directly on the electrospun niches (Fig. 6a). Cell outgrowth was studied after 2 and 3 weeks of culture. The morphology of the cells coming out from the explants was assessed by fluorescence microscopy and SEM (Fig. 6b and 6e). Cells were positive for CK3 and P63 staining (Fig. 6c and 6f). Fig. 6d shows a confocal Z-stack of an explant placed on an electrospun niche where the cells coming out from the explant were BrdU positive. BrdU is a thymidine analogue which is taken up specifically by dividing cells and is retained for longer by slowly dividing cells or stem-like cells. The cells coming out from the rabbit limbal explants retained BrdU after 2 weeks showing that the cells in the niche were proliferative after this period.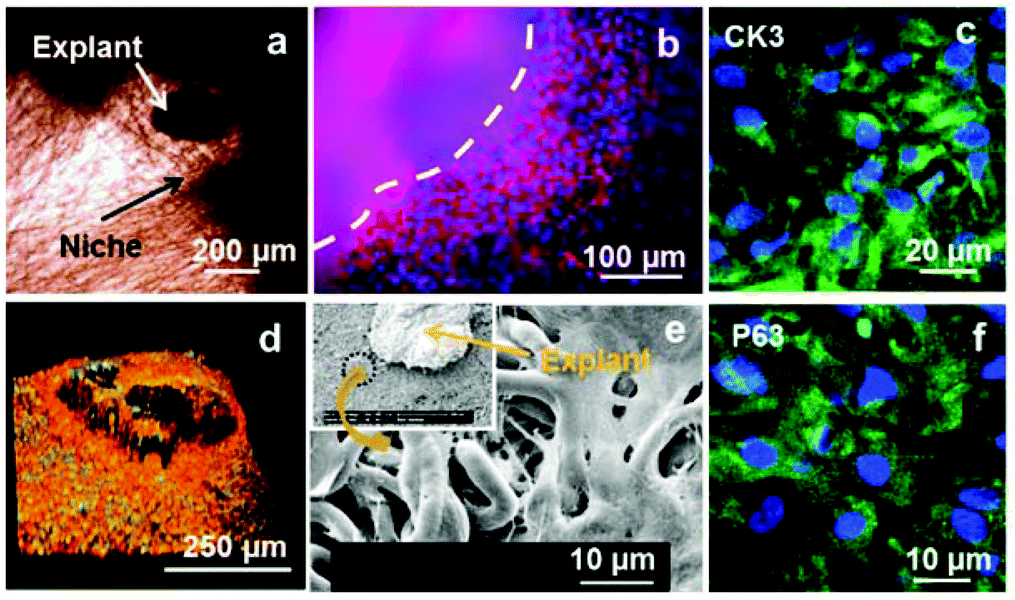 | ||
| Fig. 6 Panel (a) is an optical microscopy image of a fibrin glue treated niche with a limbal explant located on it. Panels (b) and (e) show cell outgrowth from the explant; panel (b) shows phalloidin–TRITC (red) and DAPI staining (blue) and panel (e) is an SEM image of the cell outgrowth. Panels (c) and (f) show positive staining for CK3 and P63 of cells growing out from the explants. Panel (d) is a confocal Z-stack 3D reconstruction of an explant on an electrospun niche showing cell outgrowth; cells coming out from the explant were positive for BrdU staining (green) indicating cell proliferation (the red colour corresponds to propidium iodide counterstained nuclei). Panels (d) and (e) correspond to explants kept in culture for 2 weeks. Panels (b), (c) and (f) correspond to explants kept in culture for 3 weeks. | ||
Cell outgrowth and transfer from PLGA electrospun rings to the cornea of a 3D organ culture model
PLGA rings seeded with limbal epithelial cells were kept in organ culture for 4 weeks as previously described. The samples were sectioned and analysed by histology and immunohistochemistry. Epithelial cell transfer from PLGA rings to the deliberately denuded corneas was achieved both by placing cells facing downwards and also facing upwards and by submerging the whole cornea or keeping the organ cultured at an air–liquid interface. In all cases cells transferred from the electrospun PLGA membranes to the cornea. Panel (A) in Fig. 7 compares a fresh rabbit corneal epithelium (Fig. 7a) with the cell transfer achieved by a PLGA membrane with epithelial cells facing upwards on a denuded cornea kept at an air–liquid interface (f). Cells seeded in the pockets were able to migrate towards the centre of the cornea, starting to form a new epithelium. The cells transferred to the denuded cornea were corneal epithelial cells as demonstrated by immunocytochemistry. Fig. 7c shows CK3 staining (green) counterstained by DAPI (blue, nuclei) in a fresh rabbit cornea. Fig. 7g shows the same staining for epithelial cells seeded on a PLGA scaffold and transferred onto a denuded cornea. P63 staining was negative for the transfer experiment. Fig. 7d shows positive staining for P63 in a fresh rabbit cornea (nuclear staining); Fig. 7h shows negative staining for P63 for the cells transferred to the cornea. Fig. 7h′ was taken in the same area of Fig. 7h and is a DAPI image that confirms the presence of cells in the P63 immunolabelled sample. | ||
| Fig. 7 Panel (A) compares a fresh rabbit cornea with tissue engineered corneas using both cell suspension (schematic e) and tissue explants (schematic i). Panel (a) shows H&E staining of a fresh rabbit cornea and panels (f) and (j) show tissue engineered corneas as a result of epithelial cell transfer (f) and explant outgrowth (j) from an electrospun ring to a previously wounded cornea. CK3 staining of a fresh rabbit cornea (green) using DAPI (blue) as counterstaining (c). CK3 positive staining for the tissue engineered cornea using cell suspension (g) and using limbal explants (k). Panel (d) shows P63 staining of a fresh rabbit cornea (nuclear staining; green). Panel (h) shows P63 negative staining for the tissue engineered cornea using a cell suspension; (h′) shows DAPI counterstaining for the same area shown in panel (h). Panel (l) shows positive staining for P63 using limbal explants. Panel (B) shows H&E staining for tissue engineered corneas using limbal explants. Panels (m) and (n) correspond to transfer experiments with explants facing the denuded cornea (down) without and with fibrin glue coating. Panels (o) and (p) correspond to transfer experiments with explants facing upwards without and with fibrin glue coating. Panel (C) is a schematic of the extent of cell outgrowth achieved after 4 weeks by placing a single explant on a micropocket in a wounded cornea model. Panel (q) is a combination of H&E figures taken across the rabbit cornea. Measurements of the total length of the cornea and the length of the outgrowth are shown in grey dotted lines. Panel (r) is a high magnification of the limbal explant placed on the PLGA micropocket. | ||
Transfer experiments were also performed using limbal explants. Electrospun rings with explants were placed on wounded corneas both facing upwards/downwards and using fibrin glue. Cell outgrowth was observed for both conditions (Fig. 7n and 7p). H&E staining showed the formation of a new epithelium all along the cornea; in some cases the regenerated epithelium was very similar to the multilayer epithelium presented by an intact rabbit cornea (Fig. 7j). Cell outgrowth proved to be better when coating the membrane with a thin layer of fibrin glue. Panel (B) in Fig. 7 shows H&E figures for transfer experiments with limbal explants (facing upwards or downwards) coated with fibrin and uncoated.
Histological sections were immunostained with CK3 (Fig. 6k) demonstrating that the cells coming out from the explants were corneal epithelial cells. It is important to highlight that P63 staining was positive in proximity to the explants (Fig. 7l) and it was negative when cells were seeded as a cell suspension (Fig. 7h). The extent of cell outgrowth from a single explant was quantified using a combination of H&E figures taken along the whole cornea. A single explant was placed on a PLGA micropocket and situated in the limbal region of a deliberately wounded cornea. After 4 weeks, outgrowth from the explant was observed; cells migrated from the explant covering the denuded cornea in approximately 50% of its length. A schematic of the quantification can be seen in panel (C) of Fig. 7.
Discussion
In this study we have demonstrated the potential of using microfabricated biodegradable membranes as epithelial cell delivery carriers for corneal repair. These membranes mimic to some extent the morphology and distribution of limbal stem cell niches in the eye and they can be custom-designed as previously described by our group using a combination of microstereolithography and electrospinning.20 Results indicated that the artificial micropockets and the overall structure of the PLGA ring play an important role in cell migration and directionality. Moreover, we studied the expression of putative stem cell markers (P63) using both tissue explants and cell suspension concluding that the fact of keeping cells at high densities has an effect on stem cell maintenance.An in-depth study of the morphology and fibre distribution of the membranes was carried out using OCT and SEM; four different areas of the rings were identified showing differences in fibre density and fibre orientation (Fig. 2 and 3). Fibre alignment was measured inside the micropockets showing a high degree of alignment (0–5° angular difference) in 61% of the analysed fibres. Differences in fibre density were studied by both OCT imaging and mechanical testing (nanoindentation). Mechanical characterisation was performed in areas inside and outside the microfeatures. The differences in the load/unload curves support the fact that these areas are different in both fibre density and orientation. The niche or pocket presented no resistance to the indentation.
Rabbit corneal cells specifically seeded into the areas of the micropockets were viable in the membranes after 6 days and no differences in viability (MTT assay) were observed between cells cultured on microfabricated membranes or plain PLGA membranes. These findings are in agreement with previous data recently published by our group.20 Cells residing in the pockets have the propensity to move towards the centre of the corneas following the path dictated by the aligned fibres in the area joining the niche and the central membrane demonstrating that the complexity of our 3D ring structure influences the directionality of cell migration. This fact will be further investigated using epithelial cells and limbal explants.
In this study we demonstrated that the underlying structure provided by the electrospun fibres plays an important role in cell morphology for both rabbit corneal fibroblasts and epithelial cells. However no differences were observed in the expression of CK3 and P63 markers when epithelial cells were studied in different areas of the scaffold. Cells inside the niche presented an elongated morphology whilst cells attached to the areas with random fibre orientation showed a more polygonal shape (Fig. 5). Different groups have reported the importance of the morphology of corneal epithelial cells in the corneal regeneration process.31,32 Cells surrounding wounds and damaged areas of tissue frequently become elongated and migratory. In this work we have shown the ability of the membrane design to confine groups of cells within specific microenvironments defined by low fibre density and high degree of fibre alignment; the cells confined within those pockets presented an elongated morphology which was guided by the underlying structure. A previous study by Yan and coworkers33 reported differences in attachment and proliferation between corneal fibroblasts and corneal epithelial cells concluding that aligned substrates were suitable for stromal regeneration but not for epithelial cell growth. In this respect our findings differ from the outcomes reported by Yan and coworkers. However, it is important to note that the nature of the scaffold and the solvents used in both studies were different. Certainly, in the current study, both epithelial and stromal cells did well on our aligned substrates. Epithelial cells which were seeded into the pockets and into their periphery were able to migrate and regenerate to a certain degree the epithelium of deliberately damaged corneas (panel A in Fig. 7). The membranes also supported explant outgrowth and they allowed cell transfer, achieving in some cases a multi-layered epithelium very similar to the epithelium observed in fresh rabbit corneas (panel A in Fig. 7).
It is important to notice that P63 (putative stem cell marker) was found to be expressed by the cells coming from tissue explants placed on the niches and not by the cells seeded as cell suspensions. We hypothesize that the niche is providing a physically protected environment and encouraging cells to stay in a more stem-cell like state as they do under physiological conditions. We believe that this is related to the inclusion of a high density of cells (limbal explant) within an enclosed microenvironment (artificial pocket). Jahoda and coworkers recently reported research towards the use of 3D cultures (dermal papilla spheroids) for inducing the creation of hair follicles in human skin.34 Similarly, in this work we highlight the importance of keeping cells “together” within limbal explants for encouraging corneal regeneration. The presence of native stromal cells within the explants also plays an important role in limbal stem cell maintenance. In this sense, Levis and coworkers35 recently reported research towards the fabrication of collagen-based corneal membranes equipped with biomimetic corneal crypts. They showed highly organised human corneal epithelial cells within the collagen constructs highlighting the expression of P63 in the basal layers. Human corneal fibroblasts were also seeded in these constructs and they were observed in the proximity of the crypts in the underlying engineered stroma. In the same way, we suggest that the presence of native fibroblasts within the limbal explants placed on our PLGA microfeatures probably contributed to the maintenance of a population of slow-cycling cells expressing P63.
This paper considers whether the membrane design can assist in corneal regeneration, particularly from limbal explants. The possibility of using limbal explants on the microfabricated membranes would be of great help for surgeons. In operations where only one eye is damaged, surgeons can take a biopsy from the other eye and place the explants on the microfabricated membrane in theatre. Thus the pockets combined with tissue explants could potentially be more effective than explants placed on plain scaffolds. Having scaffolds available as an off-the-shelf cost effective product combined with tissue explants also avoids the need for initial cell expansion under clean room conditions and banking procedures to access the amniotic membrane. The concept of cornea regeneration from limbal explants was recently confirmed by Sangwan et al. who reported the use of limbal explants on amniotic membrane carriers as a new treatment for unilateral limbal stem cell deficiency.36 The use of explants on PLGA plain membranes in in vitro and ex vivo studies has been also recently reported by our group.19 In this work we compared PLGA ring scaffolds coated with fibrin glue and those uncoated. Panel (B) in Fig. 7 shows how the fibrin coating encourages cell outgrowth from the explants. We recently reported a study of cell outgrowth from human and rabbit limbal explants using PLGA plain membranes and different coatings such as fibrin, collagen and laminin.19 In this study fibrin glue also provided the best outgrowth results. Fibrin glue is used in the clinic to attach the amniotic membrane to the cornea and to attach limbal tissue explants to the amniotic membrane,34 so this will aid in clinical translation.
In this work we explored one of the key aspects that define the stem cell niche: “the niche as a well-defined physical and protective space”. We hypothesize that the inclusion of the micropockets in our membranes provides explants with some kind of physical protection enhancing stem-cell maintenance. More work needs to be done to understand the contribution made by the artificial microfeatures on subjecting them to physical trauma and mild flow conditions. Our current constructs were able to keep certain slow-cycling state cells; this was evident for tissue explants and not for cultured cells. In essence, we have designed a cell delivery membrane containing protected reservoirs of migratory cells which can aid in corneal regeneration. In this work we have chosen to explore the protective nature of the niche but we are aware of the importance of studying other aspects such as combinations of different cell types and ECM proteins. Our group has recently reported research towards the development of polyethylene glycol-based fibronectin-treated niches showing that the use of this protein stimulates limbal cell outgrowth and migration.17 Other groups have reported the use of proteins such as vitronectin for studying self-renewing maintenance.35 We suggest that these micropockets can now be studied in combination with ECM proteins to determine how each contributes to stem cell behaviour.
Conclusions
In this study we have characterised the physical structure of PLGA membranes containing microfabricated pockets as delivery membranes for delivering cultured cells and cells grown out from limbal explants to wounded corneas. The results show the complexity of the scaffold with parallel, less dense fibres within the niches compared to the denser random fibre structure elsewhere. Examination of the morphology of the cells shows them responding to the underlying fibrous scaffold and cells leaving the microfabricated pockets more readily than cells placed on the random scaffolds. This characterisation of the physical nature of the scaffold represents a key step in designing membranes with microfabricated niches within them. Finally, we compared cell outgrowth from cell suspensions and tissue explants placed on the pockets suggesting that cell density plays a very important role in stem-cell maintenance. Future work will concern the study of the ability of the microfabricated niches to offer physical protection to cells as well as functionalization of the niche area with ECM proteins.Acknowledgements
We thank the EPSRC Landscape Fellowship program (EP/I017801/1) and the Wellcome Trust Foundation (091128/B/10/Z) for funding this research. We gratefully acknowledge contributions from the Electrospinning Company Ltd, Ms Dawn Bussey and Mr Thomas Paterson to these experiments and we also acknowledge Dr Pallavi Deshpande and Dr Farshid Sefat for their support.Notes and references
- B. Ebato, J. Friend and R. A. Thoft, Invest. Ophthalmol. Vis. Sci., 1987, 28, 1450–1456 CAS.
- H. S. Dua, V. A. Shanmuganathan, A. O. Powell-Richards, P. J. Tighe and A. Joseph, Br. J. Ophthalmol., 2005, 89, 529–532 CrossRef CAS PubMed.
- A. J. Huang and S. C. Tseng, Invest. Ophthalmol. Vis. Sci., 1991, 32, 96–105 CAS.
- K. R. Kenyon and S. C. Tseng, Ophthalmology, 1989, 96, 709–722 CrossRef CAS ; discussion 722–703.
- D. J. Coster, R. K. Aggarwal and K. A. Williams, Br. J. Ophthalmol., 1995, 79, 977–982 CrossRef CAS.
- E. J. Holland and G. S. Schwartz, Cornea, 1996, 15, 549–556 CrossRef CAS PubMed.
- H. S. Dua and A. Azuara-Blanco, Br. J. Ophthalmol., 2000, 84, 273–278 CrossRef CAS.
- H. S. Dua and A. Azuara-Blanco, Br. J. Ophthalmol., 1999, 83, 414–419 CrossRef CAS.
- K. Nishida, M. Yamato, Y. Hayashida, K. Watanabe, K. Yamamoto, E. Adachi, S. Nagai, A. Kikuchi, N. Maeda, H. Watanabe, T. Okano and Y. Tano, New Eng. J. Med., 2004, 351, 1187–1196 CrossRef CAS PubMed.
- S. C. G. Tseng, P. Prabhasawat, K. Barton, T. Gray and D. Meller, Arch. Ophthalmol., 1998, 116, 431–441 CrossRef CAS.
- R. J.-F. Tsai, L.-M. Li and J.-K. Chen, New Eng. J. Med., 2000, 343, 86–93 CrossRef CAS PubMed.
- A. J. Shortt, G. A. Secker, M. S. Rajan, G. Meligonis, J. K. Dart, S. J. Tuft and J. T. Daniels, Ophthalmology, 2008, 115, 1989–1997 CrossRef PubMed.
- T. R. M. Henderson, D. J. Coster and K. A. Williams, Br. J. Ophthalmol., 2001, 85, 604–609 CrossRef CAS.
- S. Basu, A. Mohamed, S. Chaurasia, K. Sejpal, G. K. Vemuganti and V. S. Sangwan, Am. J. Ophthalmol., 2011, 152, 917–924 CrossRef PubMed.
- S. Dravida, S. Gaddipati, M. Griffith, K. Merrett, S. Lakshmi Madhira, V. S. Sangwan and G. K. Vemuganti, J. Tissue Eng. Regen. Med., 2008, 2, 263–271 CrossRef CAS PubMed.
- G. Sitalakshmi, B. Sudha, H. N. Madhavan, S. Vinay, S. Krishnakumar, Y. Mori, H. Yoshioka and S. Abraham, Tissue Eng., Part A, 2008, 15, 407–415 CrossRef PubMed.
- I. Ortega, P. Deshpande, A. A. Gill, S. MacNeil and F. Claeyssens, Biofabrication, 2013, 5, 1758–5082 CrossRef PubMed.
- P. Deshpande, R. McKean, K. A. Blackwood, R. A. Senior, A. Ogunbanjo, A. J. Ryan and S. MacNeil, Regen. Med., 2010, 5, 395–401 CrossRef CAS PubMed.
- P. Deshpande, C. Ramachandran, F. Sefat, I. Mariappan, C. Johnson, R. McKean, M. Hannah, V. S. Sangwan, F. Claeyssens, A. J. Ryan and S. MacNeil, Biomaterials, 2013, 34, 5088–5106 CrossRef CAS PubMed.
- Í. Ortega, A. J. Ryan, P. Deshpande, S. MacNeil and F. Claeyssens, Acta Biomater., 2013, 9, 5511–5520 CrossRef PubMed.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949–955 CrossRef CAS PubMed.
- R. Truckenmüller, S. Giselbrecht, M. Escalante-Marun, M. Groenendijk, B. Papenburg, N. Rivron, H. Unadkat, V. Saile, V. Subramaniam, A. Berg, C. Blitterswijk, M. Wessling, J. Boer and D. Stamatialis, Biomed. Microdevices, 2012, 14, 440–448 CrossRef PubMed.
- I. Wheeldon, A. F. Ahari and A. Khademhosseini, JALA (Charlottesville Va.), 2010, 15, 440–448 CAS.
- H.-C. Moeller, M. K. Mian, S. Shrivastava, B. G. Chung and A. Khademhosseini, Biomaterials, 2008, 29, 752–763 CrossRef CAS PubMed.
- O. Z. Fisher, A. Khademhosseini, R. Langer and N. A. Peppas, Acc. Chem. Res., 2010, 43, 419–428 CrossRef CAS PubMed.
- M. T. Raimondi, S. M. Eaton, M. Laganà, V. Aprile, M. M. Nava, G. Cerullo and R. Osellame, Acta Biomater., 2013, 9, 4579–4584 CrossRef CAS PubMed.
- J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, Biomacromolecules, 2002, 3, 232–238 CrossRef CAS PubMed.
- E. Zussman, A. Theron and A. L. Yarin, Appl. Phys. Lett., 2003, 82, 973–975 CrossRef CAS PubMed.
- C. Vaquette and J. J. Cooper-White, Acta Biomater., 2011, 7, 2544–2557 CrossRef CAS PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564–1583 CrossRef CAS.
- H. Ichijima, W. M. Petroll, P. A. Barry, P. M. Andrews, M. Dai, H. D. Cavanagh and J. V. Jester, Invest. Ophthalmol. Vis. Sci., 1993, 34, 2803–2812 CAS.
- A. H. Neufeld, M. M. Jumblatt, E. D. Matkin and G. M. Raymond, Invest. Ophthalmol. Vis. Sci., 1986, 27, 1437–1442 CAS.
- J. Yan, L. Qiang, Y. Gao, X. Cui, H. Zhou, S. Zhong, Q. Wang and H. Wang, J. Biomed. Mater. Res. Part A, 2012, 100A, 527–535 CrossRef CAS PubMed.
- C. A. Higgins, J. C. Chen, J. E. Cerise, C. A. B. Jahoda and A. M. Christiano, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19679–19688 CrossRef CAS PubMed.
- H. J. Levis, I. Massie, M. A. Dziasko, A. Kaasi and J. T. Daniels, Biomaterials, 2013, 34, 8860–8868 CrossRef CAS PubMed.
- V. S. Sangwan, S. Basu, S. MacNeil and D. Balasubramanian, Br. J. Ophthalmol., 2012, 96, 931–934 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |