A collagen-based corneal stroma substitute with micro-designed architecture
Cemile
Kilic
abc,
Alessandra
Girotti
d,
J. Carlos
Rodriguez-Cabello
d and
Vasif
Hasirci
*abce
aDepartment of Biological Sciences, METU, Ankara, Turkey
bDepartment of Biotechnology, METU, Ankara, Turkey
cBIOMATEN, Center of Excellence in Biomaterials and Tissue Engineering, METU, Ankara, Turkey
dGIR Bioforge, University of Valladolid, CIBER-BBN, Valladolid, Spain
eDepartment of Biomedical Engineering, METU, Ankara, Turkey. E-mail: vhasirci@metu.edu.tr
First published on 1st November 2013
Abstract
A 3D corneal stroma substitute with micro-level patterns was constructed from a stack of 4 micro patterned collagen or collagen–elastin like recombinamer (Col:ELR) blend layers. The transparency of all of the films was quite high with the uncrosslinked (UXL) films and dehydrothermally treated (150 °C, 24 h) Col:ELR films yielding the best results. Human corneal keratocytes (HK) could be attached and proliferated equally well on the single films of Col and Col:ELR. However, for the multilayer constructs the proliferation was higher on Col than on Col:ELR. The cells were found to align along the patterns (microchannels with a 39 μm groove depth, 8 μm groove width, 3.3 μm ridge width, and 54.7° inclination angle) of the films, while no significant alignment was observed on the unpatterned films. The transparency of the seeded Col:ELR films was superior to the Col films over a 30 day incubation period and was quite close to that of a native human cornea. It was concluded that the Col and Col:ELR films and their 3D constructs have significant potential for use as corneal stroma substitutes.
Introduction
The cornea is the transparent exterior part of the eye and is about 500 μm thick. It protects the eye from external objects and functions as the principle optical element, refracting 70% of any incoming light.1 The stroma is the thickest part of the cornea, which consists of 200–400 lamellae that are parallel to the cornea surface, but orthogonal to adjacent lamellae, forming a plywood-like structure that is populated by keratocytes. The organization of the lamellae and fibrils is essential for the biomechanical and optical properties of the cornea2,3 and is made up of collagen types I and V, and proteoglycans. Any disruption of the organization of collagen or proteoglycans, like keratocan and lumican, leads to a decrease in the transparency of the cornea.4–6Corneal diseases and wounds are the next major causes of blindness after cataracts. There are about 27.9 million blind people worldwide and of these 4.9 million have bilateral corneal blindness.7 Today, cornea transplantation is the only effective treatment for corneal blindness and it is more successful than other solid organ transplantations due to the avascular nature of the cornea.8 However, a shortage of donor corneas and the risks of disease transmission and immune rejection make the replacement of a damaged cornea with an artificial substitute a promising alternative. At present, keratoprostheses (KPro) that use synthetic polymers like PHEMA9 and PMMA10 are the only options for patients with a history of several corneal graft rejections and problems including calcification, infection and retinal detachment. Osteo-odonto KPro (OOKP), Boston KPro, and AlphaCor KPro are the three most commonly used keratoprostheses. A common property of these keratoprostheses is their core-and-skirt design, which aims to achieve good integration between the prosthesis and the host tissue and a higher clinical success rate.9,11 However, the development of glaucoma, retinal detachment and necrosis are still remaining problems that can be associated with keratoprostheses.12 Tissue engineered products that can simulate the organization of a natural cornea are expected to help eliminate the problems associated with transplantation and keratoprostheses. Several tissue engineering approaches have been used to reconstruct the cornea involving natural and/or synthetic materials. Most of these studies have focused on the construction of split thickness corneas, composed of one layer that substitutes for either the epithelial,13,14 stromal15,16 or endothelial layers17,18, or two layer hemi-corneas, composed of epithelial and stromal layers.19,20 Several other attempts have been made to construct a full thickness cornea using tissue engineering.21,22 Foams, hydrogels and meshes may provide optimal conditions for cell growth and can be transparent enough, however, none of these scaffolds are able to properly mimic the natural microstructure and organization of the stroma, which is quite important for its biaxial stiffness and transparency. Orthogonal alignment of the keratocytes and collagen layers in the stroma is crucial for both the transparency and mechanical strength of the cornea and this can be imposed on the keratocytes by the surface upon which they are grown. It has been reported in several studies that cell and tissue growth can be greatly influenced by surface properties such as the topography of the substrate. As cells react with a surface, their orientation and organization change, while their movements are polarized, the strength of their adhesion changes, and signalling pathways are activated.23,24 Zorlutuna et al. (2007) oriented cells using micropatterned films that were prepared by solvent casting blends of poly(3-hydroxybutyric acid-co-3-hydroxyvaleric acid) (PHBV) and poly(L-lactic acid-co-D,L-lactic acid) (P(L/DL)LA).25 Gil et al. (2010) employed the same method to obtain a series of dimensionally different surface patterned silk films and studied the effect of the patterns on cell attachment and alignment.26 These studies demonstrated that deeper and narrower groove structures led to greater orientation of the cells, while the DNA content remained unchanged. In another study, Vrana et al. (2008) investigated the behaviour of corneal keratocytes using micropatterned collagen films prepared by a solvent casting method. They reported that the cells reached confluency on the films within 7 days and almost all of them were aligned along the patterns.27 Builles et al. (2010) attempted to create lamellar structures by applying a magnetic field,20 then carried out in vivo studies on rabbits using the resulting cell-free scaffolds. They reported that only one out of the five operated corneas retained its scaffold, but the lamellar organization of all of the corneas was interspersed with keratocytes. In another study, Gil et al. (2010) reported the helicoidal organization of stroma forming multilayers from micropatterned silk films and then carried out in vitro studies.26 These studies demonstrated that the keratocytes were well oriented, but the exact orientation angle of the layers, the spacing between layers, and the number of the stacks still remain to be determined. Even though all of these studies have mimicked the organization of the stroma well, no corneal equivalent has passed the in vivo stage and none is available for clinical use.
In addition to the organization achieved by the topography, the chemistry of the material is also crucial. Elastin like recombinamers (ELRs) are artificial proteins which are very attractive due to their amino acid sequences, which are tailored to have strong interactions with many types of cell including bone and endothelial cells.28 RGD (R: L-arginine, G: glycine, and D: L-aspartic acid) and REDV (R: L-arginine, E: L-glutamic acid, D: L-aspartic acid, and V: L-valine) were the first two sequences produced as synthetic proteins of this nature.29,30 ELRs have been used in many tissue engineering and other biomaterial applications including drug delivery,31 culture surfaces,32 and in the engineering of tissues like cartilage,33 bone,34 eye,35 liver,36 and oral mucosa.37 YIGSR (Y: L-tyrosine, I: L-isoleucine, G: glycine, S: L-serine, and R: L-arginine) is another sequence that has been shown to enhance binding, proliferation, and the spreading of various cell types especially endothelial and38 epithelial cells,39 as well as fibroblasts.40 To the best of our knowledge, the present study is the first to use YIGSR carrying ELRs blended with collagen type I for the construction of micropatterned films that can be used to improve the adhesion and proliferation of stromal keratocytes. In the current study, the natural organization of the stroma was mimicked by manufacturing micropatterned collagen and collagen:ELR films that helped to align the cells. These films were then used to construct multi-layered 3D scaffolds, in which each micropatterned film was stacked perpendicular to the adjacent film. Following crosslinking via dehydrothermal (DHT) treatment, the in vitro performance of the scaffolds was studied.
Materials and methods
Materials
4′,6-Diamine-2-phenylindole dihydrochloride (DAPI), amphotericin B, and collagenase type II from Clostridium histolyticum were purchased from Sigma-Aldrich (USA). Newborn calf serum, Dulbecco's Modified Eagle Medium–Ham's nutrient mixture F12 (DMEM–HAM's F12, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with and without phenol red, trypsin–EDTA (0.25%), penicillin/streptomycin, RPMI-1640 and fetal bovine serum were obtained from HyClone, Thermo Scientific (USA). Human basic fibroblast growth factor (hFGF basic/FGF2) with carrier was purchased from Cell Signaling Technology, Inc. (USA). Spectra™ Multicolor broad range protein ladder was obtained from Fermentas, Thermo Scientific (USA). Dimethyl sulfoxide (DMSO) and Triton X-100 were bought from AppliChem (USA). Alamar Blue was purchased from Invitrogen, Inc. (USA). NucleoCasettes were obtained from ChemoMetec (Denmark).
:1) with and without phenol red, trypsin–EDTA (0.25%), penicillin/streptomycin, RPMI-1640 and fetal bovine serum were obtained from HyClone, Thermo Scientific (USA). Human basic fibroblast growth factor (hFGF basic/FGF2) with carrier was purchased from Cell Signaling Technology, Inc. (USA). Spectra™ Multicolor broad range protein ladder was obtained from Fermentas, Thermo Scientific (USA). Dimethyl sulfoxide (DMSO) and Triton X-100 were bought from AppliChem (USA). Alamar Blue was purchased from Invitrogen, Inc. (USA). NucleoCasettes were obtained from ChemoMetec (Denmark).
Methods
000) using a phosphate buffer (pH 7.2). The obtained white precipitate of collagen was centrifuged, then sodium chloride was added (5%) and the solution was dialyzed again after further centrifugation. The white precipitate was centrifuged and the final pellet was sterilized with 70% ethanol, before being frozen at −80 °C (Sanyo MDF-U53865, Japan) and lyophilized (Labconco Freezone 6, USA). SDS-PAGE analysis was used to determine the purity of the collagen.
000 rpm) at 4 °C for 30 min and then incubated at 40 °C. Following centrifugation at 40 °C, the pellet was resuspended and the cold and warm centrifugation steps were repeated twice more. The protein was frozen at −24 °C and then lyophilized.
 | ||
| Fig. 1 Monomer composition of the elastin-like recombinamer (ELR), YIGSR. | ||
SDS-PAGE. SDS-PAGE was performed in order to assess the purity of YIGSR produced. 6 μL of YIGSR solution (1 mg mL−1 in Milli Q) was loaded into a polyacrylamide gel (separating gel: 12% acrylamide/bisacrylamide, and stacking gel: 4% acrylamide/bisacrylamide). The presence and purity of the polymer were determined by an intense band around 90 kDa.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry was also used to determine the purity and molecular weight of YIGSR. A Voyager STR mass spectrometer (Applied Biosystems) was used in linear mode and bovine serum albumin (BSA) was used for external calibration.
 | ||
| Fig. 2 Silicon master shape and dimensions. | ||
The Col and Col:ELR films. Solvent casting was used to prepare the micropatterned films. A collagen solution (15 mg mL−1 in 0.5 M acetic acid) was prepared at 29 °C using continuous stirring until complete dissolution occurred. 1 mL of the collagen solution was poured onto a patterned PDMS secondary template and allowed to dry under air for 2 days. The dry film was removed from the template and its thickness was determined to a sensitivity of 0.1 μm, before being stored at 4 °C until use.
In order to prepare the Col:ELR films, collagen and ELR solutions of the same concentration were prepared in acetic acid (0.5 M) and PBS (10 mM, pH 7.4), respectively, and mixed in a 5:1 ratio (v/v). The procedure was then completed as mentioned above.
Unpatterned (smooth) Teflon sheets (2 cm × 2 cm) were used as templates to prepare the unpatterned collagen films, which were processed as described above.
Multilayer scaffold preparation. A 4 layer stack of collagen or collagen–ELR films was prepared by placing the film patterns perpendicular to each other, then gluing them together using droplets of collagen solution placed at the 4 corners.
Crosslinking of the constructs. The single or multilayered film stacks were stabilized by physical crosslinking using a dehydrothermal treatment (DHT). In order to achieve this, the films were incubated under vacuum at 105 °C (24 h), 140 °C (24 h), or 150 °C (24 h) and the stabilities of the resulting films were then tested in PBS solution (37 °C, pH 7.4) and type II collagenase.
Enzymatic degradation with collagenase. The stability of the films towards enzymatic degradation was determined using collagenase type II as described previously.46 Briefly, the pre-weighed films were incubated in collagenase type II solution (0.1 mg mL−1 in PBS pH 7.4) for 2 h, rinsed with distilled water 3 times, lyophilized, and then weighed. The remaining weight was calculated as follows:
where Wo is the original weight and Wd is the weight after treatment.

In situ degradation tests. In order to study their stability, the crosslinked films were incubated in 10 mM PBS (pH 7.4) at 37 °C with continuous stirring and examined at 1, 2, 3 and 4 weeks. After removal from the solution each film was rinsed with distilled water 3 times, lyophilized and then the remaining weight determined.
Cell seeding on the scaffolds. Human keratocytes (passages 5 to 13) and 3T3 cells (passages 15 to 20) were detached from the surface of a TCPS flask using trypsin–EDTA solution (1
:1, 0.25% EDTA–PBS, pH 7.4) at 37 °C for 5 min, and then centrifuged (3000g for 5 min). After suspension in the medium, the cell number was determined with a NucleoCounter (ChemoMetec, Denmark). Cells were seeded onto UV sterilized films (15 min, each side) at a density of 1 × 104 cells per cm2. For the multilayer scaffolds, 1 × 104 cells per cm2 were seeded onto the top and on each of the other layers using an insulin syringe. The cell seeded and unseeded (control) scaffolds were incubated in a CO2 incubator at 37 °C. The medium used to suspend the keratocytes contained Dulbecco's Modified Eagle Medium–Ham's nutrient mixture F12 (DMEM–HAM's F12, 1:1), newborn calf serum (10%), amphotericin B (1 μg mL−1), penicillin (100 UI mL−1), and streptomycin (100 μg mL−1) and the medium used for the 3T3 cells was RPMI 1640 supplemented with 10% fetal bovine serum, penicillin (100 UI mL−1), and streptomycin (100 μg mL−1). The medium was changed every two days.
Alamar Blue cell viability assay. Proliferation of the cells on the scaffolds was determined using an Alamar Blue assay. The scaffolds were washed twice with DMEM–HAM's F12 (1
:1, colorless) and incubated in 10% Alamar Blue solution in DMEM–F12 (1:1) growth medium for 1 h at 37 °C and under a 5% CO2 atmosphere. After incubation, 200 μL of the reduced solution was transferred to a 96 well plate. The scaffolds were washed twice with the colorless medium, then the keratocyte growth medium was added and the solution was incubated again, in order to determine the cell number at different time points. The absorption intensity of the reduced dye at 570 and 595 nm was determined using an ELISA plate reader (Molecular Devices, USA). The absorbances were converted into reduction percentage values and then to cell numbers using a calibration curve.
SEM. The films (1 cm2) were coated with Au–Pd under vacuum and studied using a scanning electron microscope (JEOL, JSM-6400, USA) equipped with a NORAN System 6 X-Ray Microanalysis System and with a SEM (QUANTA, 400F Field Emission SEM, USA and SEC, Mini-SEM, South Korea) at 5–20 kV.
The seeded scaffolds were fixed with 2.5% v/v glutaraldehyde in cacodylate buffer (pH 7.4) for 2 h at room temperature. After washing several times with cacodylate buffer and distilled water, the samples were lyophilized and then examined with SEM as described above.
The samples used for fluorescence microscopy analysis were washed with distilled water and studied with SEM after lyophilization.
Fluorescence microscopy – DAPI staining. The single layer films were stained as they were, while the multilayer films were separated into single layer films and then stained. Briefly, the growth medium of the cells was discarded and the samples were then fixed directly with 4% (w/v) paraformaldehyde at room temperature for 30 min and the cell membrane was permeabilized with Triton X-100 (1% v/v in PBS, pH 7.4) at room temperature for 5 min. After washing three times with PBS (pH 7.4), the samples were incubated in DAPI (1
:3000 w/v, in 0.1% BSA) for 5 min at 37 °C. The samples were washed with PBS three times and then stored in PBS solution until being examined using a Zeiss Axio Imager M2 (Germany) fluorescence microscope.
Transparency of the films
In order to determine their transparency, the light transmittance of the films was determined in the range 250–700 nm with a UV-Vis spectrophotometer (Thermo Scientific, Multiscan Spectrum, Finland) for up to 30 days.Statistical analysis
Statistical analysis was carried out using the Student's t-test, in which p values smaller or equal to 0.05 were considered to be statistically significant.Results and discussion
Characterization of collagen type I isolated from rat tails
SDS-PAGE analysis was employed in order to determine the purity of the collagen type I isolated from the rat tails (Fig. 3). The first lane shows a protein ladder with bands at 260 kDa, 140 kDa, 100 kDa, and 70 kDa (top to bottom). Collagen type I exhibits doublets in the molecular weight ranges 115–130 kDa and 215–235 kDa.47 The isolated collagen and commercial collagen type I both exhibit doublets in these regions, indicating that they are collagen type I. The absence of any other bands indicates that the collagen isolated from the rat tails is pure. | ||
| Fig. 3 SDS-PAGE of collagen isolated from rat tail tendons. | ||
Isolation of elastin like recombinamers
YIGSR ELRs were isolated and purified from the bacterial lysate by using their temperature responsiveness. Below their transition temperature (Tt) (24.0 °C in PBS, pH 7.4) they are soluble in water, but they precipitate above this temperature. MALDI-TOF and SDS-PAGE tests were carried out to determine the purity of the YIGSR ELRs (Fig. 4). The theoretical mass of the polymer was calculated to be 89366 Da and the peak at 89455 Da in the MALDI-TOF spectrum shows that the mass of the produced polymer is very close to this theoretical value, while the peak at 44707 is due to the doubly charged species. An intense band around 89 kDa in the SDS-PAGE analysis of the same sample also confirms the purity of the polymer produced.
 | ||
| Fig. 4 MALDI-TOF and SDS gel electrophoresis analysis results for YIGSR-ELR. The theoretical molecular weight is 89.366 kDa. (A) The MALDI-TOF spectrum and (B) the SDS gel electrophoresis. | ||
Film characterization and degradation
The collagen fibrils of the cornea are more ordered and densely packed than in any other tissue. The cornea should be transparent in order to perform its function and the ordered structure and avascularity of the cornea lead to it being transparent. Corneal stromal keratocytes play an important role in collagen metabolism, both in the synthesis and degradation of the collagen fibrils. Matrix degrading enzymes and matrix metalloproteinases are both produced by corneal keratocytes.48 Thus, in order to preserve the transparency and other vital functions of the cornea, the organization of the collagen in the scaffold should not be disrupted by enzymes in the body until new extracellular matrix (ECM) is secreted and this compensates for the degraded collagen. In this study, in order to test the stability of the films against the activity of early proteolytic enzymes the films were incubated with collagenase type II, according to the procedure described by Vrana et al. (2007)46 (Fig. 5). It was observed that the uncrosslinked films (UXL) and the films crosslinked using a dehydrothermal treatment at 105 °C for 24 h (DHT105) totally degraded during the test with the collagenase, but the films crosslinked at 140 °C (DHT140) and 150 °C (DHT150) for 24 h resisted degradation to some extent. 28% of the weight of the DHT140 film remained, while 88% remained of the DHT150 film. | ||
| Fig. 5 Degradation profiles of the crosslinked patterned collagen films in PBS (pH 7.4) and collagenase type II solution (0.1 mg mL−1 in PBS, pH 7.4) at 37 °C after 2 h. | ||
In a parallel test in the absence of the enzyme, PBS (pH 7.4) was used as the degradation medium. The films, with the exception of UXL, retained most of their weight in PBS during the 2 h incubation. The DHT140 and DHT150 films were the best among all of the samples tested and the difference in their weight losses was not statistically significant, and it was significantly lower than observed under other treatment conditions (p ≤ 0.05). Thus, the experiments in the degradation medium demonstrate that the in situ degradation of the films during the 2 h incubation period was negligible.
Enzymatic degradation of the DHT150 Col:ELR film was almost at the same level as its collagen counterpart and the difference was not statistically significant (p ≤ 0.05), which shows that the presence of ELR in the structure did not affect the susceptibility of the collagen to enzymatic attack. The UXL Col:ELR film, on the other hand, was as totally degraded after the 2 h enzyme treatment as the Col film.
The effect of the proteolytic enzymes on the patterns was also studied using scanning electron microscopy (SEM). Fig. 6A shows the untreated state of DHT150. It was observed that the UXL and DHT105 films were totally degraded (not shown). Similarly, the patterns of the DHT140 film were highly deteriorated and the collagen was fragmented (Fig. 6B). The patterns on the treated DHT150 film were conserved and no fragmentation was observed, however the pattern features were not as sharp (Fig. 6C).
 | ||
| Fig. 6 SEM micrographs of the collagen films. (A) The untreated DHT150 film, and the (B) DHT140 and (C) DHT150 films after 2 h treatment with collagenase. Scale bars: 200 μm. The main and inset magnifications are ×500 and ×2000, respectively. | ||
The degradation profiles of the scaffolds under the culture conditions and in vivo are important because the rates of degradation and tissue formation should be matched. If degradation occurs too fast, then structural support may not be provided due to an insufficient number of cells and protein matrix ingrowth from the neighboring healthy tissue. On the other hand, if degradation proceeds too slowly, the scaffold is treated as a foreign material, inflammation is initiated and this can result in scaffold rejection.49
In order to mimic the culture conditions, the samples were incubated in PBS (pH 7.4, 37 °C) for 4 weeks (Fig. 7). It was observed that the UXL films degraded completely in one week. The DHT150 sample was the most stable; in the first week it lost only 15% of its weight with an overall loss of 18% after 4 weeks. DHT140 was the second most stable; it lost 28% of its weight in the first week with an overall loss of 38% at the end of 4 weeks. DHT105 was the least stable of the crosslinked films losing 60% and 85% after 1 week and 4 weeks, respectively. The films containing ELR behaved in a similar fashion with DHT150 Col:ELR losing 20% of its weight in the first week and 33% overall after 4 weeks.
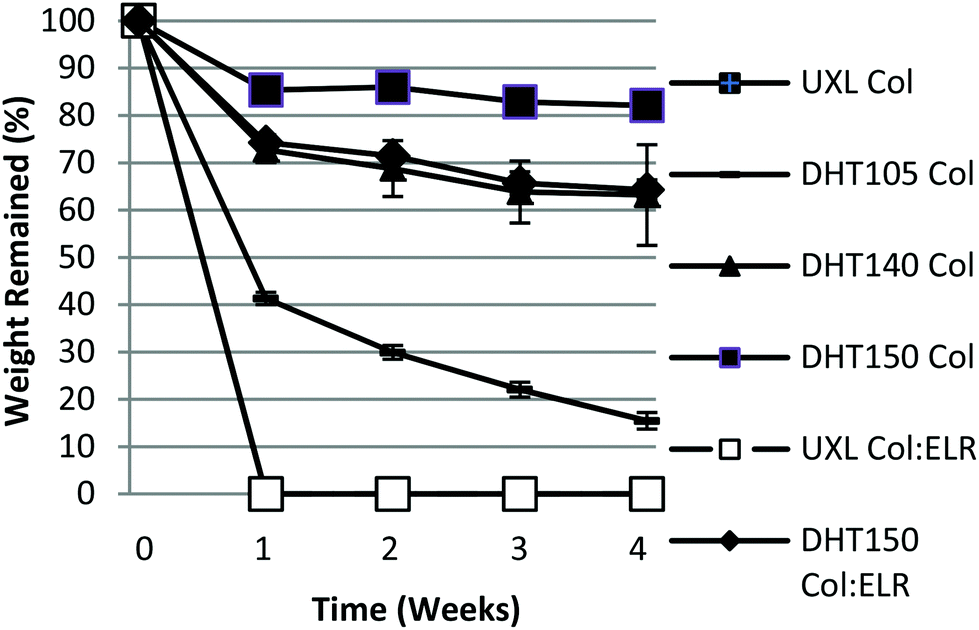 | ||
| Fig. 7 In situ degradation profiles of the patterned films incubated for 28 days in PBS (pH 7.4, 37 °C). | ||
The UXL form of Col:ELR film totally degraded in the first week just like the pure Col version. The reason for the DHT150 Col:ELR film having a higher degradation rate than the pure Col film may be because the collagen molecules could not effectively be crosslinked due to the ELR molecules, which cannot be crosslinked with DHT treatment, being homogeneously blended. Moreover, the shorter ELR chains may have gradually leached out into the water leaving behind the hollow collagen. Therefore, the UXL form of the Col:ELR film was as completely degraded in the first week as the Col film.
Transparency of the films
The cornea is composed of an avascular tissue and it is transparent. It transmits more than 90% of the light of the visible spectrum,50 therefore, any corneal substitute should be equally transparent. In order to study their transparency, the light transmittance of the films was measured in the range 250–700 nm. All of the films showed very high light transmittance (77–85%) (Fig. 8). The UXL Col film had the highest light transmittance (85%) and this value was statistically significantly different from the others (p ≤ 0.05). The DHT105, DHT140 and DHT150 films exhibited transmittance values of about 80%. The small difference in the light transmittance of the films could be explained by the difference in the hydration of the films, which was caused by the different extents of crosslinking. Additionally, upon increasing the temperature to 140 °C and 150 °C a slight yellowish tinge (probably a result of oxidation) was observed in the films, which may have decreased the transmittance. Incorporation of ELRs into the structure enhanced its light transmittance. The light transmittance of the Col:ELR film was 83% at 700 nm, which was very close to the UXL Col film transparency at 85%. | ||
| Fig. 8 The transparency of the micropatterned films in the visible and ultra violet range. | ||
The transparency of the films was also assessed using stereomicroscopy and it was observed that as the number of the films increased, the transparency decreased. A yellowish tint was observed more distinctly in the stacks (Fig. 9). DHT150 Col:ELR films had better transparency than DHT150 Col films as shown by the sharper letters.
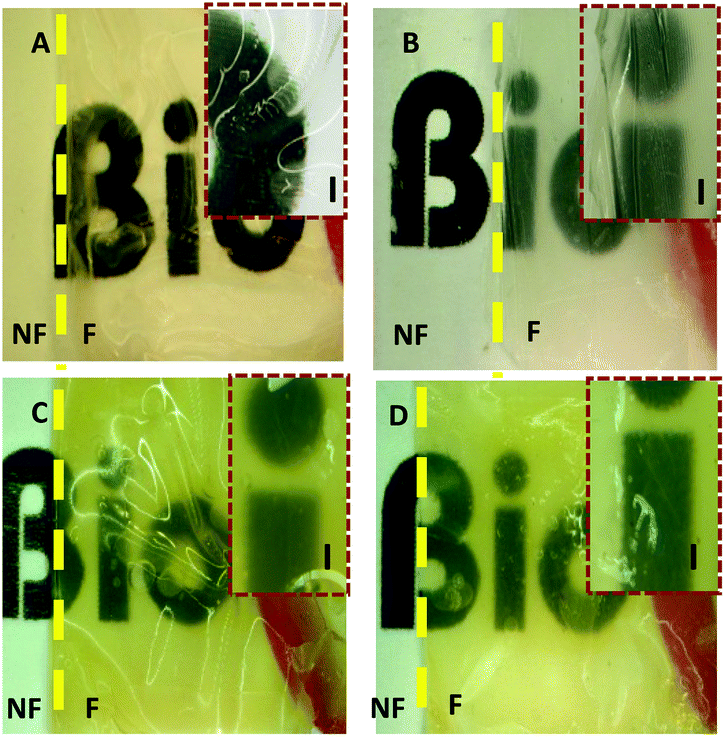 | ||
| Fig. 9 Stereomicrographs showing the transparency of the films. (A) The single layer UXL Col film, (B) the 4-layer UXL Col scaffold, (C) the 4-layer DHT150 Col scaffold, and (D) the 4-layer Col:ELR scaffold. NF: no film, F: film, I: inset. Magnification: ×3. Inset magnification: ×8. | ||
In vitro studies
 | ||
| Fig. 10 The proliferation of keratocytes on TCPS, and the Col and Col:ELR films over 3 weeks. Initial seeding density: 4 × 104 cells per sample. | ||
Cell proliferation on the multilayer scaffolds is shown in Fig. 11. On the first day, the cell numbers of the two scaffolds (Col and Col:ELR) were not significantly different from each other (p ≤ 0.05) and were lower than the seeded amount. The number of cells increased significantly in the following 3 weeks. The results show that the cells proliferated better on the Col film than on the Col:ELR film at each time point. At the end of the 21 day incubation period, the number of cells on the Col film was twice as high as on the Col:ELR film. On the multilayer scaffold, the number of cells increased 7 fold in 3 weeks, but it increased 17 fold on the single Col film. The decrease in the rate for the multilayer structure was more dramatic for the Col:ELR film, where the increase after 21 days was 2.5 fold for the multilayer, while it was 17 fold for the single layer Col:ELR film. Interestingly, however, the plateau in the cell number observed during the second week for the single layer films was also observed for the multilayer films. Thus, the cellular behavior does not apparently change when the number of layers is increased. The main reason for the lower rate of proliferation may be the poorer oxygen and nutrient levels between the layers of the scaffold, which lead to poorer metabolic activity.
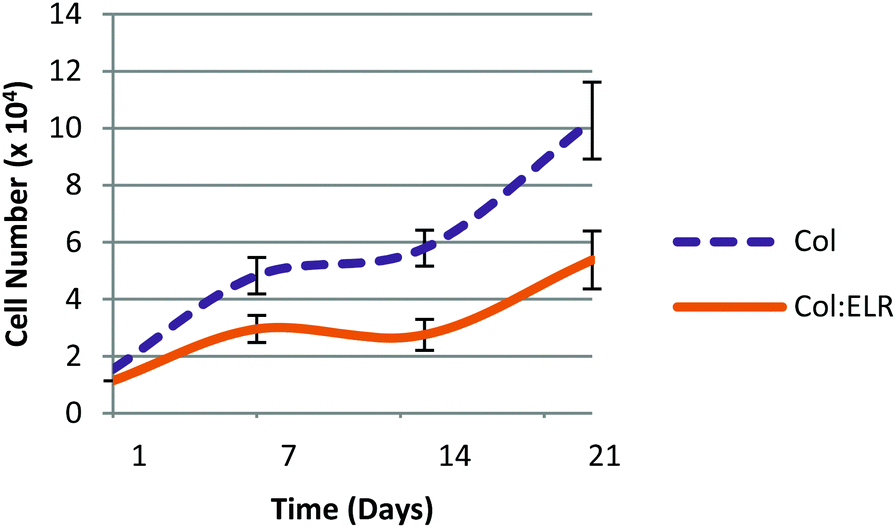 | ||
| Fig. 11 Cell proliferation on the multilayer Col and Col:ELR scaffolds after 21 days of incubation. | ||
Microscopy
The human keratocyte seeded patterned collagen films were stained with DAPI and the cell nuclei were studied using fluorescence microscopy (Fig. 12). The keratocytes responded to the 8 μm wide channels on the films and aligned along the direction of the grooves on day 1. This alignment was maintained on days 7 and 21. Cell alignment on the equally treated Col:ELR films was similar to that of the Col films (data not shown). Within the alignment it can be observed that the nuclei are also aligned in the direction of the channel axis. The nuclei are slightly more elliptical than usual, as shown by their aspect ratios, which are significantly higher at each time point than those on the unpatterned films (Table 1) (p ≤ 0.05). The aspect ratios show that the nuclei of the cells on the unpatterned surfaces also gradually became significantly more elliptical over the 3 weeks (p ≤ 0.05). However, this elliptical shape was seen in isolated cells (which were not grouped). In the second week and the following weeks, the cells on the patterned films were detected at various depths. This could be due to ECM secreted by the cells, which might have filled the grooves and allowed stacked layers of cells to form. For the keratocytes on the unpatterned Col films, no special alignment was observed during the three weeks of incubation (Fig. 13C and D). The effect of the pattern on the cell alignment can be observed in the SEM micrographs, even on day 1. The red arrows in the figures point to the random orientation seen in the smooth regions found between the patterned fields (Fig. 12 and 13).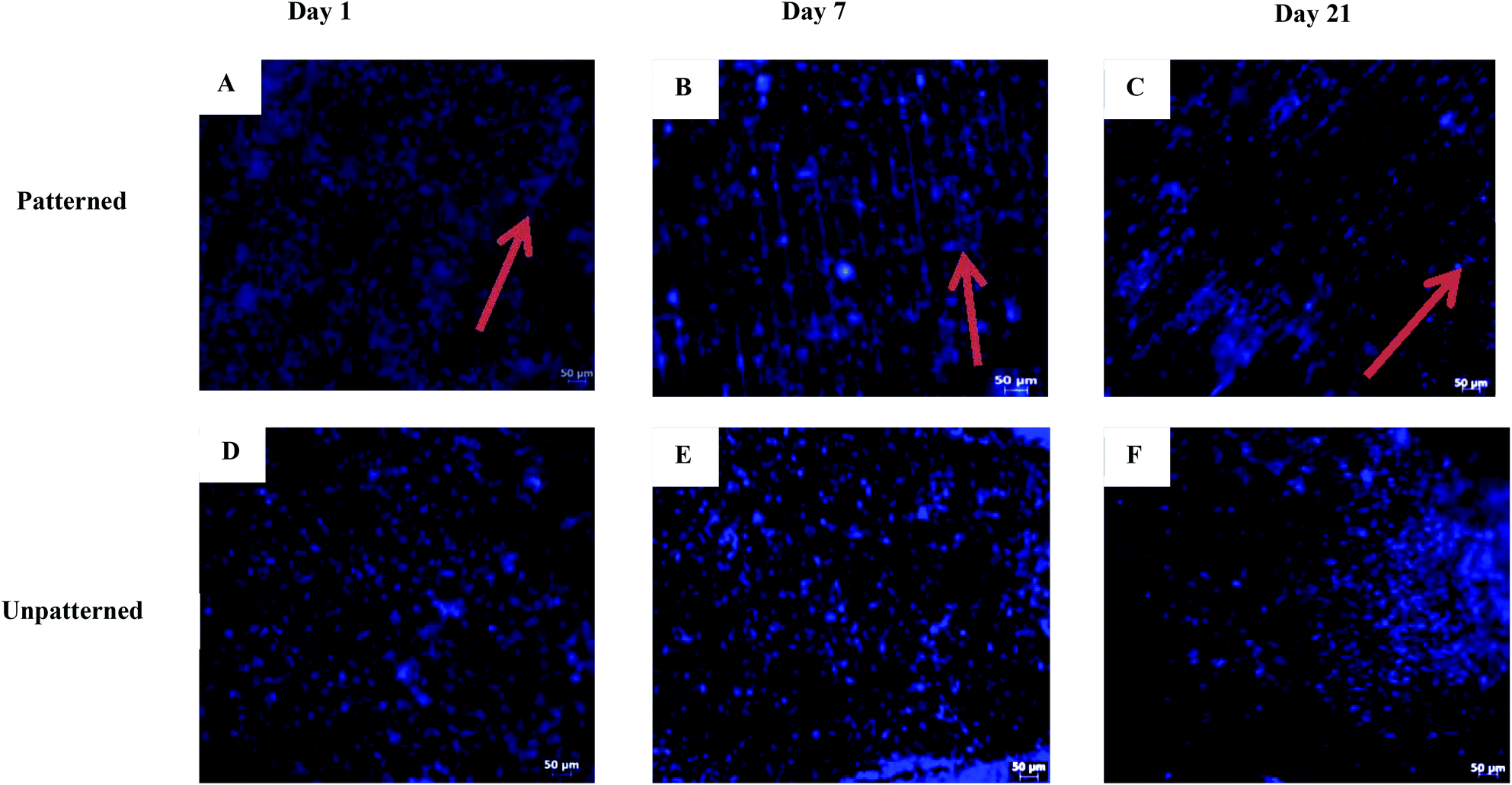 | ||
| Fig. 12 Fluorescence micrographs of DAPI stained human keratocytes on the collagen films. (A–C) The patterned and (D–F) unpatterned films. The time of incubation (days): (A, D) 1, (B, E) 7, and (C, F) 21. Scale bars: 50 μm. The arrows show the pattern directions. | ||
 | ||
| Fig. 13 SEM micrographs of human corneal keratocytes on (A, B) the patterned and (C, D) unpatterned Col films. Time of incubation (days): (A, C) 1, and (B, D) 7. Magnification: ×50. The arrow shows an unpatterned region on the micro-grooved film. | ||
| Time (days) | Aspect ratio of the cell nuclei | |
|---|---|---|
| Unpatterned film | Patterned film | |
| 1 | 1.31 ± 0.18 | 1.66 ± 0.28 |
| 7 | 1.64 ± 0.21 | 2.41 ± 0.42 |
| 21 | 2.17 ± 0.43 | 3.01 ± 0.60 |
Transparency of the cultured films
Transparency is very important for the artificial cornea constructs in order to fulfil their function properly. ECM contributes to the transparency due to its proteoglycan content and the organization of the collagen fibrils and keratocytes of the stroma keep the cornea transparent via the continuous synthesis and remodeling of the proteoglycans and crystalline proteins.4,52 Thus, the transparency of the any cornea equivalent that is prepared in the lab is expected to be improved over time by the corneal keratocytes. Patterned Col and Col:ELR films were prepared and seeded with keratocytes, before testing in vitro for one month (Fig. 14). After the first day, the wet Col film exhibited 80% transmittance at 700 nm, a wavelength in the visible range. This value was better for the Col:ELR film (85%). In the following four weeks the transmittance of the Col film increased to 92%. The transparency of the Col:ELR film was slightly higher than that of the Col film, reaching 93% after 4 weeks. Both these values are comparable to that of a native cornea (98% at 700 nm).53,54 This indicated that the transmittance of the collagen based patterned films with keratocytes, was similar to that of a native cornea and could be gradually improved in the incubation medium both by degradation and the activity of the cells. Incorporation of ELR into the structure also slightly enhanced the transparency of the collagen constructs. The increase in the transparency of both films was due to an increase in the organization of the films and the alignment of the cells within the grooves. In order to see the effect of the cells on the transparency of the films, unseeded and 3T3 seeded Col films were prepared as positive and negative controls, respectively (Fig. 15). The transparency of the unseeded film was significantly lower (p ≤ 0.05) than the seeded ones at each time point. After day 1, the transmittance was decreased compared to the day 0 data (Fig. 8), most probably due to serum accumulation. 3T3 cells are fibroblastic cells like stromal keratocytes and they respond to the patterned surfaces in a similar fashion.55 Thus, this proves that the increase in the transparency is not only due to the alignment of the cells, but also due to the keratocytes, which contribute to the transparency via proteoglycan and crystalline protein synthesis. The transmittance of the film seeded with the 3T3 cells was lower than those of the films seeded with keratocytes at each time point, which strongly indicates the importance of the keratocytes for good transparency. The crosslinking method, DHT, probably also contributed to the transparency of the films, since studies conducted with other crosslinking agents like glutaraldehyde, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), and cyanamide resulted in opaque films with much higher degrees of swelling than those observed using DHT crosslinking.56 Another important parameter for the transparency of the stroma is the regular and orthogonal alignment of the collagen fibers, which prevents light scattering.4 The unpatterned collagen films seeded with keratocytes and the unseeded control film were used to demonstrate the effect of the pattern on the transparency (Fig. 15). Transmittance of the films on day 0 (unseeded) was 72% (data not shown) and this did not change significantly (p ≤ 0.05) during the 30 day culture period. The seeded unpatterned films transmit light less than their unseeded counterparts at each time point, but the difference is not significant (p ≤ 0.05). The transmittance of the unpatterned seeded films was 68% at 700 nm on day 30, which is significantly lower than the transmittance of the Col and Col:ELR films (which are 92% and 93%, respectively). Thus, these results indicate that it is not only the keratocytes and their secretions that contribute to the transparency of the film, but also the topography of the surface on which they grow, which can have an extensive effect. Additionally, the transparency of the unpatterned films was higher than the transparency of the Col film seeded with 3T3 cells, which again shows the importance of the keratocytes and the ECM and crystalline proteins secreted by them. | ||
| Fig. 14 Transparency of the Col and Col:ELR films: (a) the native cornea, (b) Col:ELR, (c) Col, (d) Col unseeded, and (e) Col seeded with 3T3. The native cornea data are adapted from Meek et al. (2003).54 | ||
 | ||
| Fig. 15 Transparency of the unpatterned films: (a) native cornea, (b) Col unseeded, and (c) Col seeded. The native cornea data are adapted from Meek et al. (2003).54 | ||
Conclusions
In this study a 3D scaffold was constructed from Col and Col:ELR patterned films in an attempt to mimic the natural structure and organization of the corneal stroma.The Col and Col:ELR films and their constructs were shown to support cell attachment and proliferation. Alignment of the cells on the patterned films was achieved even after 1 day of incubation and their transparency increased significantly over a 30 day period. Both the patterns and the keratocytes have been shown to contribute greatly to the transparency of the films. The constructs appear to have the potential for use as stroma equivalents and in future studies their performance will be enhanced before in vivo trials are carried out. The amount of ELR in the structure needs to be increased or optimized in order to show its effect more distinctly. The layers will be stacked on top of each other using bioadhesives. Bioreactors will also be used to achieve better culture conditions for tissue formation and then the samples will be tested in vivo on rabbits using rabbit corneal keratocytes, before attempting clinical trials.
Acknowledgements
We (C. K. and V. H.) gratefully acknowledge the financial support of METU (through BAP 07.02.2012-101) and the METU International Cooperations Office for a 2011 Erasmus Summer Internship Program Scholarship. J. C. R.-C. acknowledges financial support from the Spanish Minister of Economy and Competitiveness (MAT2009-14195-C03-03, MAT-2010-15982, MAT2010-15310, IT2009-0089, ACI2009-0890 and PRI-PIBAR-2011-1403), the Junta de Castilla y León-JCyL (projects VA034A09 and VA0049A11-2), CIBER-BBN (project CB06-01-1038), and the JCyL and the Instituto de Salud Carlos III under the “Network Center of Regenerative Medicine and Cellular Therapy of Castilla and León”.Notes and references
- R. W. Buerman and L. Pedroza, Microsc. Res. Tech., 1996, 33, 320 CrossRef.
- K. M. Meek, Biophys. Rev., 2009, 1, 83–93, DOI:10.1007/s12551-009-0011-x.
- C. R. Ethier, M. Johnson and J. Ruberti, Annu. Rev. Biomed. Eng., 2004, 6, 249–273, DOI:10.1146/annurev.bioeng.6.040803.140055.
- D. M. Maurice, J. Physiol., 1957, 136, 263–286 CAS.
- K. M. Meek and N. J. Fullwood, Micron, 2001, 32, 261–272, DOI:10.1016/S0968-4328(00)00041-X.
- K. M. Meek and C. Boote, Exp. Eye Res., 2004, 78, 503–512, DOI:10.1016/j.exer.2003.07.003.
- M. S. Oliva, T. Schottman and M. Gulati, Indian J. Ophthalmol., 2012, 60, 423–427, DOI:10.4103/0301-4738.100540.
- T. V. Chirila, C. R. Hicks, P. D. Dalton, S. Vijayasekaran, X. Lou, Y. Hong, A. B. Clayton, B. W. Ziegelaar, J. H. Fitton, S. Platten, G. J. Crawford and I. J. Constable, Prog. Polym. Sci., 1998, 23, 447–473, DOI:10.1016/S0079-6700(97)00036-1.
- T. V. Chirila, Biomaterials, 2001, 22, 3311–3317, DOI:10.1016/S0142-9612(01)00168-5.
- B. Strampelli, Ber. Zusammenkunft – Dtsch. Ophthalmol. Ges., 1972, 71, 322–335 CAS.
- D. Myung, P. E. Duhamel, J. R. Cochran, J. Noolandi, C. N. Ta and C. W. Frank, Biotechnol. Prog., 2008, 24, 735–741, DOI:10.1021/bp070476n.
- G. Falcinelli, B. Falsini, M. Taloni, P. Colliardo and G. Falcinelli, Arch. Ophthalmol., 2005, 123, 1319–1329, DOI:10.1001/archopht.123.10.1319.
- L. J. Bray, K. A. George, S. L. Ainscough, D. W. Hutmacher, T. V. Chirila and D. G. Harkin, Biomaterials, 2011, 32, 5086–5091, DOI:10.1016/j.biomaterials.2011.03.068.
- T. S. Jiang, L. Cai, W. Y. Ji, Y. N. Hui, Y. S. Wang, D. Hu and J. Zhu, Mol. Vision, 2010, 16, 1304–1316 Search PubMed.
- J. Torbet, M. Malbouyres, N. Builles, V. Justin, M. Roulet, O. Damour, Å. Oldberg, F. Ruggiero and D. J. S. Hulmes, Biomaterials, 2007, 28, 4268–4276, DOI:10.1016/j.biomaterials.2007.05.024.
- N. E. Vrana, N. Builles, H. Kocak, P. Gulay, V. Justin, M. Malbouyres, F. Ruggiero, O. Damour and V. Hasirci, J. Biomater. Sci., Polym. Ed., 2007, 18, 1527–1545 CAS.
- R. Watanabe, R. Hayashi, Y. Kimura, Y. Tanaka, T. Kageyama, S. Hara, Y. Tabata and K. Nishida, Tissue Eng., Part A, 2011, 17, 2213–2219, DOI:10.1089/ten.TEA.2010.0568.
- J. Y. Lai, K. H. Chen and G. H. Hsiue, Transplantation, 2007, 84, 1222–1232, DOI:10.1097/01.tp.0000287336.09848.39.
- P. Zorlutuna, A. Tezcaner, I. Kiyat, A. Aydinli and V. Hasirci, J. Biomed. Mater. Res., Part A, 2006, 79A, 104–113, DOI:10.1002/jbm.a.30772.
- N. Builles, H. Janin-Manificat, M. Malbouyres, V. Justin, M. Rovère, G. Pellegrini, J. Torbet, D. J. S. Hulmes, C. Burillon, O. Damour and F. Ruggiero, Biomaterials, 2010, 31, 8313–8322, DOI:10.1016/j.biomaterials.2010.07.066.
- C. R. McLaughlin, M. C. Acosta, C. Luna, W. Liu, C. Belmonte, M. Griffith and J. Gallar, Biomaterials, 2010, 31, 2770–2778, DOI:10.1016/j.biomaterials.2009.12.031.
- N. E. Vrana, N. Builles, V. Justin, J. Bednarz, G. Pellegrini, B. Ferrari, O. Damour, D. J. S. Hulmes and V. Hasirci, Invest. Ophthalmol. Vis. Sci., 2008, 49, 5325–5331, DOI:10.1167/iovs.07-1599.
- A. Curtis and C. Wilkinson, Biomaterials, 1997, 18, 1573–1583, DOI:10.1016/S0142-9612(97)00144-0.
- N. Zhang, H. Yan and X. Wen, Brain Res. Rev., 2005, 49, 48–64, DOI:10.1016/j.brainresrev.2004.11.002.
- P. Zorlutuna, N. Builles, O. Damour, A. Elsheikh and V. Hasirci, Biomaterials, 2007, 28, 3489–3496, DOI:10.1016/j.biomaterials.2007.04.013.
- E. S. Gil, B. B. Mandal, S. Park, J. K. Marchant, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2010, 31, 8953–8963, DOI:10.1016/j.biomaterials.2010.08.017.
- N. E. Vrana, N. Builles, M. Hindie, O. Damour, A. Aydinli and V. Hasirci, J. Biomed. Mater. Res. A, 2008, 84, 454–463, DOI:10.1002/jbm.a.31442.
- J. C. Rodríguez-Cabello, L. Martín, M. Alonso, F. J. Arias and A. M. Testera, Polymer, 2009, 50, 5159–5169, DOI:10.1016/j.polymer.2009.08.032.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798, DOI:10.1016/j.progpolymsci.2007.05.017.
- J. M. Pachence, M. P. Bohrer and J. Kohn, in Principles of Tissue Engineering, ed. R. Lanza, R. Langer and J. Vacanti, Academic Press, Burlington, 3rd edn, 2007, pp. 323–339 Search PubMed.
- G. L. Bidwell III, I. Fokt, W. Priebe and D. Raucher, Biochem. Pharmacol., 2007, 73, 620–631, DOI:10.1016/j.bcp.2006.10.028.
- N. Ozturk, A. Girotti, G. T. Kose, J. C. Rodríguez-Cabello and V. Hasirci, Biomaterials, 2009, 30, 5417–5426, DOI:10.1016/j.biomaterials.2009.06.044.
- H. Betre, A. Chilkoti and L. A. Setton, Eng. Med. Biol., 2002, 1, 829–830, DOI:10.1109/IEMBS.2002.1137096.
- S. S. Amruthwar and A. V. Janorkar, Dent. Mater., 2013, 29, 211–220, DOI:10.1016/j.dental.2012.10.003.
- H. Martínez-Osorio, M. Juárez-Campo, Y. Diebold, A. Girotti, M. Alonso, F. J. Arias, J. C. Rodríguez-Cabello, C. García-Vázquez and M. Calonge, Curr. Eye Res., 2009, 34, 48–56, DOI:10.1080/02713680802542053.
- A. V. Janorkar, P. Rajagopalan, M. L. Yarmush and Z. Megeed, Biomaterials, 2008, 29, 625–632, DOI:10.1016/j.biomaterials.2007.10.022.
- B. Kinikoglu, J. C. Rodriguez-Cabello, O. Damour and V. Hasirci, J. Mater. Sci. Mater. Med., 2011, 22, 1541–1554, DOI:10.1007/s10856-011-4315-6.
- M. H. Fittkau, P. Zilla, D. Bezuidenhout, M. P. Lutolf, P. Human, J. A. Hubbell and N. Davies, Biomaterials, 2005, 26, 167–174, DOI:10.1016/j.biomaterials.2004.02.012.
- S. P. Massia, S. S. Rao and J. A. Hubbell, J. Biol. Chem., 1993, 268, 8053–8059 CAS.
- J. H. Yoon, J. Kim, H. Lee, S. Y. Kim, H. Jang, S. H. Ryu, B. J. Kim and T. G. Lee, Biochem. Biophys. Res. Commun., 2012, 428, 416–421, DOI:10.1016/j.bbrc.2012.10.070.
- H. Franke, H. Galla and C. T. Beuckmann, Brain Res. Protoc., 2000, 5, 248–256, DOI:10.1016/S1385-299X(00)00020-9.
- P. Zorlutuna, A. Elsheikh and V. Hasirci, Biomacromolecules, 2009, 10, 814–821, DOI:10.1021/bm801307y.
- S. Ber, G. Torun Köse and V. Hasırcı, Biomaterials, 2005, 26, 1977–1986, DOI:10.1016/j.biomaterials.2004.07.007.
- A. Girotti, J. Reguera, F. J. Arias, M. Alonso, A. M. Testera and J. C. Rodriguez-Cabello, Macromolecules, 2004, 37, 3396–3400, DOI:10.1021/ma035603k.
- D. T. Mcpherson, C. Morrow, D. S. Minehan, J. Wu, E. Hunter and D. W. Urry, Biotechnol. Prog., 1992, 8, 347–352, DOI:10.1021/bp00016a012.
- N. E. Vrana, A. Elsheikh, N. Builles, O. Damour and V. Hasirci, Biomaterials, 2007, 28, 4303–4310, DOI:10.1016/j.biomaterials.2007.06.013.
- E. J. Miller and R. Kent Rhodes, in Methods in Enzymology, ed. Anonymous, Academic Press, 1982, pp. 33–64 Search PubMed.
- J. Hao, T. Nagano, M. Nakamura, N. Kumagai, H. Mishima and T. Nishida, Exp. Eye Res., 1999, 68, 565–572, DOI:10.1006/exer.1998.0637.
- S. F. Williams, D. P. Martin, D. M. Horowitz and O. P. Peoples, Int. J. Biol. Macromol., 1999, 25, 111–121, DOI:10.1016/S0141-8130(99)00022-7.
- J. V. Jester, Semin. Cell Dev. Biol., 2008, 19, 82–93, DOI:10.1016/j.semcdb.2007.09.015.
- B. D. Lawrence, J. K. Marchant, M. A. Pindrus, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2009, 30, 1299–1308, DOI:10.1016/j.biomaterials.2008.11.018.
- J. W. Ruberti, J. D. Zieske and V. Trinkaus-Randall, in Principles of Tissue Engineering, ed. R. Lanza, R. Langer and J. Vacanti, Elsevier/Academic Press, Amsterdam, Boston, 2007, p. 1307 Search PubMed.
- A. Shah, J. Brugnano, S. Sun, A. Vase and E. Orwin, Pediatr. Res., 2008, 63, 535–544, DOI:10.1203/PDR.0b013e31816bdf54.
- K. M. Meek, D. W. Leonard, C. J. Connon, S. Dennis and S. Khan, Eye, 2003, 17, 927–936 CrossRef CAS PubMed.
- S. Zhang, L. Yan, M. Altman, M. Lässle, H. Nugent, F. Frankel, D. A. Lauffenburger, G. M. Whitesides and A. Rich, Biomaterials, 1999, 20, 1213–1220, DOI:10.1016/S0142-9612(99)00014-9.
- R. A. Crabb and A. Hubel, Tissue Eng., Part A, 2008, 14, 173–182, DOI:10.1089/ten.a.2007.0139.
| This journal is © The Royal Society of Chemistry 2014 |