Alanine-rich amphiphilic peptide containing the RGD cell adhesion motif: a coating material for human fibroblast attachment and culture†
V.
Castelletto
*a,
R. M.
Gouveia
a,
C. J.
Connon
a,
I. W.
Hamley
a,
J.
Seitsonen
b,
A.
Nykänen
b and
J.
Ruokolainen
b
aSchool of Chemistry, Food Science and Pharmacy, University of Reading, Whiteknights, Reading RG6 6AD, UK. E-mail: v.castelletto@reading.ac.uk
bDepartment of Applied Physics, Aalto University School of Science, P.O. Box 15100, FI-00076 Aalto, Finland
First published on 20th November 2013
Abstract
We studied the self-assembly of peptide A6RGD (A: alanine, R: arginine, G: glycine, D: aspartic acid) in water, and the use of A6RGD substrates as coatings to promote the attachment of human cornea stromal fibroblasts (hCSFs). The self-assembled motif of A6RGD was shown to depend on the peptide concentration in water, where both vesicle and fibril formation were observed. Oligomers were detected for 0.7 wt% A6RGD, which evolved into short peptide fibres at 1.0 wt% A6RGD, while a co-existence of vesicles and long peptide fibres was revealed for 2–15 wt% A6RGD. A6RGD vesicle walls were shown to have a multilayer structure built out of highly interdigitated A6 units, while A6RGD fibres were based on β-sheet assemblies. Changes in the self-assembly motif with concentration were reflected in the cell culture assay results. Films dried from 0.1–1.0 wt% A6RGD solutions allowed hCSFs to attach and significantly enhanced cell proliferation relative to the control. In contrast, films dried from 2.5 wt% A6RGD solutions were toxic to hCSFs.
Introduction
Surfactant-like peptides (SLPs)1–3 comprise a headgroup, consisting of a short sequence of charged peptide residues, attached to a tailgroup of neutral amino acids. For bio-functional purposes, the headgroup is usually designed from hydrophilic biologically-active epitopes, while a hydrophobic tail group drives peptide self-assembly in aqueous medium. The bio-functionality and bioactivity afforded by sequences of amino acids, together with their amphiphilicity, lead to the remarkable potential of SLPs in the development of novel biomaterials.4Zhang and coworkers pioneered the study of amphiphilic peptides including those with poly-alanine sequences such as A6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5–8 (A: alanine). The self-assembly of peptides such as A6K (K: lysine) with a cationic lysine headgroup has been studied by several groups.9–16 In a previous work we described the self-assembly of the amphiphilic peptide A6R (R: arginine) into ultrathin free-floating nanosheets in dilute aqueous solution.17 The sheets comprise opposed dimers of A6R molecules that are closely-packed but not hydrogen-bonded into β-sheets. We showed that A6R self-assembly is driven by the amphiphilic sequence design of the peptide with the unique conformational and electrostatic properties of the cationic R headgroup. At high concentration, A6R forms helically wrapped ribbons coexisting with nanotubes, and β-sheet formation occurs concomitantly.17
5–8 (A: alanine). The self-assembly of peptides such as A6K (K: lysine) with a cationic lysine headgroup has been studied by several groups.9–16 In a previous work we described the self-assembly of the amphiphilic peptide A6R (R: arginine) into ultrathin free-floating nanosheets in dilute aqueous solution.17 The sheets comprise opposed dimers of A6R molecules that are closely-packed but not hydrogen-bonded into β-sheets. We showed that A6R self-assembly is driven by the amphiphilic sequence design of the peptide with the unique conformational and electrostatic properties of the cationic R headgroup. At high concentration, A6R forms helically wrapped ribbons coexisting with nanotubes, and β-sheet formation occurs concomitantly.17
Here, we functionalise the A6 sequence by attaching the RGD (G: glycine, D: aspartic acid) tripeptide. More than three decades ago the peptide epitope RGD was first shown to promote cell adhesion18 and it is now widely used as a model cell adhesion domain. Direct interactions occur between the RGD motif and integrins, namely those composed of the α5, α8, or αV subunits. Integrins are a family of α,β-heterodimeric transmembrane receptors that interlace transmembrane ties between the extracellular matrix or cell surface molecules and the cytoskeleton,19 thus triggering intracellular signalling pathways involved in the regulation of mitosis and cell proliferation.20,21 Several SLPs have been functionalized with a RGD sequence over the past three decades.22–30 The great potential in the development of novel biomaterials, presented by a broad range of RGD-SLPs, has been widely explored in academic research.3,19–21,31–35
In this work we studied the self-assembly of A6RGD in water and its use as films to create coated surfaces for cell attachment and growth. We first used a combination of spectroscopic, microscopic, and X-ray scattering techniques to investigate the self-assembly of A6RGD in water. We then evaluated the biocompatibility of A6RGD, as well as its ability to promote adhesion of human cornea stromal fibroblasts (hCSFs) when used as a dry film coating.
Experimental
Materials
Peptides A6RGD, C16-ETTES (E: glutamic acid, T: threonine, S: serine) and cRGD (cyclo [RGD(f)-NMe-V], V: valine, f: D-phenylalanine, NMe: methylated amide group) were custom-synthesized by CS Bio (Menlo Park, USA) as TFA salts. The purity of the peptides was determined by analytical HPLC in a TFA-water–acetonitrile gradient, while the molecular weight (MW) was obtained by electrospray-mass spectrometry. A6RGD purity was 99.15%, with analyzed MW = 771.9 Da (expected MW = 772.83 Da). In this work, weighed amounts of A6RGD were dissolved in ultrapure water from a Barnstead Nanopure system to the desired concentration. C16-ETTES and cRGD were used for cell culture assays. Purity was 96.51% and 97.58% for C16-ETTES and cRGD, respectively, with corresponding MW = 803.4 and 588.15 Da (expected MW = 803.94 and 588.66 Da, respectively).:1:2) for a day before imaging.
:10 resazurin reagent in culture media for 4 h, after which samples were taken and culture media replaced. Samples were analysed by spectrofluorometry (λex = 540 nm, λem = 590 nm), and fluorescence signals plotted to give a fluorescence vs. cell number calibration curve. For each assay, data statistics from three independent replicates was performed using two-way analysis of variance (ANOVA) with Bonferroni's post-hoc test.
Results and discussion
A6RGD consists of a hydrophobic A6 block and the cell adhesion peptide motif RGD, and was designed to simultaneously ensure solubility in water and specific binding to cells. The good solubility of A6RGD in water was quantified by an octanol–water partition coefficient log P = −5.757 calculated using the software Molinspiration.38The isoelectric point of A6RGD in water was calculated as pH 6.78 using Innovagen software.39 ESI Fig. 1† shows the measured pH dependence with concentration for the samples studied in this work, together with the chemical structure of A6RGD. Our study focused on solutions with pH below the isoelectric point, and therefore the SLP was expected to be positively charged in solution due to the arginine residue.
Fig. 1 shows ThT and Pyr fluorescence emission assays performed to determine the critical concentration for the formation of A6RGD aggregates. The emission fluorescence spectra (λex = 440 nm) for A6RGD solutions diluted in ThT at 5.0 × 10−3 wt% showed a broad peak centred at ∼486 nm (Fig. 1a, inset). The dependence of I486 on A6RGD concentration is shown in Fig. 1a. ThT fluorescence showed a dramatic increase of I486 emission band intensity upon amyloid formation.40,41 In agreement with that information, the critical aggregation concentration (c.a.c.) was measured as 2.1 wt% A6RGD from the inflection point of the data in Fig. 1a.
 | ||
| Fig. 1 Determination of c.a.c., from (a) ThT and (b) Pyr fluorescence assays. The insets show representative fluorescence emission spectrum for (a) ThT and (b) Pyr. | ||
A Pyr fluorescence assay was performed to provide additional information about the c.a.c. The emission fluorescence spectra (λex = 339 nm) for samples containing A6RGD diluted in Pyr at 1.3 × 10−5 wt% displayed a band centred at ∼374 nm (Fig. 1b, inset). The dependence of the intensity I374 with the peptide concentration is shown in Fig. 1b. The inflection point for the data in Fig. 1b denoted a change of environment for the Pyr molecule, and was identified as the c.a.c. Indeed, Fig. 1b shows that, for concentrations higher than 1.4 wt% A6RGD, the Pyr molecule was surrounded by a hydrophobic environment corresponding to the core of the peptide aggregate. The averaged value for A6RGD c.a.c. = 1.8 ± 0.5 wt% was calculated from the c.a.c. values reported in Fig. 1 using ThT and Pyr assays.
We further investigated the amyloid-like properties of A6RGD aggregates by measuring the binding to another amyloid dye, Congo red. Congo red alone had an absorbance maximum at 498 nm (Fig. 2). Upon increasing the A6RGD concentration in the Congo red solution, the absorbance maximum progressively red-shifted, reaching a stable value of ∼549 nm for 0.07–2.0 wt% A6RGD (Fig. 2). This result indicated that, for concentrations below the c.a.c. and as low as 0.07 wt%, A6RGD formed peptide oligomers.
 | ||
| Fig. 2 Representative results for the absorption spectrum of Congo red in the presence of 0, 0.01 and 2 wt% A6RGD. Inset: dependence of the Congo red absorption peak position as a function of the A6RGD concentration. | ||
The products of A6RGD aggregation were analyzed using native polyacrylamide gel electrophoresis (PAGE). Fig. 3 shows the results obtained for A6RGD solutions at 0.1, 1.0 and 2.5 wt% run by PAGE in non-reducing, non-denaturing conditions. During the run, a large smear was observed in the front of migration in lanes corresponding to 1.0 and 2.5 wt%, but not for 0.1 wt% A6RGD. After de-staining, a diffuse group of bands corresponding to high molecular weight (>80 kDa) species were resolved for A6RGD at 1.0 and 2.5 wt% (Fig. 3). In particular, 2.5 wt% A6RGD migrated as ∼250 kDa species, whereas bands from 1.0 wt% were resolved within the 80–250 kDa regions. In contrast, no bands were observed for 0.1 wt% A6RGD. Although A6RGD aggregates of smaller sizes (20–60 kDa) were probably responsible for the smears occurring during electrophoresis, no bands were observed at this molecular weight range after de-staining. A6RGD at 0.1 wt% did not form stained aggregates in the resolution range of the polyacrylamide gel (>20 kDa) (Fig. 3).
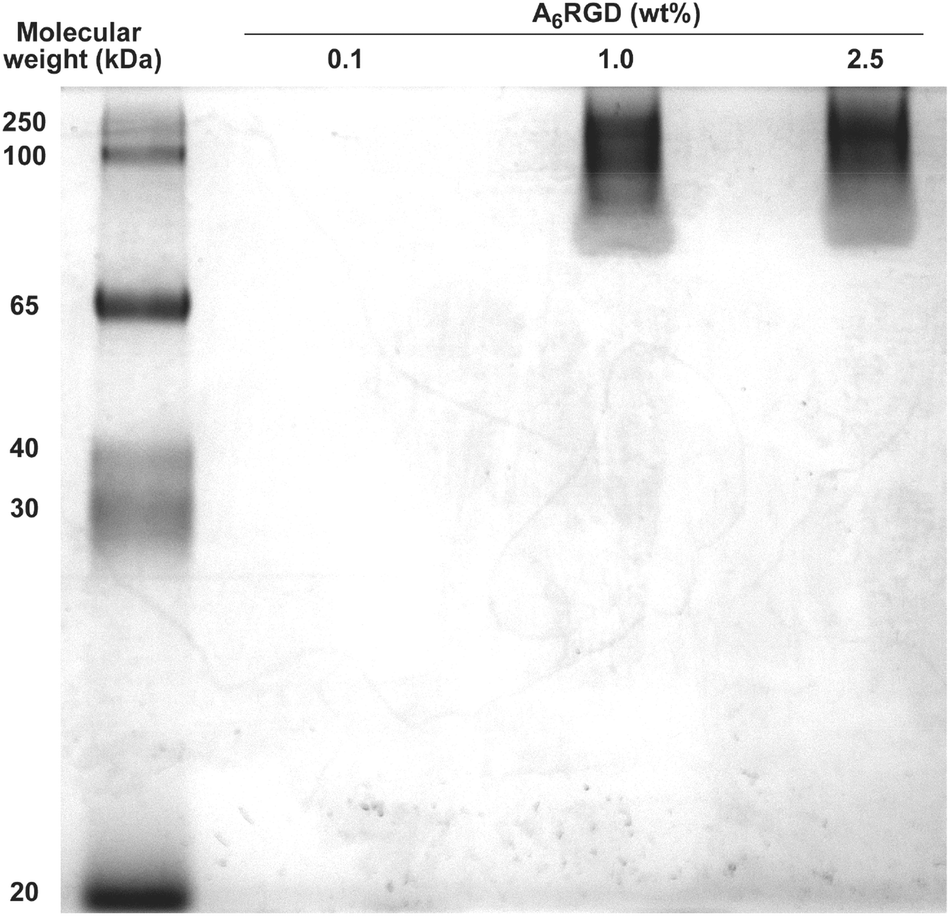 | ||
| Fig. 3 Non-reducing native PAGE of A6RGD. | ||
Taken together, the results shown in Fig. 2 and 3 were consistent, although obtained using very different analytical methods. The self-assembly of A6RGD into large fibrillar aggregates takes place at ∼1 wt% of A6RGD, while lower concentrations in the 0.07–1.0 wt% range are dominated by oligomeric species.
CD was used to access the secondary structure of A6RGD in solution below and above the c.a.c. CD spectra measured for 0.5 and 5.0 wt% A6RGD, as displayed in ESI Fig. 2.† The CD spectra suggest contributions from several secondary structures. The spectrum for 0.5 wt% A6RGD was characterized by a negative band at ∼194 nm and a weak positive band at ∼217 nm (ESI Fig. 2†), characteristic of a predominant polyproline II conformation.42 However, the spectrum for 5 wt% A6RGD showed a much shallower minimum and maximum, which indicates the presence of mainly disordered conformation.43
FTIR experiments were also performed to probe the secondary structure of A6RGD in solution. The amide I region of the FTIR spectra measured for A6RGD at 0.5–15.5 wt% is displayed in ESI Fig. 3.† The spectra contain two main bands at 1672 and 1649 cm−1. The band at 1672 cm−1 is due to the TFA dissolved in the solution,44,45 while the contribution of the band centred at 1649 cm−1 indicate a disordered component46 in the A6RGD structure, in good agreement with the CD data in ESI Fig. 1.†
Fibre X-ray diffraction was performed to investigate the self-assembled morphology at the level of the β-sheet structure. The one-dimensional integration of the 2D XRD spectra was performed in order to quantify peak positions. ESI Fig. 4† shows the profile obtained for a stalk dried from a 10 wt% A6RGD sample, and for a 12 wt% A6RGD solution. The wide peak with spacing d = 4.7 Å, measured for the stalk indicated a distribution of β-sheet spacing.47,48 The XRD pattern for 12 wt% A6RGD (ESI Fig. 4†) presented eight spacings d = 27.7, 13.2, 11.1, 9.3, 7.2, 5.5, 4.8, and 4.5 Å. Possibly, d = 27.7, 13.2, and 9.3 Å corresponded to the first, second and third order of a lamellar structure with cell parameter do = 27.7 Å. The spacings d = 4.8 and 4.5 Å from A6RGD at 12 wt% were assigned to a β-sheet spacing and the spacing of alanine residues in non β-sheet structures, respectively.17,49 The latter is consistent with the substantial disordered component observed by FTIR (ESI Fig. 3†). In addition, and based on the indexation of similar XRD patterns previously reported by us for A6K, A6R, and A12R2 in water,13,14,17,49 the d = 5.5 Å spacing found for 10 wt% A6RGD was assigned to the stacking distance between β-sheets. The relatively small β-sheet stacking distance is due to the efficient packing of alanine residues50 and is consistent with a compact structure as observed for other alanine-rich peptides.50 The XRD from the stalk presented a lower number of reflections than that from A6RGD in solution (ESI Fig. 4†), probably due to a loss of order caused by the drying process.
The self-assembled structures of A6RGD at 0.1, 1.0, 2.5, and 15 wt% were further investigated by cryo-TEM (Fig. 4). Cryo-TEM images for 0.1 wt% A6RGD did not show the formation of aggregates (results not shown). Indeed, 0.1 wt% A6RGD presented oligomeric species in solution, which might be extremely difficult to visualize using cryo-TEM. The images in Fig. 4 suggest that the self-assembled structure was dependent on concentration. Images obtained for 1.0 wt% A6RGD (Fig. 4a) showed the formation of short fibrillar structures. At higher concentrations, a network of fibres ∼11 ± 3 nm thick was observed for 2.5 wt% A6RGD (Fig. 4b). Cryo-TEM images for 15 wt% A6RGD (Fig. 4c,d) indicated the co-existence of structures consisting of fibrils and vesicles with variable sizes. The peptide fibrils were 14 ± 3 nm thick (Fig. 4c). On the other hand, A6RGD vesicles had very thin walls, and were highly polydisperse in size, with an average diameter of 135 ± 62 nm (Fig. 4d). According to these results, A6RGD formed fibres at lower concentrations, while vesicles co-exist with fibrils at higher concentrations.
 | ||
| Fig. 4 Cryo-TEM images measured for (a) 1, (b) 2.5 and (c–d) 15 wt% A6RGD. (c) shows the co-existence of fibrils with vesicles for 15 wt% peptide, while (d) corresponds to a region in the 15 wt% A6RGD grid, where only A6RGD vesicles were present. | ||
The concentration-dependent morphology of the peptide aggregates is probably due to different mechanisms of aggregation. Changes in the concentration, correlated to the number of peptide clusters seeding the aggregation process, determine the mechanism governing fibrillization and the final morphology of the peptide aggregates. SAXS curves measured for 2, 5, and 10 wt% A6RGD (Fig. 5) were used to identify the self-assembly motifs of the A6RGD in water.
 | ||
| Fig. 5 Models for the (a) Gaussian bilayer and (b) long cylinder fitted to the SAXS curves. (c) SAXS data for 2, 5 and 10 wt% A6RGD, fitted according to a co-existence of Gaussian bilayers and long cylinders. (d) Representative bimodal distribution for 10 wt% A6RGD obtained from the fits shown in (c), corresponding to the radius of the cylinder in (b) and the thickness of the bilayers in (a). I1 and I2 in (d) correspond to the bilayer and cylinder contributions to the form factor. The inset in (d) displays the dependence of I2/I1 with concentration. | ||
The SAXS data were fitted to a co-existence of long cylinders and a bilayer structure, using the software SASfit.51 The fitted SAXS curves are displayed in Fig. 5c. The parameters obtained from the SAXS model, already described in the Experimental section, are listed in ESI Table 1.† The length of the cylinder was fixed to 106 Å for all the fits and it behaved as a scale factor because R ≫ L in the form factor.
Fig. 5d displays the bimodal Gaussian distribution function weighted by R3 (R = intra-particular distance) used to model the SAXS data for 10 wt% A6RGD in Fig. 5c. I1 and I2 in Fig. 5d corresponds to the intensities for the distributions in bilayer thickness and cylinder radius, respectively. The inset in Fig. 5d shows the values obtained for I2/I1 as a function of A6RGD concentration. According to the inset in Fig. 5d, the fraction of fibres relative to bilayers decreases with increasing A6RGD concentration.
On the basis of the poor water solubility of the A6 block, we expect that the Gaussian bilayers and long cylinders will be built from A6RGD bilayers, with highly interdigitated hydrophobic A6 ‘tails’ and positively charged RGD groups exposed to water. Similarly, the cylinders will comprise an A6 core and RGD corona. A similar bilayer structure has been proposed by us for free-floating sheets of A6R17 and A12R2 nanotapes49 in aqueous solution. This idea was confirmed by the calculated layer spacing listen in ESI Table 1,†lT = 37 Å ≪ 2 × lE (lE: length of the A6RGD molecule in a β-sheet conformation = 9 × 3.4 Å = 30.6 Å; 3.4 Å = distance between residues comprising the β-sheet52), which corresponds to bilayers of interdigitated A6 blocks.
The multilayer cell parameter do = 27.7 Å measured from the XRD data (ESI Fig. 4†) is within the experimental error of the bilayer spacing lT (ESI Table 1†). We propose that the multilayer order deduced from the XRD data for the A6RGD solution (ESI Fig. 4†) is associated to the ordering at the wall of the SLP vesicles. According to these results, the lamellar order was lost after the drying process and only the β-sheet order prevailed for the 10 wt% A6RGD stalk.
Cryo-TEM images for a 2.5 wt% A6RGD solution did not show the formation of vesicles, probably because the population of bilayers was ∼88 times lower than the population of fibres (Fig. 5d, inset).
To test the use of A6RGD in cell culture, solutions of peptide at 0.01–2.5 wt% were drop-spotted on polystyrene cell culture plates and allowed to dry, forming thin films (Fig. 6a) that were then tested for their properties as biocompatible promoters of cell adhesion and proliferation. Human cornea stromal fibroblasts (hCSFs) seeded onto surfaces produced by 0.01–1.0 wt% A6RGD adhered strongly to the surface and maintained high viability of cultured cells. Cells growing on 1 wt% A6RGD films acquired a fusiform morphology with a substantial number of process extensions (Fig. 6b), which constituted an indication of viable cells with appropriate phenotype.
 | ||
| Fig. 6 Adhesion and morphology of human cornea stromal fibroblasts (hCSFs) cultured on A6RGD coatings. A6RGD in solution at 0.01, 0.1, 1 and (a) 2.5 wt% were drop-spotted and dried overnight to create dense films (f; inset) that stably-coated the underlying cell culture polystyrene surface (u). Cells seeded and grown for three days on 1.0 wt% A6RGD films (b) adhered to the coating, whereas those seeded onto 2.5 wt% A6RGD films (c) failed to attach, and assumed a round morphology. Scale bars = 100 μm (inset = 50 μm). | ||
In addition, cells growing on 1.0 wt% A6RGD films achieved a significantly (p < 0.001) higher cell density when compared to uncoated controls, corresponding to a 1.2-fold increase in cell number at day 5 (Fig. 7). This is also equivalent to values recently obtained with the same cells on fibronectin-coated surfaces routinely used in tissue culture.53
 | ||
| Fig. 7 Bioactivity of A6RGD. Human cornea stromal fibroblasts (hCSFs) were grown on films produced from 0.01–2.5 wt% A6RGD solutions or uncoated polystyrene culture plate controls (0 wt% A6RGD) for five days. Cells were quantified (mean ± S.D.) using the Alamar Blue assay (n = 3; *** corresponded to p < 0.001; n.d., not determined due to absence of cells). | ||
However, cells grown on 2.5 wt% A6RGD films assumed a round morphology (Fig. 6c) and adhered poorly compared to the uncoated control (Fig. 7), both constituting evident signs of cytotoxicity. Furthermore, after five days in culture, no viable cells were found on films produced from A6RGD at 2.5 wt%, in contrast to the still-proliferating hCSFs on control uncoated surfaces (Fig. 6 and 7).
To verify the specificity of cell adhesion to the RGD motif, inhibition assays were performed using cRGD or anti-integrin αV antibody as soluble factors to block the predominant integrins expressed at the surface of hCSFs (Fig. 8). Cells were incubated with soluble anti-integrin αV antibody or cRGD prior to seeding onto 1.0 wt% A6RGD films coating ultra-low attachment plates, while soluble anti-mouse IgG-HRP antibody or the peptide amphiphile C16-ETTES were used as corresponding mock treatments. Results showed that, 24 h post-seeding, the number of integrin-blocked hCSFs attached to A6RGD coatings was significantly (p < 0.001) reduced to 27 ± 4% (+Antibody) and 27 ± 11% (+cRGD) of that from corresponding mock treatments (Fig. 8). In addition, no cells were shown to attach to uncoated low-attachment surfaces. These results demonstrated that the enhanced cell adhesion observed for the A6RGD coating was a specific effect, and involved the direct interaction between the RGD motif and integrins.
 | ||
| Fig. 8 A6RGD inhibition assays. Human cornea stromal fibroblasts (hCSFs) treated with soluble anti-integrin αV antibody or cRGD (blockers) and corresponding mock substitutes were seeded on 1 wt% A6RGD films coating ultra-low attachment surfaces. Cells were quantified (mean ± S.D.) 24 h post-seeding using the Alamar Blue assay (n = 3; *** corresponded to p < 0.001). | ||
These results indicate that films produced from A6RGD solutions containing up to 1.0 wt% SLP promote adhesion and enhanced proliferation of hCSFs, probably through mechanisms mediated by integrin clustering and downstream signaling.54 However, 2.5 wt% A6RGD induces cytotoxicity that limited cell adhesion and prevents subsequent proliferation. This effect can be explained by either a direct or indirect action of the RGD motif on mechanisms of cell apoptosis. Previous studies have shown that incubation with 1 × 10−3 M of soluble RGD-containing linear peptides directly activates caspase signaling, inducing apoptosis and reducing proliferation of human lymphocytes35 and umbilical vein endothelial cells.34 Alternatively, cell death could result from the uptake of SLP vesicles able to integrate cell membranes and causing formation of pores, thus increasing lipid bilayer conductance and causing membrane disruption and apoptosis.55,56 Long fibers formed from 2.5 wt% A6RGD provide a compact coating of the cell culture plates (Fig. 6a). Excess A6RGD molecules might then be released from the SLP coating to the media, thus blocking integrin-mediated cell attachment and inducing caspase activation and apoptosis. However, it is unlikely that apoptosis is caused by the presence of SLP vesicles, due to the small population of vesicles present in a ∼2.5 wt% A6RGD solution (inset Fig. 5d).
Our results showed that the self-assembly motif of A6RGD depended on its concentration. In particular, we propose that 0.1 wt% A6RGD self-assembles into oligomers, while at 1.0 and 2.5 wt% A6RGD forms short and long peptide fibrils respectively. Both 0.1 and 1.0 wt% SLP promoted adhesion and enhanced proliferation of hCSFs, while higher A6RGD concentrations were toxic to the cells.
Conclusions
In this work we studied the nanostructure of A6RGD in water. The self-assembly motif was shown to depend on the concentration of SLP. Initial fluorescence assays, together with Congo red absorption experiments and native PAGE assays confirmed the presence of A6RGD oligomers for 0.1 wt% SLP, short peptide fibrils for 1.0 wt% SLP and a network of long peptide fibres for 2.5 wt% A6RGD. Cryo-TEM and SAXS confirmed that a small population of vesicles co-exists with fibres from 2 to 15 wt% A6RGD. The population of vesicles relative to fibres increases with concentration. This is, to our knowledge, the first study to report the formation of vesicles by an alanine-rich SLP in solution.A6RGD vesicle walls are built out of highly interdigitated A6 blocks, presumably with positively charged RGD epitope groups exposed to water. An efficient packing of alanine residues into β-sheets was also revealed by XRD. A6RGD fibres are also likely to present an A6 core surrounded by RGD groups exposed to water.
Changes in the A6RGD self-assembly motif with concentration have implications in the use of this SLP to fabricate substrates for cell culture assays. At concentrations below the c.a.c., films of A6RGD are compatible with cell viability and proliferation. Films produced from A6RGD solutions at 0.1–1.0 wt%, cell adhesion proliferation was enhanced relative to uncoated controls.
As such, A6RGD was shown to constitute a promising SLP for the manufacture of biocompatible dry film substrates for tissue engineering and cell biology applications.
Acknowledgements
This work was supported by EPSRC grants EP/F048114/1 and EP/G026203/1 and BBSRC BB/I008187/1. We would like to acknowledge T. Narayan for support during the beamtime at ID02 (Project Number SC-3235). We would like to acknowledge the University of Reading (UK) for access to the Chemical Analysis Facility.Notes and references
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355 CrossRef CAS PubMed.
- S. Santoso, W. Hwang, H. Hartman and S. Zhang, Nano Lett., 2002, 2, 687 CrossRef CAS.
- I. W. Hamley, Soft Matter, 2011, 7, 4122 RSC.
- S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171 CrossRef CAS PubMed.
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355 CrossRef CAS PubMed.
- G. von Maltzahn, S. Vauthey, S. Santoso and S. G. Zhang, Langmuir, 2003, 19, 4332 CrossRef CAS.
- R. G. Ellis-Behnke, Y. X. Liang, S. W. You, D. K. C. Tay, S. G. Zhang, K. F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054 CrossRef CAS PubMed.
- R. Das, P. J. Kiley, M. Segal, J. Norville, A. A. Yu, L. Y. Wang, S. A. Trammell, L. E. Reddick, R. Kumar, F. Stellacci, N. Lebedev, J. Schnur, B. D. Bruce, S. G. Zhang and M. Baldo, Nano Lett., 2004, 4, 1079 CrossRef CAS.
- C. X. Chen, F. Pan, S. Z. Zhang, J. Hu, M. W. Cao, J. Wang, H. Xu, X. B. Zhao and J. R. Lu, Biomacromolecules, 2010, 11, 402 CrossRef CAS PubMed.
- S. Bucak, C. Cenker, I. Nasir, U. Olsson and M. Zackrisson, Langmuir, 2009, 25, 4262 CrossRef CAS PubMed.
- A. Nagai, Y. Nagai, H. J. Qu and S. G. Zhang, J. Nanosci. Nanotechnol., 2007, 7, 2246 CrossRef CAS PubMed.
- U. Khoe, Y. L. Yang and S. G. Zhang, Macromol. Biosci., 2008, 8, 1060 CrossRef CAS PubMed.
- V. Castelletto, D. Nutt, I. W. Hamley, S. Bucak, C. Cenker and U. Olsson, Chem. Commun., 2010, 46, 6270 RSC.
- D. A. Middleton, J. Madine, V. Castelletto and I. W. Hamley, Angew. Chem., Int. Ed., 2013, 52, 10537 CrossRef CAS PubMed.
- C. X. Chen, F. Pan, S. Z. Zhang, J. Hu, M. W. Cao, J. Wang, H. Xu, X. B. Zhao and J. R. Lu, Biomacromolecules, 2010, 11, 402 CrossRef CAS PubMed.
- H. Xu, J. Wang, S. Y. Han, J. Q. Wang, D. Y. Yu, H. Y. Zhang, D. H. Xia, X. B. Zhao, T. A. Waigh and J. R. Lu, Langmuir, 2009, 25, 4115 CrossRef CAS.
- I. W. Hamley, A. Dehsorkhi and V. Castelletto, Chem. Commun., 2013, 49, 1850 RSC.
- M. D. Pierschbacher and E. Ruoslahti, Nature, 1984, 309, 30 CrossRef CAS.
- T. Pakalns, K. L. Haverstick, G. B. Fields, J. B. McCarthy, D. L. Mooradian and M. Tirrelli, Biomaterials, 1999, 20, 2265 CrossRef CAS.
- N. Oku, C. Koike, Y. Tokudome, S. Okada, N. Nishikawa, H. Tsukada, M. Kiso, A. Hasegawa, H. Fujii, J. Murata and I. Saiki, Adv. Drug Delivery Rev., 1997, 24, 215 CrossRef.
- V. Marchi-Artzner, B. Lorz, C. Gosse, L. Jullien, R. Merkel, H. Kessler and E. Sackmann, Langmuir, 2003, 19, 835 CrossRef CAS.
- V. Castelletto, I. W. Hamley, C. Stain and C. J. Connon, Langmuir, 2012, 28, 12575 CrossRef CAS PubMed.
- G. Cheng, V. Castelletto, R. R. Jones, C. J. Connon and I. W. Hamley, Soft Matter, 2011, 7, 1326 RSC.
- V. Castelletto, C. M. Moulton, G. Cheng, I. W. Hamley, M. R. Hicks, A. Rodger, D. E. López-Pérez, G. Revilla-López and C. Alemán, Soft Matter, 2011, 7, 11405 RSC.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646 CrossRef CAS PubMed.
- M. Zhou, A. M. Smith, A. K. Das, N.W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523 CrossRef CAS PubMed.
- M. O. Guler, L. Hsu, S. Soukasene, D. A. Harrington, J. F. Hulvat and S. I. Stupp, Biomacromolecules, 2006, 7, 1855 CrossRef CAS PubMed.
- T. Muraoka, C. Y. Koh, H. Cui and S. I. Stupp, Angew. Chem., Int. Ed., 2009, 48, 5946 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684 CrossRef CAS PubMed.
- H. W. Jun, S. E. Paramonov, H. Dong, N. Forraz, C. McGuckin and J. D. Hartgerink, J. Biomater. Sci., Polym. Ed., 2008, 19, 665 CrossRef CAS PubMed.
- M. J. Webber, J. Tongers, M.-A. Renault, J. G. Roncalli, D. W. Losordo and S. I. Stupp, Acta Biomater., 2010, 6, 3 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352 CrossRef CAS PubMed.
- M. S. Aguzzi, C. Giampietri, F. De Marchis, F. Padula, R. Gaeta, G. Ragone, M. C. Capogrossi and A. Facchiano, Blood, 2004, 103, 4180 CrossRef CAS PubMed.
- C. D. Buckley, D. Pilling, N. V. Henriquez, G. Parsonage, K. Threlfall, D. Scheel-Toellner, D. L. Simmons, A. N. Albar, J. M. Lord and M. Salmon, Nature, 1999, 397, 534 CrossRef CAS PubMed.
- G. Pabst, M. Rappolt, H. Amenitsch and P. Laggner, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 4000 CrossRef CAS.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-rays, John Willey & Sons, Inc., New York, 1955 Search PubMed.
- Molinspiration Cheminformatics, Software package Molinspiration, 2010 Search PubMed.
- Innovagen, Software package Innovagen's Peptide Property Calculator, 2012 Search PubMed.
- H. LeVine, Protein Sci., 1993, 2, 404 CrossRef CAS PubMed.
- M. R. Nilsson, Methods, 2004, 34, 151 CrossRef CAS PubMed.
- A. F. Drake, G. Siligardi and W. A. Gibbons, Biophys. Chem., 1988, 31, 143 CrossRef CAS.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291 CrossRef CAS PubMed.
- H. Gaussier, H. Morency, M. C. Lavoie and M. Subirade, Appl. Environ. Microbiol., 2002, 68, 4803 CrossRef CAS.
- J. T. Pelton and L. R. McLean, Anal. Biochem., 2000, 277, 167 CrossRef CAS PubMed.
- P. Haris and D. Chapman, Biopolymers, 1995, 37, 251 CrossRef CAS PubMed.
- L. C. Serpell, Biochim. Biophys. Acta, Bioenerg., 2000, 1502, 16 CrossRef CAS.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi, V. Castelletto, J. Seitsonen, J. Ruokolainen and H. Iatrou, Soft Matter, 2013, 9, 4794 RSC.
- O. Rathore and D. Y. Sogah, J. Am. Chem. Soc., 2001, 123, 5231 CrossRef CAS PubMed.
- J. Kohlbrecher and I. Bressler, Software package SASfit for fitting small-angle scattering curves, 2011 Search PubMed.
- T. E. Creighton, Protein Folding, W.H. Freeman, New York, 1992 Search PubMed.
- R. M. Gouveia, V. Castelletto, S. G. Alcock, I. W. Hamley and C. J. Connon, J. Mater. Chem. B, 2013, 1, 6157 RSC.
- F. G. Giancotti and E. Ruoslahti, Science, 1999, 285, 1028 CrossRef CAS.
- R. Kayed, Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, J. E. Hall and C. G. Gable, J. Biol. Chem., 2004, 279, 46363 CrossRef CAS PubMed.
- R. Jelinek and T. Sheynis, in Lipids and Cellular Membranes in Amyloid Diseases, ed. R. Jelinek, Wiley-VCH Verlag & Co., Germany, 2011, ch. 12 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Displays chemical formula and contains pH, CD, FTIR and XRD data. Parameters obtained from the fittings to the SAXS data. See DOI: 10.1039/c3bm60232j |
| This journal is © The Royal Society of Chemistry 2014 |