Hydrogelation of dextran-based polyampholytes with cryoprotective properties via click chemistry†
Minkle
Jain
ab,
Robin
Rajan
ab,
Suong-Hyu
Hyon
c and
Kazuaki
Matsumura
*a
aSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: mkazuaki@jaist.ac.jp; Fax: +81-761-51-1149; Tel: +81-761-51-1680
bM. Tech (CSPT), Department of Chemistry, University of Delhi, Delhi-110007, India
cCenter for Fiber and Textile Science, Kyoto Institute of Technology, Matsugasaki, Kyoto 606-8585, Japan
First published on 25th November 2013
Abstract
Hydrogels are promising substrates for tissue engineering applications because of their unique biocompatibility, flexible methods of synthesis, range of constituents, and desirable physical characteristics. Cryopreservation of cell-containing constructs using such hydrogel scaffolds is in high demand in tissue-engineering applications for the production of “off-the-shelf” tissue-engineered products. However, cryopreservation of regenerated tissues including cell sheets and cell constructs is not easy compared to the preservation of cell suspensions, even when cryoprotectants are used. Here, we report a dextran-based polyampholyte hydrogel that itself shows cryoprotective properties, which could be useful for cell encapsulation and tissue engineering applications involving hydrogel formation. Amination was performed by introducing poly-L-lysine onto azide groups conjugated with dextran, and a portion of the amino groups was converted into carboxyl groups. These dextran-based polyampholytes showed good cryoprotective properties for mammalian cells, and the addition of dextran substituted with dibenzylcyclooctyne acid induced in situ hydrogel formation via Cu-free click chemistry with high biocompatibility. Cells encapsulated with such in situ hydrogels can be cryopreserved well without the addition of any cryoprotectants. Thus, these hydrogels can serve as scaffolds with cryoprotective properties that also provide structural integrity to tissue constructs.
1. Introduction
Hydrogels have been the material of choice for many applications in regenerative medicine because of their unique biocompatibility, flexible methods of synthesis, range of constituents, and desirable physical characteristics.1,2 They can be used as scaffold materials for drug delivery vehicles, as engineered tissue replacements, and in a variety of other applications.3 For tissue engineering applications, injectable hydrogels are important tools because of their ability to homogeneously encapsulate the cells, their easily manipulated physical and chemical properties, and their ability to be administered in a minimally invasive manner.4–6 They are also potential substrates for tissue engineering, because drugs and cells can be readily integrated into the gelling matrix.Cell encapsulation with such hydrogels provides cells with a three-dimensional environment that is similar to the in vivo conditions. Previously, we developed a poly(vinyl alcohol) cryogel with many crosslinking points that encapsulated vascular endothelial cells and smooth muscle cells7 through physical hydrogel formation via crystallization during freezing.8 In that report, we found that cells could be stored for the desired period using the cryogelation process, where gelation proceeds after thawing occurs. When this cryogel is used to encapsulate cells in a freezing process, cryoprotectants (CPAs) must be added to avoid freezing damage to the cells. “CPA” is the functionally derived term coined to describe “any additive that can be provided to cells before freezing that yields a higher post-thaw survival than can be obtained in its absence”.1,9,10 However, in general, CPAs are cytotoxic11,12 and need to be removed after thawing. It is likely to be difficult to completely remove CPAs from inside the hydrogel. Therefore, if a hydrogel can be developed that itself has cryoprotective properties, the problem of the removal of CPAs could be solved.
In addition to these challenges, the success of tissue engineering applications in regenerative medicine requires further advances in low-temperature preservation. Storage of tissues and tissue engineering products is an important technique for the commercialization of tissue engineering.13–15 However, cryopreservation of regenerated tissues including cell sheets and cell constructs is not easy compared to the cryopreservation of cell suspensions, and conventional freezing methods destroy the membranous structures of cultured sheets during the freezing and thawing processes.16 Cryopreservation of cell-containing constructs is in high demand for tissue engineering applications such as producing “off-the-shelf” tissue-engineered products. A hydrogel that has cryoprotective properties could be a good alternative for the storage of tissue-engineered constructs or cell-based systems.
In spite of these problems, the application of cryopreservation to living cells and tissues has revolutionized the areas of biotechnology, plant and animal breeding programs, and modern medicine. Using this technique, cells from prokaryotic and eukaryotic organisms can be recovered from temperatures down to almost 200 °C below the freezing point of water. This has been made possible by the presence of CPAs, as mentioned above. Currently, 10% dimethyl sulphoxide (DMSO) solution is the most efficient CPA and is commonly used for cell preservation in cell banks worldwide,17,18 in spite of its cytotoxicity and its effects on cell differentiation.19
As an alternative to the use of cytotoxic CPAs, we have shown in our previous study that carboxylated poly-L-lysine (COOH-PLL), which is classified as a polyampholyte, yields excellent post-thaw survival efficiency.7,20–23 Cryoprotective properties are generally found in polyampholytes, and the balance of positive and negative charges is very important.20,24 However, the detailed mechanisms by which non-membrane-penetrating polymers can have good cryoprotective properties are still not clear. Nevertheless, we have shown that the polyampholytes were adsorbed on the cell membrane during freezing23 and hypothesized that the mechanisms are related to the protection of the membrane from outside. The results of this previous report were the seeds from which we conceived of the idea that a hydrogel that is formed with a polymeric CPA could itself have cryoprotective properties.
Among the natural polymers suitable for such a purpose is dextran, a hydrophilic, biocompatible, and nontoxic polysaccharide composed of linear α-1,6-linked D-glucopyranose residues. From a structural point of view, dextran has reactive hydroxyl groups that can be modified to introduce positive and negative charges and also to introduce functional groups with which to form an in situ hydrogel.25 Because dextran is naturally resistant to protein adsorption and cell adhesion, modification of its polymer backbone allows the development of a hydrogel with specific characteristics.26
In this study, we attempted to make a polyampholyte based on dextran because of its ease of chemical modification, and we showed that the dextran-based polyampholytes have good cryoprotective properties. We also intend to modify the dextran to develop a hydrogel-forming dextran-based polyampholyte CPA as a building block for cell scaffolds with cryoprotective properties for tissue engineering applications. We have shown that hydrogels form from dextran substituted with azide and alkyne groups via Cu-free click chemistry25 when there is an appropriate ratio of carboxyl and amino groups. These hydrogels yield excellent post-thaw survival rates of more than 90% of murine fibroblasts. This is the first report of successful cryopreservation of cells encapsulated in a hydrogel with its own cryoprotective properties, and such approaches could open new avenues for tissue engineering research and application of polymeric hydrogels.
2. Experiments
2.1. General
Dextran with a molar mass of 70 kDa (dextran, Mw = 70 kDa, Meito Sangyo Co., Ltd, Nagoya, Japan) was dried in vacuum oven at 50 °C for 24 h before use. p-Pyrrolidinopyridine (PYP, Wako Pure Chemical Industries Ltd, Osaka, Japan) was recrystallized from distilled ethyl acetate and stored over a silica gel at 0 °C. All reaction solvents were purified by distillation and stored over a 4A molecular sieve. Standard solvent F consisted of 50% HCONH2, 45% N,N-dimethylformamide, and 5% CH2Cl2 (v/v).272.2. Azide-amino-dextran preparation
First, 3-azidopropanol and 3-azidopropyl carbonylimidazole (AP-CI) were synthesized following the procedure in the literature.28 3-Chloropropanol (Wako, 5 mL, 5.66 g, 59.7 mmol), sodium azide (Tokyo Chemical Industry Co., Ltd (TCI), 7.83 g, 119 mol), and tetrabutylammonium hydrogen sulphate (TCI, 0.167 g) were dissolved in 20 mL of water and stirred at 80 °C for 24 h, then stirred overnight at room temperature. The solution was extracted three times with 100 mL ether and dried over sodium sulphate. The ether was removed by rotary evaporation, and 3-azidopropanol was obtained as a liquid and purified by vacuum distillation (boiling point 62 °C at 3–4 mbar). Next, mixing 13.77 g (84.93 mmol) of 1,1′-carbonyldiimidazole (CDI) and 150 mL of ethyl acetate yielded a turbid suspension, to which 5.72 g (56.9 mmol) 3-azidopropanol was added dropwise under vigorous stirring while the reaction mixture turned into a clear solution. After 2 h of reaction at room temperature, the solution was extracted three times with 150 mL of water. The organic layer was dried over magnesium sulphate. After the magnesium sulphate was filtered off, the solvent was evaporated by rotary evaporation and AP-CI was obtained as a liquid.To synthesize the azide-amino-substituted dextran (azide-amino-Dex), first the azide substitution was performed using AP-CI and triethylamine.28 Dex (3 g) and AP-CI (544 mg, 2.8 mmol) were dissolved in DMSO (33 mL) for 20 h at 50 °C. After 20 h, the hydroxyl groups of azide-dextran (azide-Dex) were activated by CDI (0.27 g, 0.09 eq. per sugar unit) (Tokyo Chemical Industry Co., Ltd, Japan) for 2 h at 50 °C. For this reaction, the required amount of ε-poly-L-lysine (PLL) (JNC Corporation, Tokyo, Japan) was added after activation, and the reaction was run for 36 h at 50 °C. Azide-amino-Dex was purified by dialysis (cutoff molecular weight: 14 kDa) against water for 48 h and then freeze-dried for 24 h. The amount of amino groups in the azide-amino-Dex was determined using the 2,4,6-trinitrobenzenesulfonate (TNBS) method.29 Briefly, 0.3 mL of 250 mg mL−1 sample solution, 1 mL of 1.0 mg mL−1 TNBS solution, and 2 mL of 40 mg mL−1 sodium bicarbonate aqueous solution containing 10 mg mL−1 sodium dodecyl sulphate (pH 9.0) were mixed and incubated at 37 °C for 2 h. After the mixture was cooled at 25 °C, the absorbance was measured at 335 nm using glycine as the standard sample.
2.3. Polyampholyte preparation
To synthesize the carboxylated azide-amino-dextran (azide-dextran polyampholyte, azide-Dex-PA), azide-amino-Dex and succinic anhydride (SA) (Wako) in 0%–90% mol ratios (SA/NH2-dextran amino groups) were mixed and reacted at 50 °C for 3 h to convert the amino groups into carboxyl groups. The amount of amino groups that were converted into carboxyl groups was determined using the TNBS assay.202.4. Preparation of alkyne-substituted dextran
Alkynes with different degrees of substitution (DSs) were prepared. To synthesize alkyne-substituted dextran, dibenzylcyclooctyne (DBCO) acid (Click Chemistry Tools, Scottsdale, AZ, USA) was used. In this reaction, CDI was first dissolved in solvent F, and then DBCO acid dissolved in solvent F was added and allowed to react at 25 °C for 2 h. Subsequently, dextran and PYP dissolved in solvent F were added to the reaction mixture dropwise, and the reaction was run for 18 h. After the reaction was over, the product, DBCO-substituted dextran (DBCO-Dex), was purified by multiple precipitation using methanol.272.5. Hydrogel preparation
Hydrogels were formed when azide-Dex-PA and DBCO-Dex were dissolved in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St Louis, MO) without foetal bovine serum (FBS). For the preparation of different hydrogels, the desired amounts of azide-Dex-PA and DBCO-Dex were dissolved in the culture medium. The hydrogel began to form after the azide-Dex-PA and DBCO-Dex were mixed, depending upon the concentration of the two reactants. Hydrogel formation was evaluated using the test-tube-tilting method, where a test tube containing the gel was inverted.2.6. Fluorescein isothiocyanate (FITC) labelling of Dex-PLL-PA
For fluorescent labelling, 10% Dex/DMSO solution was treated with FITC at a 1/100 molar ratio to Dex for 16 h at 50 °C. The FITC-Dex was purified by dialysis (molecular weight cutoff, 10 kDa; Spectra/Por, Spectrum Laboratories, CA, USA) against water for 72 h. The amination and succination reactions were performed to obtain FITC-labelled Dex-PA. The interaction between the FITC-labelled Dex-PA and the L929 cells following cryopreservation was observed using confocal laser-scanning microscopy (FV1000-D; Olympus, Tokyo, Japan).2.7. Nuclear magnetic resonance (NMR) spectroscopy
The 1H-NMR (400 MHz) spectra of the synthesized chemicals were recorded at 25 °C on a Bruker AVANCE III 400 spectrometer (Bruker BopSpin Inc., Switzerland) in CDCl2 for AP-CI and D2O for dextran derivatives.2.8. Scanning electron microscope observation
Prior to scanning electron microscope observation, the freeze-dried hydrogels were sputter-coated with gold for 240 s (E1030 Ion sputter). Then, images of the surface and interior of the hydrogel were recorded by a scanning electron microscope (Hitachi model 4200) at 15.0 kV.2.9. Cell culture
L929 cells (American Type Culture Collection, Manassas, VA, USA) were cultured in DMEM supplemented with 10% FBS. The cell culture was carried out at 37 °C under 5% CO2 in a humidified atmosphere. When the cells reached 80% confluence, they were removed by 0.25% (w/v) of trypsin containing 0.02% (w/v) of ethylenediaminetetraacetic acid in phosphate-buffered saline without calcium and magnesium (PBS(−)) and seeded on a new tissue culture plate for the subculture.2.10. Cryopreservation with azide-Dex-PA
Azide-Dex-PA cryopreservation solutions were prepared as follows. Azide-Dex-PA samples with various succination ratios were dissolved in DMEM at concentrations of 7.5%–15% (w/w), and the pH was adjusted to 7.4 using HCl or NaOH. The osmotic pressure was adjusted by addition of 10% (w/w) NaCl aqueous solution using an Osmometer 5520 (Wescor, Inc., UT, USA). The L929 cells were counted and resuspended in 1 mL of azide-Dex-PA solution at a density of 1 × 106 cells per mL in 1.9 mL cryovials and stored in a −80 °C freezer for 24 h. These vials were then transferred into liquid nitrogen for 1 week until it was time for thawing. Individual vials were thawed at 37 °C in a water bath with gentle shaking, and the thawed cells were diluted 10-fold in 4 °C DMEM. After centrifugation, the supernatant was removed and the cells were resuspended in 5 mL of medium. All the cells were counted using a haemocytometer and a trypan blue staining method. The reported values are the ratios of living cells to total cells.2.11. Cryopreservation in the hydrogel
Azide-Dex-PA was dissolved in 10%–12% (w/w) DMEM, and the pH was adjusted to 7.4 using HCl or NaOH. The osmotic pressure was adjusted by addition of 10% (w/w) NaCl aqueous solution to about 580 mOsm using an Osmometer. L929 cells were counted and resuspended in 1 mL of azide-Dex-PA solution at a density of 1 × 103 cells/0.1 mL in a 96-well plate, and to this was added DBCO-Dex dissolved in DMEM at the required concentration. After this, the hydrogel formation occurred within 4–5 min. This plate was stored in a −80 °C freezer overnight without any other cryoprotectants. The 96-well plate was thawed after 1 day, and then the viability of the cells was determined using a live–dead assay kit (Life Technologies Corp., NY, USA). The reported values are the ratios of living cells to total cells. As a control, cells were encapsulated in a collagen hydrogel (Cellmatrix Type I-A, Nitta Gelatin, Inc., Osaka, Japan), in a hydrogel formed by azide-Dex (non-ionic hydrogel), and in an azide-amino-Dex without any succination and cryopreserved in the same way as for the polyampholyte hydrogel.2.12. Rheological measurements of the mechanical properties of hydrogels
Rheological measurements were conducted using a strain-controlled rheometer (TA Instruments Model TA AR2000ex). A cone-plate geometry with a cone diameter of 25 mm and an angle of 4° was employed. Hydrogels for the rheological studies were prepared in the same fashion as those for the cryoprotective studies. In each measurement, 250 μL of a mixed solution of azide-Dex-PA and DBCO-Dex was loaded onto the plate by a micropipette. The dynamic viscoelastic properties of the polymer solutions during hydrogelation were measured by oscillatory shear experiments performed at a fixed frequency of 1 Hz at room temperature.2.13. Statistical analysis
All data are expressed as the mean ± standard deviation (SD). Measurements for post-thaw viability were collected with n = 5. All experiments were conducted in triplicate. Data among the different groups were compared using a one-way analysis of variance (ANOVA) with a post-hoc Tukey–Kramer test.3. Results and discussion
3.1. Synthesis of azide-substituted polyampholyte (PA) and its cryoprotective properties
The NMR spectrum of 3-azidopropyl carbonylimidazole (AP-CI) is given in Fig. S1 in the ESI.† Azide groups were introduced into the dextran by treatment with AP-CI (Scheme 1a), and dextran samples with different DSs of azide groups were synthesized. The DS was calculated from the 1H-NMR spectra (Fig. S2†) using the following formula:| DS(%) = [(Imethylene/2)/(Ianomeric proton) × 1.04] × 100 |
 | ||
| Scheme 1 Schematic representation of the synthesis of dextran polyampholyte (Dex-PA). (a) Substitution of azide in dextran. Dextran and 3-azidopropyl carbonylimidazole reacted to form dextran azopropyl carbonate (azide-Dex, A). (b) Amination of azide-dextran. A was activated with 1,1′-carbonyldiimidazole (CDI), and poly-L-lysine was grafted onto azide-Dex (azide-amino-Dex, B). (c) Succinylation of azide-amino-dextran. Succinic anhydride (SA) reacted with the azide-amino-Dex, yielding the carboxylated azide-amino-Dex (azide-Dex-PA, C). | ||
Here, Imethylene and Ianomeric proton are the integrals of the methylene peaks of the introduced azide propanol located at 2.0 ppm and of the anomeric proton of dextran located at 4.9 ppm, respectively, and 1.04 is the correction factor for the average 4% of α-1,3 linkages in dextran.30,31
Amino groups were introduced into dextran by treatment with PLL (Scheme 1b), and it was possible to control the NH2 introduction rates per sugar unit of azide-amino-Dex from 18% to 40%, as calculated from the TNBS assay. The amino groups were converted into carboxyl groups by treatment with succinic anhydride (SA) (Scheme 1c). The ratio of carboxylation shown in parentheses, e.g., azide-Dex-PA(0.65), indicates that 65% of the amino groups introduced into dextran have been converted into carboxyl groups by SA addition. The ratio of carboxylation was well controlled by the reaction with SA, and the values were proportional to the amount of SA added.
The cell-cryoprotection properties of azide-Dex-PA depend on the polymer concentration, the amount of amino groups introduced into the dextran, the ratio of carboxylation, and the osmolarity of the solution. Fig. 1a shows the cell viabilities obtained immediately after thawing for solutions of azide-Dex-PA with 10% polymer concentrations and various COOH introduction ratios, with an osmotic pressure of 580 mOsm. The cell viability increased with the percentage of introduced carboxyl groups up to 69%. When L929 cells were preserved with 10% azide-Dex-PA with carboxylation ratios of 0.50, 0.54, 0.57, 0.62, 0.66, and 0.69, the viability after thawing was similar to that when 10% DMSO without FBS was used. This result shows that the PLL polyampholytes introduced into the dextran backbone with azide groups still have good cryoprotective properties like those of DMSO. When azide-Dex-PA(0.69) was used for cryopreservation with an osmotic pressure of 580 mOsm, concentrations of 10% resulted in highest viability (Fig. 1b). Fig. 1c shows the cell viabilities immediately after thawing for solutions of azide-Dex-PA(0.69) with 10% polymer concentrations and various NH2 introduction ratios, with an osmotic pressure of 580 mOsm. In this figure, the calculated amount of NH2 groups introduced via introduction of PLL is shown as the number of NH2 groups for each dextran sugar unit. The cell viability obtained with 10% azide-Dex-PA(0.69) in which the NH2 group introduction rate was 39.5% was significantly higher than that for the NH2 introduction rate of 18.8%. The cell viability after cryopreservation tends to increase with increasing numbers of NH2 groups introduced by the PLL. This indicates that a certain amount of charged groups is required to provide the cryoprotective function. We controlled the osmotic pressure of the 10% azide-Dex-PA(0.69) solutions to about 580 mOsm based on the results of a viability assay against the osmotic pressure of azide-Dex-PA(0.69). In this assay, azide-Dex-PA(0.69) solutions with osmotic pressures of 300–700 mOsm, which are higher than that of the living body, exhibited better cryopreservation properties (Fig. 1d) than those with lower pressures. It seems that the cells were dehydrated under relatively high osmotic pressures during cooling and thus avoided damage from intracellular freezing.32,33 More than 90% viability was obtained after cryopreservation with 10% azide-Dex-PA(0.69) with 578 mOsm.
 | ||
| Fig. 1 Cryoprotective properties of dextran polyampholytes (Dex-PAs). The viability of L929 cells cryopreserved with various azide-Dex-PAs as cryoprotectant agents (CPAs) was measured immediately after thawing. (a) L929 cells were cryopreserved with 10% DMSO and 12% (w/w) azide-Dex-PAs with different ratios of introduced COOH. (b) L929 cells were cryopreserved with various concentrations of azide-Dex-PA(0.65). (c) L929 cells were cryopreserved with 10% azide-Dex-PA(0.65) with various ratios of NH2 introduced by ε-poly-L-lysine (PLL). (d) L929 cells were cryopreserved with 10% Dex-PA(0.65) under various osmotic pressures. The osmotic pressures were adjusted with 10% (w/w) NaCl aqueous solution. Data are expressed as the mean ± SD. ***P < 0.001. ###P < 0.001 vs. 10% DMSO. NS: not significant. | ||
These results agree well with our previous reports,20,21 which showed that polyampholytes could have cryoprotective properties for living cells. We have previously discussed the mechanisms leading to the cryoprotective properties of polyampholytes.23 To cryopreserve cells for long-term storage, intracellular ice formation has to be avoided. During freezing, the increasing osmotic pressure due to the concentrated, partially frozen extracellular solution dehydrates the cell. The speed and degree of dehydration both influence the cryoprotection. If polyampholytes, with their charged moieties, interact with salt molecules, which are ordinarily concentrated during freezing, the degree of dehydration could be controlled. An appropriate level of dehydration would therefore prevent intracellular ice formation. Although further investigation should be done, we proposed that polyampholytes act as cryoprotective agents by protecting cells from stresses such as drastic changes in the soluble space size and osmotic pressure.23,34 In this study, we confirmed that a high-molecular-weight (over 70 kDa) polyampholyte-based polysaccharide, namely, dextran with azide groups, also protected cells from freezing damage and that cryoprotection can be a property not only of polylysine derivatives but also of polyampholytes in general.
The affinity of dextran-based polyampholytes for the cell surface during freezing was evaluated with a confocal laser microscope. After cryopreservation with 10% Dex-PA(0.69) labelled with FITC, adsorption of Dex-PA(0.69) onto the cells was observed. Dex-PA(0.69) molecules appeared to be adsorbed onto the cell membrane immediately after thawing (Fig. 2). As shown in Fig. 2, penetration of Dex-PA was prevented by the high molecular weight and high charge density. FITC labelling does not affect the cryoprotective properties of Dex-PA or the cell membrane interactions because of the low molar ratio of FITC. This polymer adsorbed to the cell surface during freezing, which may have then protected the cell membrane in the same way as COOH-PLL.23 Although the mechanisms by which the cell membrane is protected during freezing need to be investigated in future research, we aimed to develop a hydrogel with its own cryoprotective properties by taking advantage of this extracellular cell protection.
 | ||
| Fig. 2 Confocal microscopic images of L929 cells cryopreserved with FITC-labelled Dex-PA(0.65) taken immediately after thawing. (a) Dark and (b) bright fields at the same point. The bar is 10 μm. | ||
3.2. Synthesis of alkyne-substituted dextran and hydrogel formation via Cu-free click chemistry
Hydrogels derived from Cu-free processes have many potential applications in tissue engineering. For example, they can be used in controlled drug delivery systems and in 3D cell culturing.35 Hydrogels developed using Cu-free click chemistry also shed light on the possibility of cytotoxicity due to Cu.For this purpose, DBCO acid was introduced into the dextran as an alkyne substituent, as shown in Scheme 2a, to synthesize dextran samples with different DSs of alkynes (Table 1). The DS of DBCO per sugar unit was calculated from the 1H-NMR results (Fig. S3†) using the formula given below:
| DS(%) = [(Iaromatic protons/8)/(Ianomeric proton) × 1.04] × 100 |
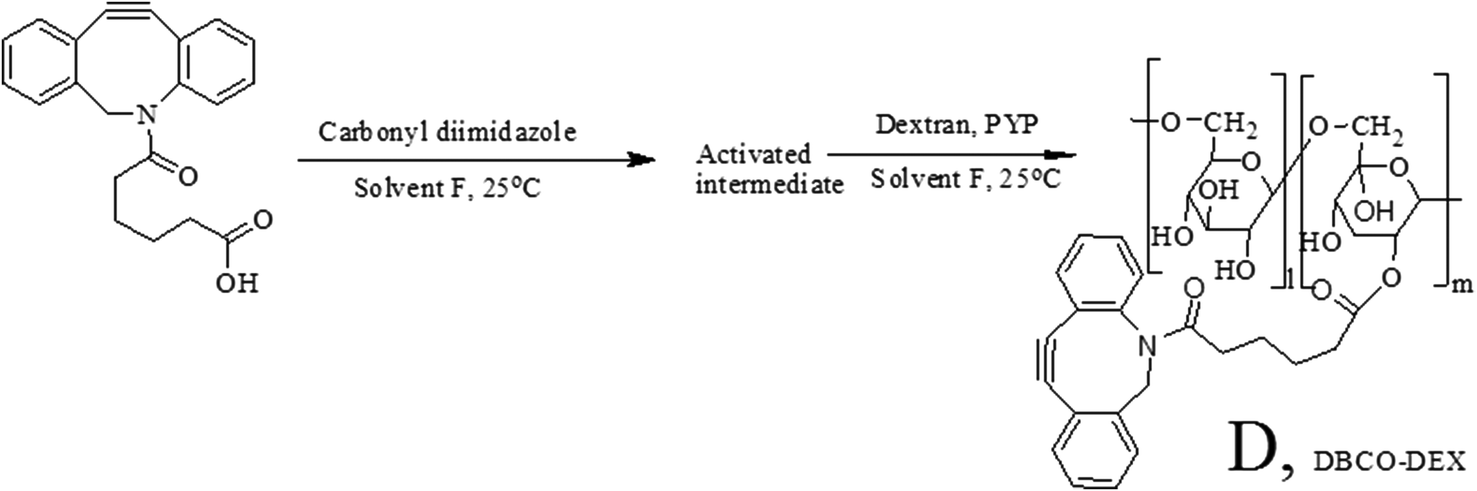 | ||
| Scheme 2 Synthesis of DBCO-Dex. DBCO was activated with CDI in solvent F, and addition of dextran yielded DBCO-substituted dextran (DBCO-Dex). | ||
| Degree of substitution of alkyne in dextran per sugar unit (%) | Water solubility | Hydrogel formation (azide-Dex-PA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DBCO-Dex [w/w]) :DBCO-Dex [w/w]) |
|---|---|---|
| 1.5 | Soluble | No (1:1) |
| 3.0 | Soluble | Yes (2:1, 3:1) |
| 4.8 | Soluble | Yes (4:1) |
| Yes (6:1) |
||
| 5.4 | Soluble | Yes (4:1, 6:1) |
| 14.3 | Insoluble | No |
Here, Iaromatic protons and Ianomeric proton are the integrals of the methylene peaks of the introduced DBCO acid located at around 7.5 ppm and of the anomeric proton of dextran at 4.9 ppm, respectively, and 1.04 is the correction factor for the average 4% of α-1,3 linkages in dextran.30,31 Dextran with a higher DS of alkynes is water-insoluble, probably because of the increase in hydrophobicity.
Hydrogels were formed by reacting DBCO-Dex and azide-Dex PA (Scheme 3a) via strain-promoted azide–alkyne cycloaddition (SPAAC) click chemistry.36,37 SPAAC has already been utilized for live-cell imaging38 and in vivo metabolic labelling, as well as hydrogel formation, because of its significant advantages in terms of cytocompatibility and biocompatibility compared with Cu-catalysed click chemistry. This click-chemistry-derived hydrogel also does not require any toxic crosslinking agents. Hydrogel formation via SPAAC between DBCO-terminated polyethylene glycol (PEG) and azide-substituted PEG has been reported previously for tissue engineering applications.39 In this previous report, the viability of cells encapsulated in situ with the Cu-free SPAAC-derived hydrogel was significantly higher than that of the cells encapsulated by the photo-crosslinked PEG hydrogel because of the high cytocompatibility of SPAAC.37 Our purpose is to encapsulate cells in situ with a polyampholyte hydrogel in order to form a cell-cryoprotective scaffold; hence, bioorthogonal and cytocompatible reactions such as SPAAC will be useful.
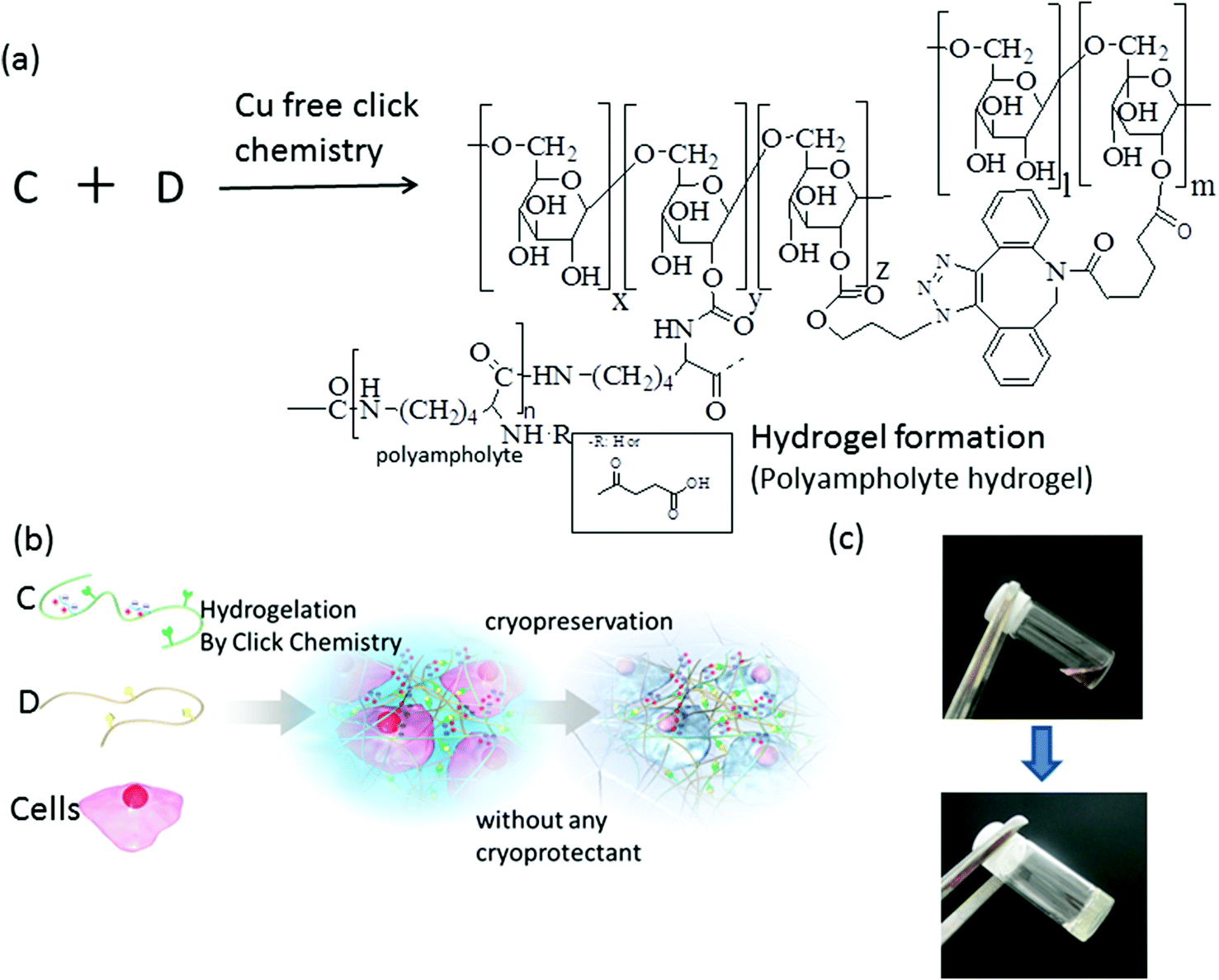 | ||
| Scheme 3 Synthesis of hydrogel and cell encapsulation. (a) Schematic representation of hydrogel formation using azide-Dex-PA and DBCO-Dex. (b) Depiction of cell encapsulation and cryopreservation by in situ hydrogelation via SPAAC click chemistry. (c) Representative picture of cell-hydrogel construct (10% polymer concentration, azide-Dex-PA(0.69) (DS of azide 11.7%):DBCO-Dex (DS of DBCO 6.5%) ratio = 4:1 (wt:wt), 570 mOsm, pH 7.4, cell density = 1 × 104 mL−1). | ||
3.3. Rheological characterization of hydrogels
The strength of the hydrogels was investigated by performing rheological tests in order to establish the degree to which the variations in concentrations of DBCO-Dex and azide-Dex-PA influence crosslinking. In Fig. 3, the dynamic mechanical properties of different hydrogels are shown. Different hydrogels were formed at different concentrations of DBCO-Dex and azide-Dex-PA, so the strength of the hydrogel can be tuned by varying the concentrations of the two reacting species. The storage modulus gradually increased with time because crosslinking points were generated by the click chemistry reaction between the azides and alkynes. The gelation time and storage modulus 600 s after mixing of the two components are summarized in Table 2. The mechanical strength of the hydrogels can easily be varied from 6 Pa to 2.7 kPa. These values are consistent with the mechanical properties of tissue-engineered hydrogels and the extracellular matrix.40 When we mixed azide-Dex-PA and DBCO-Dex in weight ratios of 1:1 and 1:2.5, both the gelation time and storage modulus were observed to increase with the total polymer concentration.
 | ||
| Fig. 3 Storage moduli for different hydrogels with varying total polymer concentrations. Hydrogels composed of azide-Dex (DS of azide 11.7%) and DBCO-Dex (DS of DBCO 6.5%). | ||
| Azide-Dex-PA:DBCO-Dex (wt:wt) |
Total polymer concentration/% | Gelation time/s | Storage modulus at 600 s after mixing/Pa |
|---|---|---|---|
| 1:1 |
2.0 | 560 | 53.6 |
| 1:1 |
2.5 | 480 | 134.1 |
| 1:1 |
3.0 | 120 | 243.8 |
| 1:1 |
3.5 | 60 | 595.9 |
| 1:2.5 |
5.0 | 120 | 308.0 |
| 1:2.5 |
6.8 | 60 | 915.8 |
| 1:2.5 |
8.6 | 50 | 2612.0 |
| 6:1 |
11.7 | 540 | 6.46 |
| 4:1 |
7.3 | 480 | 19.6 |
Hydrogels formed with higher amounts of alkynes and lower amounts of azides are stronger than hydrogels formed with larger amounts of azides and lower amounts of alkynes. However, when the amounts of DBCO-Dex and azide-Dex-PA were similar, intermediate strengths were obtained. This is reasonable, because the DS of azide groups is higher than that of alkynes in dextran. Hence, when the amount of azide groups increases relative to the amount of alkynes, a substantial amount of azide groups will be unreacted during crosslinking.
Furthermore, the gelation time for hydrogel formation can be tuned by varying the concentrations of azide groups and alkynes in the dextran. After some preliminary tuning, concentrations at which the hydrogel formation time is around 5–10 min were selected for cryopreservation purposes.
Scanning electron microscopy (SEM) was also performed to evaluate the physical and chemical nature of the crosslinked hydrogels. SEM images of the hydrogels showed densely crosslinked networks with varying porosities and microstructures (Fig. 4). The pore size of the hydrogels was found to be approximately 50–150 μm, which is larger than the size of a cell. This observation suggests that during cryopreservation, the cells are inside the cavities of the hydrogel.
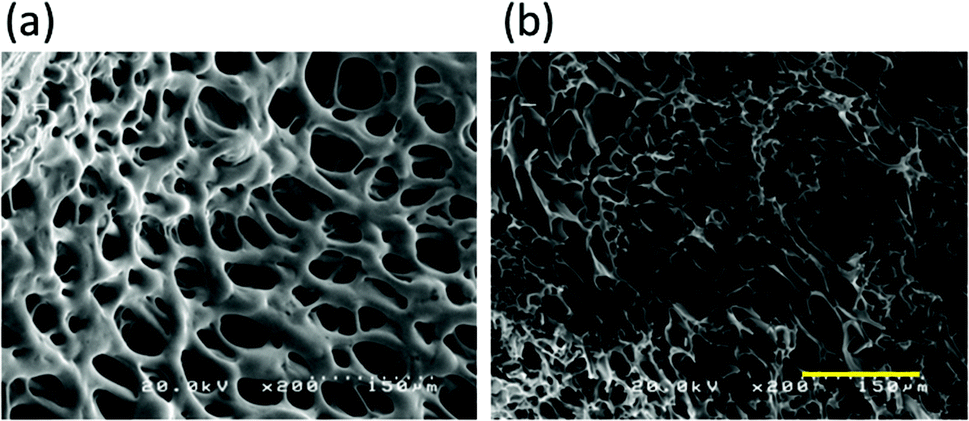 | ||
| Fig. 4 SEM image of lyophilized hydrogel composed of azide-Dex-PA (DS of azide 11.7%) (12 mg mL−1) and DBCO-Dex (DS of DBCO 6.5%) (3 mg mL−1) formed via Cu-free click chemistry in a DMEM medium without FBS. (a) Surface and (b) inside of the lyophilized hydrogel. The bar is 150 μm. | ||
This result suggests that an azide-conjugated polyampholyte cryoprotectant could be used with DBCO-Dex for biomedical therapeutic technologies such as an injectable hydrogel cell-delivery system41 or orthopaedic applications.42 For example, stem cells cryopreserved with azide-Dex-PA can be mixed with DBCO-Dex immediately after thawing for injection into defect sites to form a scaffold for cell growth and tissue repair. This system does not require any pretreatment of the stem cells before injection, making cell maintenance, harvesting, and mixing with the hydrogel-forming media unnecessary. In our system, the stem cells could be preserved until just before usage, and the cells can be injected just after thawing without washing out the cryoprotectant.
3.4. Cryoprotective properties of the hydrogel
The cryoprotective properties of different hydrogels were checked using azide-Dex-PA (DS of azide is 11.7%, and DS of NH2 groups is 40%) and DBCO-Dex, which act as crosslinkers (DS of alkyne is 6.5%). L929 cells cryopreserved with these hydrogels (total polymer concentration was 10%) (Scheme 3b and c) showed a viability of more than 90% immediately after thawing (Fig. 5, Table 3). When the cells were cryopreserved without any cryoprotectants in a collagen hydrogel, which is widely used for tissue engineering applications, all cells were killed. Almost 0% viability was obtained when cells were cryopreserved within the hydrogel formed by azide-amino-Dex and azide-Dex without any succination. These results showed that the polyampholyte hydrogel was necessary to protect cells from freezing damage. | ||
| Fig. 5 Cell viability after cryopreservation with two different types of hydrogels with different ratios of azide-Dex-PA(0.65) (DS of azide 11.7%) and Dex-alkyne (DS of DBCO 6.5%) and with a collagen hydrogel. Fluorescent microphotographs of cell encapsulated in (a) 4:1 azide-Dex-PA(0.69):DBCO-Dex, (b) 6:1 azide-Dex-PA(0.69):DBCO-Dex, (c) 4:1 azide-amino-Dex:DBCO-Dex, (d) 4:1 azide-Dex:DBCO-Dex, and (e) collagen hydrogel taken using a live–dead assay kit. The bar is 50 μm. | ||
Current cryopreservation techniques have mainly been devised and optimized for cell suspensions, and their application to adherent monolayers or thick biological samples is known to be less effective.43,44 To cryopreserve larger volume cell-containing matrixes such as cell aggregates, tissue-engineered constructs, and cell sheets, a vitrification method is usually applied.13,45 Vitrification is one of the most established methods for the cryopreservation of large cells like oocytes and embryos.46 Vitrification circumvents the problems associated with ice-crystal formation by inducing glass-like solidification during rapid cooling in a high-concentration cryoprotectant solution. However, the high cytotoxicity from highly concentrated cryoprotectants is still a serious problem for maintaining tissue-like structures with high cell viability. As a substitute for a vitrification method, this study proposed a novel approach to cryopreservation of tissue-engineered constructs with a cryoprotective hydrogel. We succeeded in introducing 0.041 mg mL−1 of the cell-attachment peptide RGDS into dextran in the present study, and found that cells could attach to and proliferate on the hydrogel (ESI, Fig. S4†) during a culture over 7 days. This finding clearly demonstrates that we can introduce cell compatibility into a hydrogel formed in situ and that this system can be used to produce cell scaffolds with cryoprotective properties. Moreover, the hydrogel might be suitable as a tissue-engineering scaffold,47 because the triazole ring at the crosslinking point is connected to dextran through carbonate esters (Scheme 3), and this structure is known to be hydrolysable under physiological conditions.25 In future research, we should investigate the cryoprotection properties of tissue-engineered constructs developed using this in situ hydrogel system and further details of the scaffolds such as their degradation and cell proliferation.
4. Conclusion
In this study, we developed polyampholyte cryoprotectants with azide groups that form in situ hydrogels that can encapsulate cells along with DBCO-Dex via Cu-free click chemistry. The system showed excellent cryoprotective properties without any additional cryoprotectants. This is the first challenge in the development of cryoprotective hydrogels by means of a combination of materials science and cryobiology. In the past few years, the potential to mould the shapes and sizes of biologically relevant hydrogels has led to new directions in generating tissues. It has also provided opportunities to meet various challenges in tissue engineering such as vascularization, the formation of tissue architectures, and cell seeding. These methodologies are useful for engineering tissue architectures inside the cell-containing hydrogel. Overall, it appears that our dextran-based cryoprotective hydrogel has the potential to overcome many of the challenges that have troubled the field of tissue engineering.Disclosures
The authors have no conflicts of interest to declare.Acknowledgements
This study was supported in part by a grant from the Japan Science and Technology Agency for the Adaptable and Seamless Technology transfer Program through target-driven R&D (AS231Z03401F), in part by a Grant-in-Aid, KAKENHI (25870267) for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and in part by a grant from The Canon Foundation (K11-N-028).References
- A. M. Karow Jr. and W. R. Webb, Cryobiology, 1965, 4, 270–273 CrossRef.
- F. Brandl, F. Sommer and A. Goepferich, Biomaterials, 2007, 28, 134–146 CrossRef CAS PubMed.
- T. Huaping and G. M. Kacey, Materials, 2010, 3, 1746–1767 CrossRef.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- J. S. Tememoff and A. G. Mikos, Biomaterials, 2000, 21, 2405–2412 CrossRef.
- N. E. Vrana, K. Matsumura, S. H. Hyon, L. M. Geever, J. E. Kennedy, J. G. Lyons, C. L. Higginbotham, P. A. Cahill and G. B. McGuinness, J. Tissue Eng. Regen. Med., 2012, 6, 280–290 CrossRef CAS PubMed.
- S. H. Hyon, W. Cha and Y. Ikada, Polym. Bull., 1989, 22, 119–122 CrossRef CAS.
- A. M. Karow Jr., O. Carrie Jr. and B. R. Clower, J. Pharm. Pharmacol., 1968, 20, 297–301 CrossRef CAS.
- B. J. Fuller, Cryoletters, 2004, 25, 375–388 CAS.
- R. M. Lowenthal, D. S. Park, J. M. Goldman, R. S. Hill, G. Whyte and K. H. Th'ng, Br. J. Haematol., 1976, 34, 105–117 CrossRef CAS.
- L. Douay, N. C. Gorin, R. David, J. Stachowiak, C. Salmon, A. Najman and G. Duhamel, Exp. Hematol., 1982, 10, 360–366 CAS.
- Y. N. Wu, H. R. Yu, S. Chang, R. Magalhaes and L. L. Kuleshova, Tissue Eng., 2007, 13, 649–658 CrossRef CAS PubMed.
- W. X. Fan, X. H. Ma, T. Q. Liu and Z. F. Cui, J. Biosci. Bioeng., 2008, 106, 610–613 CrossRef CAS PubMed.
- F. F. Chen, W. J. Zhang, W. Wu, Y. Q. Jin, L. Cen, J. D. Kretlow, W. C. Gao, Z. P. Dai, J. M. Wang, G. D. Zhou, W. Liu, L. Cui and Y. L. Cao, Biomaterials, 2011, 32, 8426–8435 CrossRef CAS PubMed.
- K. Kito, H. Kagami, C. Kobayashi, M. Ueda and H. Terasaki, Cornea, 2005, 24, 735–741 CrossRef.
- N. Kotobuki, M. Hirose, H. Machida, Y. Katou, K. Muraki, Y. Takakura and H. Ohgushi, Tissue Eng., 2005, 11, 663–673 CrossRef CAS PubMed.
- Y. A. Petrenko, D. R. E. Jones and A. Y. Petrenko, Cryobiology, 2008, 57, 195–200 CrossRef CAS PubMed.
- G. S. Jiang, K. H. Bi, T. H. Tang, J. W. Wang, Y. K. Zhang, W. Zhang, H. Q. Ren and Y. S. Wang, Int. Immunopharmacol., 2006, 6, 120413 Search PubMed.
- K. Matsumura and S. H. Hyon, Biomaterials, 2009, 30, 4842–4849 CrossRef CAS PubMed.
- K. Matsumura, J. Y. Bae and S. H. Hyon, Cell Transplant., 2010, 19, 691–699 CrossRef PubMed.
- K. Matsumura, J. Y. Bae, H. H. Kim and S. H. Hyon, Cryobiology, 2011, 63, 76–83 CrossRef CAS PubMed.
- K. Matsumura, F. Hayashi, T. Nagashima and S. H. Hyon, J. Biomater. Sci. Polym. Ed., 2013, 24, 1484–1497 CrossRef CAS PubMed.
- R. Rajan, M. Jain and K. Matsumura, J. Biomater. Sci. Polym. Ed., 2013, 24, 1767–1780 CrossRef CAS PubMed.
- B. G. De Geest, W. Van Camp, F. E. Du Prez, S. C. De Smedt, J. Demeester and W. E. Hennink, Chem. Commun., 2008, 190–192 RSC.
- G. Sun, Y. I. Shen, C. C. Ho, S. Kusuma and S. Gerecht, J. Biomed. Mater. Res. A, 2010, 93, 1080–1090 Search PubMed.
- C. H. Bamford, I. P. Middleton and K. G. Al-Lamee, Polymer, 1986, 27, 1981–1984 CrossRef CAS.
- J. Zhang, C. Li, Y. Wang, R. X. Zhuo and X. Z. Zhang, Chem. Commun., 2011, 47, 4557–4559 RSC.
- A. F. Habeeb, Anal. Biochem., 1966, 14, 328–336 CrossRef CAS.
- S. G. Lévesque, R. M. Lim and M. S. Shoichet, Biomaterials, 2005, 26, 7436–7446 CrossRef PubMed.
- W. N. E. Van Dijk-Wolthuis, O. Franssen, H. Talsma, M. J. Van Steenbergen, J. J. Kettenes-van den Bosh and W. E. Hennink, Macromolecules, 1995, 28, 6317–6322 CrossRef CAS.
- P. Mazur, Cryobiology, 1997, 14, 251–272 CrossRef.
- P. Mazur, I. L. Pinn and F. W. Kleinhans, Cryobiology, 2007, 55, 158–166 CrossRef CAS PubMed.
- K. Matsumura, F. Hayashi, T. Nagashima and S. H. Hyon, Cryobiol. Cryotechnol., 2013, 59, 23–28 Search PubMed.
- C. A. DeForest, E. A. Sims and K. S. Anseth, Chem. Mater., 2010, 22, 4783–4790 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. Xu, T. M. Filion, F. Prifti and J. Song, Chem.–Asian J., 2011, 6, 2730–2737 CrossRef CAS PubMed.
- K. E. Beatty, J. D. Fisk, B. P. Smart, Y. Y. Lu, J. Szychowski, M. J. Hangauer, J. M. Baskin, C. R. Bertozzi and D. A. Tirrell, ChemBioChem, 2010, 11, 2092–2095 CrossRef CAS PubMed.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- S. Even-Ram, V. Artym and K. M. Yamada, Cell, 2006, 126, 645–647 CrossRef CAS PubMed.
- K. Nagahama, T. Ouchi and Y. Ohya, Adv. Funct. Mater., 2008, 18, 1220–1231 CrossRef CAS.
- D. Gyawali, P. Nair, H. K. Kim and J. Yang, Biomater. Sci., 2013, 1, 52–64 RSC.
- A. Sotres-Vega, J. Villalba-Caloca, R. Jasso-Victoria, J. R. Olmos-Zuniga, M. Gaxiola-Gaxiola, M. Baltazares-Lipp, A. Santibanez-Salgado and P. Santillan-Doherty, J. Invest. Surg., 2006, 19, 125–135 CrossRef PubMed.
- R. Malpique, F. Ehrhart, A. Katsen-Globa, H. Zimmermann and P. M. Alves, Tissue Eng. C, 2009, 15, 373–386 CrossRef CAS PubMed.
- R. Magalhães, B. Nugraha, S. Pervaiz, H. Yu and L. L. Kuleshova, Biomaterials, 2012, 33, 829–836 CrossRef PubMed.
- W. F. Rall and G. M. Fahy, Nature, 1985, 313, 573–575 CrossRef CAS.
- E. A. Scott, M. D. Nichols, R. Kuntz-Willits and D. L. Elbert, Acta Biomater., 2010, 6, 29–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60261c |
| This journal is © The Royal Society of Chemistry 2014 |