Poly(ester-ether)s: II. Properties of electrospun nanofibres from polydioxanone and poly(methyl dioxanone) blends and human fibroblast cellular proliferation†
Nowsheen
Goonoo
a,
Archana
Bhaw-Luximon
a,
Isaac A.
Rodriguez
b,
Daniel
Wesner
c,
Holger
Schönherr
c,
Gary L.
Bowlin
b and
Dhanjay
Jhurry
*a
aANDI Centre of Excellence for Biomedical and Biomaterials Research, MSIRI Building and University of Mauritius, Réduit, Mauritius. E-mail: djhurry@uom.ac.mu
bDepartment of Biomedical Engineering and Virginia Commonwealth University, Richmond, Virginia, USA. E-mail: glbowlin@vcu.edu
cPhysical Chemistry I, Department of Chemistry and Biology, University of Siegen, 57076 Siegen, Germany. E-mail: schoenherr@chemie.uni-siegen.de
First published on 11th November 2013
Abstract
This article deals with an in-depth study of the thermal, mechanical and degradation behaviours of nanofibres from polydioxanone (PDX) and polyDL-3-methyl-1,4-dioxan-2-one (PMeDX) and a comparison with their blend films. Varying ratios of both polymers were blended and electrospun from solution. Electrospun fibres exhibited a melting transition at 109 °C independently of the PMeDX content, which corresponds to the melting of PDX nanofibres. As a result of the drawing process, PMeDX had a reduced plasticizing effect on PDX. In general, it was observed that overall crystallinity of the fibres decreased from 53% to 36% with increasing PMeDX content and this impacted on their mechanical properties. The Young's moduli decreased as the PMeDX content of the fibres increased. However, an increase in strain at break and peak stress was noted as a result of a decrease in the fibre diameter. AFM images of the electrospun fibres showed an increasing degree of morphological heterogeneity with increasing PMeDX content. Thermal degradation studies showed that electrospun mats were thermally more stable than blend films, as confirmed by a two-fold increase in activation energy. The hydrolytic degradation of the electrospun mats conducted in phosphate buffer solution at 37 °C showed that the degradation followed a surface erosion mechanism as opposed to bulk degradation observed for blend films. Degradation of fibres was found to be mainly dependent on their diameter. On the other hand, the degradation of blend films depended on the overall crystallinity of the blends. Electrospun PDX/PMeDX nanofibrous scaffolds were also subjected to cell viability studies with human dermal fibroblasts, in which they did not show illicit response and demonstrated excellent cell attachment and proliferation.
Introduction
The cellular response to a biomaterial is often enhanced when the architecture of the scaffold resembles that of the native tissue. The structural extracellular matrix (ECM) proteins in native tissues (50–500 nm diameter fibres) are 1 to 2 orders of magnitude smaller than the cell itself, enabling the cell to be in direct contact with many ECM fibres, thereby defining its 3D orientation. The combination of the nanoscale dimensionality of scaffolds, high surface area, porosity, flexibility, and superior strength makes electrospun scaffolds suitable for tissue engineering applications.1 Indeed, electrospinning remains a preferred method due to low cost, high throughput, ease of operation and system control. It is lauded for its ability to produce non-woven meshes containing fibres ranging from tens of microns to tens of nanometers in diameter, which can mimic both the form and function of the native ECM.Polydioxanone (PDX), a biodegradable poly(ester-ether) was first electrospun by Boland et al.2 Since then, many blends of PDX with other natural or synthetic polymers have been electrospun in view of improving the mechanical properties, cellular responses and biodegradability. For instance, McClure et al.3 studied electrospun blends of PDX and elastin at various ratios. Electrospun vascular grafts composed of elastin and PDX were designed by Sell et al.4 Elastin was chosen to provide elasticity and bioactivity to the prosthetic, while PDX contributed to the mechanical integrity. Electrospun PDX had an elastic modulus of 19.98 ± 0.74 MPa, but when blended with elastin, the modulus (4.89–9.64 MPa) could be adjusted to closely match that of the native femoral artery (9–12 MPa). The authors also reported that a 50/50 PDX/elastin blend had closer elastic modulus (∼9.64 MPa) and strain at failure (∼65%) values to those of the native artery (9–12 MPa and 63–76%, respectively). McManus et al.5 studied electrospun blends of PDX and fibrinogen for potential application in urologic tissue engineering. Fibrinogen was chosen because of its innate ability to induce cellular interaction and subsequent scaffold remodeling. They found that peak stress and modulus values increased linearly with increasing PDX concentration in the blends. Recently, Thomas et al.6 designed a trilayered electrospun tubular conduit based on blends of PDX, elastin, and gelatin. The mechanical properties could be tuned through variation of blend composition. In vitro degradation studies for up to 30 days showed about 40% mass loss and increased crystallinity due to the removal of the proteins and ‘cleavage-induced crystallization’ of PDX. Madurantakam et al.7 found that PDX/hydroxyapatite (HA) scaffolds had enhanced bone mineralization potential compared to the corresponding PLGA ones. Rodriguez et al.8 presented preliminary studies evaluating the mineralization potential of electrospun PDX/nano-hydroxyapatite and fibrinogen.
Recently, Wolfe et al.9 reported for the first time on the electrospinning of random copolymers of 1,4-dioxan-2-one and D,L-3-methyl-1,4-dioxan-2-one. Both the thermal and mechanical properties could be tailored by varying the percentage incorporation of MeDX in the random copolymer. In a more recent study, we reported on blend films of PDX and PMeDX and showed that low amounts of PMeDX in the blends (of the order of 15 weight%) could act as a plasticizer to high molar-mass-PDX as confirmed by an increase in Young's modulus. Mechanical tests showed overall reduced tensile properties of the blend films. Interaction parameters from viscosity analysis and surface morphology images indicated immiscibility of the blend films over the range of compositions studied.10
In a recent review,11 we highlighted the importance of tailoring the physico-chemical properties of polymers for scaffold applications with focus on the mechanical properties, degradability characteristics, cell attachment and proliferation. The aim of this paper is precisely to assess the effect of varying compositions of PDX/PMeDX homopolymers on the physico-chemical properties of corresponding blends and their capability to promote attachment and proliferation of human dermal fibroblasts. To this end, poly(ester-ether) blend nanofibres were electrospun and fully characterized from a morphological, thermal and mechanical point of view using a broad range of tests to have a better insight into their miscibility characteristics, crystalline properties, thermal stability and stress vs. strain behaviour. In addition, a detailed hydrolytic study of these electrospun fibres has been conducted to investigate their mechanism of degradation in comparison with blend films. In a last section of this study, the cellular response of the electrospun mats was tested using human fibroblast cells.
Experimental section
Materials
Tin(II) octanoate (Alfa Aeser, 97%) was used as received. Polydioxanone (inherent viscosity 1.7 at a concentration of 0.1 g dL−1 at 30 °C in 1,1,1,3,3,3 hexafluoroisopropanol) was purchased from Evonik. The structure of PDX is given in Fig. 1. D,L-3-Methyl-1,4-dioxan-2-one was synthesized according to procedures previously described by us.12 HFIP from Apollo Scientific Limited was used as received. PBS was purchased from Sigma-Aldrich and used as received.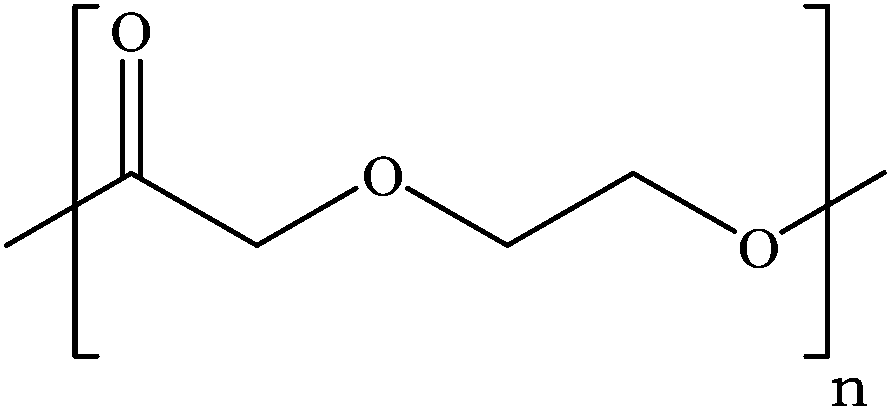 | ||
| Fig. 1 Structure of PDX. | ||
Methods
 | ||
| Fig. 2 Structure of PMeDX. | ||
 | (1) |
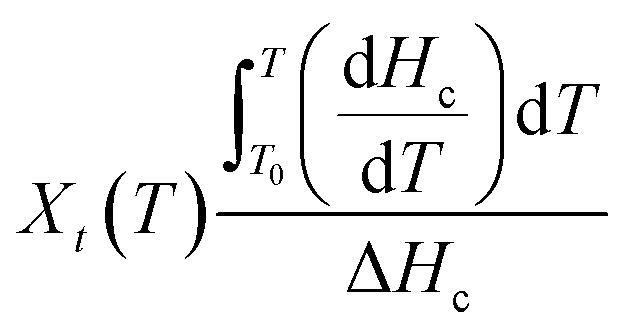 | (2) |
Considering the temperature difference between the sample and the furnace to be negligible, the relationship between time, t and temperature, T can be expressed as follows:
 | (3) |
Using eqn (3), it is possible to convert Xt = f(T) curves observed from non-isothermal DSC data into XT = f(t) curves.
| ln[ln(1 − α)−1] = Ea [Eaθ/RT2max] | (4) |
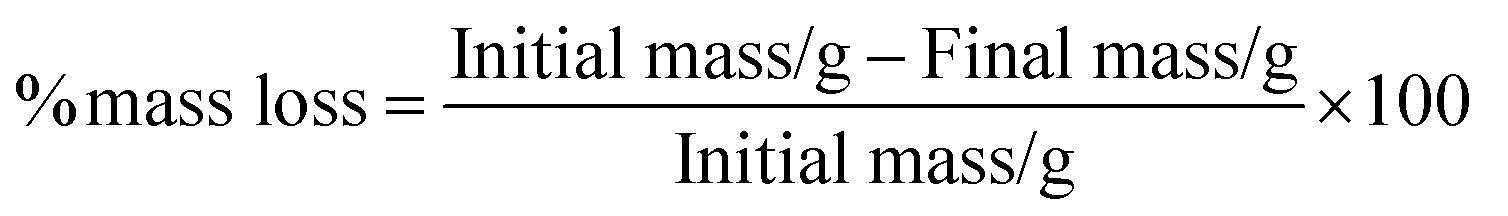 | (5) |
 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 human dermal fibroblast (HDF) cells. They were then incubated for 45 minutes to allow the cells to attach to the scaffolds. After 45 minutes, 100 μL of DMEM F-12 media (10% FBS, 1% penicillin/streptomycin) was added and cells cultured for 1 and 7 days (media replaced every 3 days). After each time point, cellularized scaffolds were immersed in 1 ml of formalin and stored at 4 °C until further use. Scaffolds were then removed from formalin, immersed in a 30% sucrose solution in DI water for 48 hours at 4 °C to ensure displacement of all air bubbles, suspended in premium frozen section compound (VWR), and frozen at −70 °C overnight. 10 μm slices were cryosectioned perpendicular to the scaffold surface to obtain cross-sections using a Cryostat (Thermo) and transferred to microscope slides. Cryosectioned samples were then stained with 4′-6-diamidino-2-phenylindole (DAPI) stain for 5 minutes and imaged using a Zeiss Axiovert fluorescent microscope to display the location of cell nuclei. Cell migration was measured and calculated from the DAPI images using the UTHSCSA Image Tool 3.0 software. Briefly, 30 length measurements from the cell seeded scaffold surface to the furthest migrated cell was measured and compared to the scaffold thickness (at the same location). Averages and standard deviations were reported as percent cell migration through the scaffold. For scanning electron microscopy (SEM), cellularized scaffolds on days 1 and 7 were removed from formalin, briefly rinsed in PBS and DI water, and subjected to ethanol dehydration (10 minute soaks in 30, 50, 70, 90, 100% ethanol, subsequently). Dehydrated scaffolds were air dried overnight, mounted on aluminum stubs, sputter coated in gold for 70 seconds, and imaged using a JEOL JSM-5610LV scanning electron microscope.
000 human dermal fibroblast (HDF) cells. They were then incubated for 45 minutes to allow the cells to attach to the scaffolds. After 45 minutes, 100 μL of DMEM F-12 media (10% FBS, 1% penicillin/streptomycin) was added and cells cultured for 1 and 7 days (media replaced every 3 days). After each time point, cellularized scaffolds were immersed in 1 ml of formalin and stored at 4 °C until further use. Scaffolds were then removed from formalin, immersed in a 30% sucrose solution in DI water for 48 hours at 4 °C to ensure displacement of all air bubbles, suspended in premium frozen section compound (VWR), and frozen at −70 °C overnight. 10 μm slices were cryosectioned perpendicular to the scaffold surface to obtain cross-sections using a Cryostat (Thermo) and transferred to microscope slides. Cryosectioned samples were then stained with 4′-6-diamidino-2-phenylindole (DAPI) stain for 5 minutes and imaged using a Zeiss Axiovert fluorescent microscope to display the location of cell nuclei. Cell migration was measured and calculated from the DAPI images using the UTHSCSA Image Tool 3.0 software. Briefly, 30 length measurements from the cell seeded scaffold surface to the furthest migrated cell was measured and compared to the scaffold thickness (at the same location). Averages and standard deviations were reported as percent cell migration through the scaffold. For scanning electron microscopy (SEM), cellularized scaffolds on days 1 and 7 were removed from formalin, briefly rinsed in PBS and DI water, and subjected to ethanol dehydration (10 minute soaks in 30, 50, 70, 90, 100% ethanol, subsequently). Dehydrated scaffolds were air dried overnight, mounted on aluminum stubs, sputter coated in gold for 70 seconds, and imaged using a JEOL JSM-5610LV scanning electron microscope.
Measurements
Differential scanning calorimetry (DSC) analysis was carried out using a Netzsch DSC 200 F3 Maia® thermal analyzer (Chennai, India). All blend samples were heated from 30 to 120 °C, cooled to −30 °C and reheated to 120 °C at 3 °C min−1. Netzsch TG 209 F3 Tarsus® analyzer (Chennai, India) was used to measure and record the sample mass change with temperature over the course of the pyrolysis reaction. Thermogravimetric curves were obtained at a heating rate of 10 °C min−1 between 25 °C and 1000 °C. Nitrogen was used as an inert purge gas to displace air in the pyrolysis zone, thus avoiding unwanted oxidation of the sample. The sample mass used in this study was approximately 10 mg. Surface morphology characterization was accomplished using scanning electron micrographs (SEM, Zeiss EVO 50 XVP, Germany). Both the inner (in contact with mandrel) and the outer surface of the electrospun mats were imaged. Blend samples were mounted on an aluminum stub and sputter coated with gold for imaging. To determine fibre and pore size, the ImageTool 3.0 image analysis software package was used (Shareware provided by University of Texas Health Science Center at San Antonio). The software was calibrated using the micron scale bar of each picture. An average fibre diameter was determined by measuring the diameter of 60 different fibres, while an average pore size was determined by measuring the diameter of 60 different pores. Pores were identified as areas of void space bounded by fibres on all sides at or near the same depth of field, while their long and short diagonal axes were measured and averaged together to serve as their diameter.13 Intermittent contact (tapping) mode AFM imaging was done on as prepared samples on an Asylum MFP-3D atomic force microscope (Asylum Research, USA) using Olympus AC160TS cantilevers (with a resonance frequency of 300 kHz and a nominal spring constant of 40 N m−1) under ambient conditions. The rms amplitude of the cantilever was adjusted to 85 nm and a set point ratio of 0.8 was chosen. Constant amplitude images were acquired, depending on the scan size, with 512 pixels × 512 pixels (up to 2024 pixels × 2024 pixels); the phase shift was recorded simultaneously. The data were processed off line using the MFP-3D software. The tensile properties of the blend films (40 × 10 × 4 mm3) were studied utilizing an Instron Tensile Tester 3343 (Instron, USA) at 27 °C and 60% relative humidity using a crosshead speed of 10 mm min−1, gauge length of 1 cm and 500 N load cell. “Dog-bone” shaped samples were punched from electrospun mats (2.75 mm wide at their narrowest point with a gauge length of 7.5 mm) and tested on a MTS Bionix 200 testing system with a 100 N load cell (MTS Systems Corp.) and an extension rate of 10.0 mm min−1. The contact angles of the modified surfaces were measured using water as a probe liquid (Milli-Q water from a Millipore Direct-Q 8 system with resistivity of 18.0 MΩ cm−1) with an OCA 15plus instrument (Data Physics Instruments GmbH, Germany). Static contact angle data based on the sessile drop method were acquired immediately after deposition of a 1 μL drop on at least three positions for each sample and are stated as arithmetic mean. A short film sequence covering several seconds before and after deposition of the droplet was taken.Statistical analysis
Three measurements were recorded and averaged for the determination of viscosity and mechanical properties of electrospun fibres. Values are reported as a mean ± SD. Error values for Young's moduli were calculated using the linear regression analysis method and errors associated with strain and stress were calculated according to eqn (7) and (8) respectively.14–16 The degree of confidence was 95%. | (7) |
 | (8) |
Results and discussion
Morphology of electrospun blend mats
PMeDX (reduced viscosity 0.34 dL g−1) was blended in different proportions (100/0, 98/2, 93/7, 90/10 and 85/15 wt/wt%) with PDX (reduced viscosity 2.11 dL g−1) and the fibres were electrospun from a solution of polymers in HFIP. Nanofibre mats were fabricated successfully. Typical images of the random electrospun mats with smooth fibres are shown in Fig. 3 (see Fig. S1 in ESI† for SEM of 93/7 and 90/10 fibre). Table 1 summarizes the outside surface fibre diameter values as determined from SEM images as well as reduced viscosities of polymer blends before and after electrospinning. Fibre diameters varied from 0.39 to 0.65 μm while the pore sizes ranged from 0.9 to 2.8 μm. The highest diameter and pore size correspond to blend composition 98/2. | ||
| Fig. 3 SEM (2000× magnification, scale bar = 10 μm) of electrospun (a) PDX, (b) 98/2, (c) 85/15 at a concentration of 100 mg ml−1 (inside surface). | ||
| Blend composition PDX:PMeDX (wt/wt)% |
Fibre diameters/μm | Pore size/μm | Reduced viscosity of blends before electrospinning, ηreda |
Reduced viscosity of electrospun fibres, ηreda |
|---|---|---|---|---|
| a Viscosity carried out in HFIP at a concentration of 0.2 g dL−1 and a temperature of 30 °C. | ||||
| 100/0 | 0.57 ± 0.23 | 2.0 ± 1.3 | 1.96 ± 0.13 | 2.11 ± 0.14 |
| 98/2 | 0.65 ± 0.25 | 2.8 ± 1.3 | 2.17 ± 0.16 | 2.40 ± 0.19 |
| 93/7 | 0.44 ± 0.18 | 0.9 ± 0.4 | 1.76 ± 0.14 | 1.87 ± 0.11 |
| 90/10 | 0.54 ± 0.20 | 2.0 ± 1.1 | 1.67 ± 0.11 | 1.72 ± 0.13 |
| 85/15 | 0.39 ± 0.20 | 1.9 ± 0.7 | 1.45 ± 0.15 | 1.51 ± 0.12 |
The determination of fibre diameter is important as it corresponds not only to molecular level orientation,17 but also affects cellular behaviour.18,19 As can be noted from Table 1, the 98/2 blend gave the highest fibre diameter but there is no clear cut trend in fibre diameters with increasing content of amorphous PMeDX in the blend. Previous findings reported on either an increase or a decrease of fibre diameter with the addition of an amorphous polymer. For instance, it decreases in the case of binary blends of incompatible polymers such as polycaprolactone/polytrimethylene carbonate due to effects caused by internal phase morphology and solution viscosity of electrospun fibres.20 During electrospinning, the application of a strong electrostatic field to the polymer solution causes the solution jet to be pulled, stretched and elongated in the axial direction. When an incompatible polymer blend is subjected to shear and/or elongation flow, the dispersed droplet is considerably elongated in the flow direction.17 If the dispersed phase in the blend has a lower viscosity, the solution jet will be stretched and elongated more by the electric forces during the electrospinning process, thus resulting in a decrease in fibre diameter. However, fibre diameters may also increase with increasing content of the amorphous phase as was the case for electrospun polycaprolactone/poly(methyl methacrylate) (PMMA) fibres.21 A clear explanation as to the lack of trend in fibre diameter in our case remains difficult at this stage and has prompted further investigations.
The reduced viscosities of the blends were recorded before electrospinning. Values listed in Table 1 show that the reduced viscosities decreased with increasing PMeDX content except in the case of the 98/2 blend which again shows an abnormal behaviour. The addition of an amorphous polymer to a semi-crystalline one generally results in a decrease in reduced viscosities mainly due to shrinkage of macromolecular coils and increased polymer chain mobility.22 The reduced viscosities of the electrospun fibres were also measured and found to be higher than the values before electrospinning. The reduced viscosity of blend 98/2 was found to be significantly higher than PDX homopolymers and to the other blends. Noteworthy is the fact that this blend composition gave rise to the highest fibre diameter.
AFM images of the electrospun scaffolds showed that the fibres had cylindrical morphologies irrespective of the blend compositions. Also, AFM confirmed the three-dimensionality of the PDX/PMeDX scaffolds, which also possessed large voids with height differences greater than 4 μm (Fig. 4).
 | ||
| Fig. 4 AFM image showing the topography of the electrospun 93/7 PDX/PMeDX scaffold (scale bar: 10 μm). | ||
The AFM and SEM images show that the PDX fibres possess a rather smooth surface compared to the blend fibres, which had heterogeneous surface morphologies as indicated by the presence of protrusions on the fibre surface (Fig. 5). The nanofibrous mats possess interstitial spaces between the fibres. No pores were observed on the fibre surface, but instead were present between the fibres on the electrospun mat. Addition of only 2 wt% PMeDX caused a significant increase in the pore size which then decreases again without a clear trend in variation. Fibre diameter and pore size are strongly correlated as reported by several studies.23,24 Generally, a decrease in fibre diameter corresponds to a decrease in pore size as we also noted. It is likely that the presence of protrusions on the fibre surfaces which increased with increasing PMeDX contents affects pore size.
 | ||
| Fig. 5 AFM phase images of electrospun (a) 100/0, (b) 93/7 and (c) 85/15 PDX/PMeDX scaffolds (scale bar: 500 nm). | ||
More in-depth AFM analysis of the homopolymer PDX fibres reveals the presence of lamellae on the fibre surface (see Fig. S2 in ESI†) in addition to isolated heterogeneities. In particular, tapping mode phase imaging reveals locally different energy dissipation of the polymer especially for the mixed fibres (Fig. 5). The degree of heterogeneity increases with increasing PMeDX content (0, 7, 15 wt%) indicating greater immiscibility of the two polymers.
In conclusion, we noted a correlation between fibre diameter and pore size although a clear cut trend for both was not observed with increasing PMeDX content. We hypothesize that this is possibly due to the presence of protrusions on the fibre surfaces as confirmed by AFM. Moreover, AFM images showed that surface roughness of electrospun PDX/PMeDX fibres increases with increasing PMeDX content. We noted the abnormal behaviour of the 98/2 electrospun fibre which exhibited the largest fibre diameter, pore size and reduced viscosity.
Thermal properties of solution cast films and electrospun mats
PDX is a semi-crystalline polymer, while PMeDX is amorphous. The melting temperature (Tm), crystallization temperature (Tc), enthalpy of fusion (ΔHm) and enthalpy of crystallization (ΔHc) of PDX and PDX/PMeDX solution cast films and electrospun mats were determined by DSC. The values obtained are listed in Table 2.| Blend composition PDX:PMeDX (wt/wt)% |
Solution cast films | |||||
|---|---|---|---|---|---|---|
| T m/°C | ΔHm/J g−1 | T c/°C | ΔHc/J g−1 | χ blend/(%) 2nd scan | X PDX /(%) 2nd scan | |
| 100/0 | 109.4 | 63.2 | 83.9 | 64.6 | 44.8 | 44.8 |
| 98/2 | 107.1 | 55.31 | 80.3 | 60.75 | 39.2 | 40.0 |
| 93/7 | 87.1 | 55.6 | 43.6 | 49.9 | 39.4 | 42.4 |
| 90/10 | 84.3 | 50.5 | 37.8 | 46.5 | 35.8 | 39.8 |
| 85/15 | 79.8 | 43.5 | 35.5 | 37.7 | 30.8 | 36.2 |
| 74/26 | 79.6 | 48.8 | 22.7 | 35.6 | 34.6 | 46.8 |
| 60/40 | 80.5 | 37.0 | 21.6 | 26.5 | 26.2 | 43.7 |
| Electrospun non-woven mats | ||||||
| 100/0 | 109.1 | 75.2 | 51.0 | 61.2 | 53.3 | 53.3 |
| 98/2 | 109.5 | 60.6 | 59.4 | 54.5 | 42.9 | 43.8 |
| 93/7 | 109.1 | 66.8 | 52.7 | 53.6 | 47.3 | 50.8 |
| 90/10 | 109.1 | 57.6 | 53.8 | 50.3 | 40.8 | 45.3 |
| 85/15 | 109.3, 99.3 | 50.4 | 80.4 | 52.0 | 35.7 | 42.0 |
The degree of crystallinity of the blends (χblend) and that of the PDX phase in the blends (χPDX) were calculated according to eqn (9)–(11) respectively:
 | (9) |
 | (10) |
 | (11) |
As can be observed from Table 2, the solution cast blend films exhibit a significant drop in melting temperature as the PMeDX content increases. Tm dropped from 109.4 °C for pure PDX to 80.5 °C for a 60/40 PDX/PMeDX blend. On the other hand, almost no change was observed in the melting temperatures of the electrospun blends independently of the PMeDX composition. This shows that the effect of PMeDX as a plasticizer is greatly reduced as the fibres are stretched during the electrospinning process. This stretching concerns both PDX and PMeDX chains. Also, in the case of electrospun 85/15 fibre, two melting transitions at 109.3 °C and 99.3 °C respectively could be observed. When PDX is heated, it undergoes partial melting and recrystallization. This can cause the appearance of an exotherm just before the broad melting peak in DSC scan as reported elsewhere.26,27 The higher temperature melting peak at 109.3 °C observed in the DSC scan of electrospun 85/15 fibre can be attributed to the melting of these reorganized crystals.
In general, the melting enthalpies, ΔHm decreased with increasing PMeDX contents in both cases. The overall crystallinity of the blends decreased with increasing weight% of amorphous PMeDX. The crystallinity (χblend) of the electrospun fibres was higher than the corresponding blend films. This increase in crystallinity could be explained by the alignment of polymer chains, which occurs during the electrospinning process as a result of the field and flow induced stretching. A drop in the crystallinity of the PDX phase is observed for the electrospun fibres, but a trend cannot be established with increasing PMeDX content. On the basis of these findings, there is a likely possibility of weak interactions occurring between the crystalline phase of PDX and that of the disentangled and stretched PMeDX. The crystallization temperatures of electrospun fibres increased from 51.0 to 80.4 °C, while their enthalpy of crystallization decreased from 61 (for PDX) to 50.3 J g−1 (fibre with 10 wt% PMeDX). In the case of blend films, both crystallization temperature and enthalpy of crystallization were composition dependent and a shift to lower temperatures is noted with increasing PMeDX content. Noteworthy is the fact that the ΔHc of a 90/10 film is 46.5 J g−1 and can decrease up to 26.5 J g−1 for a 60/40 film.
The shift in crystallization temperature was related to the overall crystallization kinetics in the cooling scan and was proportional to the density of active nuclei in the crystallizing polymeric material. The crystallization temperature of PDX in the blend films decreased from 83.9 °C to 21.6 °C, when 40% of PMeDX was added. This “anti-nucleating” effect, synonymous to a decrease in the density of active nuclei is characterized by a decrease in Tc and ΔHc.28 PMeDX does not have an anti-nucleating effect on PDX for fibres contrary to the blend films, as reported in one of our previous studies.29
Details on the theory of non-isothermal crystallization kinetics are given in the methods section. The plots of relative crystallinity against crystallization time (Fig. 6) show a sigmoid shape, indicative of a fast primary crystallization during the early stage and slow secondary crystallization in a later stage.
 | ||
| Fig. 6 Plots of relative crystallinity versus crystallization time for (a) PDX/PMeDX blend films and (b) electrospun PDX/PMeDX mats. | ||
An important parameter in the non-isothermal crystallization kinetics is the half-time of crystallization, t1/2, which is the time interval from the onset of crystallization to the time at 50% completion. From the graph of Xt against time, the half-life crystallization time, t1/2 can be obtained at X = 50%. Upon PMeDX addition, all blend films showed improved crystallization rates compared to neat PDX (Fig. 6a). From graphs of relative crystallinity against time (Fig. 6b), it could be deduced that the addition of PMeDX in the fibres accelerated the crystallization rate of PDX except in the case of the 98/2 blend. At PMeDX contents greater than 2 wt%, the latter can easily be rejected out of the crystallization front due to immiscibility effects (increased chain mobility) which results in an increase in crystallization rate. The measurement was repeated twice and is therefore not an artifact. We hypothesize that in the electrospun 98/2 fibre, PMeDX may be acting as a plasticizer, penetrating in between PDX chains, which causes its rejection to be more difficult and thereby resulting in decreased crystallization rate compared to neat PDX.
The inverse value of t1/2 signifies the bulk crystallization rate. Fig. 7 shows the variation of the bulk crystallization rate (t1/2−1) with increasing PMeDX wt% for both films and electrospun mats. The bulk crystallization rates increase and finally level off in the case of blend films. However, for electrospun blend mats, the crystallization rate increases continuously. This accelerated crystallization is due to increasing chain mobility. Moreover, this suggests that increasing PMeDX content favours its rejection out of the crystallization front, which results in an increase in the crystallization rate. In general, the rate constant depends on the nucleation mode, nucleation density and growth rate of the crystalline moieties.30 Overall crystallization depends on two factors namely the growth rates of individual spherulites and the number of spherulites growing. Comparing blends with the same PMeDX contents, for instance 90/10 blend, it can be observed that the presence of PMeDX enhanced the crystallization rate more in the case of fibres compared to films.
 | ||
| Fig. 7 Variation of the bulk crystallization rate with increasing PMeDX wt% (films and fibres). | ||
To summarize, data from thermal analysis indicate that the overall crystallinity of the electrospun mats decreases as amorphous PMeDX content increases. However, the melting transitions of electrospun PDX/PMeDX mats do not differ from that of PDX mats. This is accounted for by a stretching of polymer chains during electrospinning. Moreover, based on bulk crystallization rate values, PMeDX influences crystallization to a greater extent in electrospun fibres compared to blend films.
Thermal degradation of blend films and electrospun mats
The TG profiles showed that degradation occurred in one stage. Onset degradation temperatures, Tonset decreased with increasing PMeDX wt% for both blend films and electrospun mats, supporting immiscibility of PDX/PMeDX in both cases. However, the onset degradation temperatures for electrospun mats were significantly higher than the corresponding blend films (Table 3). This increase in thermal stability of electrospun fibres is due to the alignment of polymeric chains, which occurs during the electrospinning process and also due to the possible interactions occurring between PDX and PMeDX chains. This observation is in agreement with the studies of Freire et al.,31 whereby they noted the higher thermal stability of electrospun cellulose mats compared to the corresponding films.
| Blend composition PDX:PMeDX (wt/wt)% |
Electrospun fibres | Solution cast films | ||
|---|---|---|---|---|
| T onset/°C | T max/°C | T onset/°C | T max/°C | |
| 100/0 | 224.0 | 296.4 | 102.0 | 221.6 |
| 98/2 | 217.7 | 292.9 | 98.0 | 208.8 |
| 95/5 | — | — | 98.0 | 213.3 |
| 93/7 | 216.0 | 281.4 | 97.0 | 203.7 |
| 90/10 | 212.5 | 282.1 | 96.0 | 211.8 |
| 85/15 | 210.0 | 284.2 | 86.9 | 202.1 |
| 70/30 | — | — | 86.6 | 192.6 |
| 50/50 | — | — | 82.0 | 199.6 |
The activation energies of thermal degradation were determined from the Horowitz–Metzger plot. The activation energies of degradation decrease only slightly with increasing wt% of PMeDX for films. A more pronounced drop is observed for electrospun mats, but the values remain twice higher even at high PMeDX content (see Fig. S4 in ESI†).
In conclusion, thermal degradation studies showed that electrospun PDX/PMeDX fibres were more thermally stable compared to PDX/PMeDX films and activation energies of degradation of electrospun fibres were approximately twice that of blend films.
Mechanical performance of blend films and fibres
The mechanical property of a scaffold is an important criterion to be considered since the latter should be able to support cell growth and proliferation. The mechanical properties of a fibrous mat are determined by a combination of several factors including polymers' intrinsic properties, fibre packing density, structural morphology, average fibre diameter, fibre alignment, fibre–fibre junctions, porosity etc. The co-continuous fibre structure obtained by blend electrospinning enables the components in the fibres to be blended at the nano-level. This imparts not only new thermal but also new mechanical properties to the blended fibres.32 The mechanical properties of both PDX/PMeDX films and fibres were assessed. The Young's modulus values of the electrospun mats were found to decrease with increasing PMeDX wt% (Table 4). The elastic modulus of fibres is strongly influenced by the lamellar and amorphous fractions of chains present within fibres. Electrospinning brings about changes in terms of molecular orientations and hence, alterations in crystallinity which, in turn, impart physical uniqueness to the material and play an important role in the deformation behaviour of the fibres. Furthermore, the Young's modulus values of electrospun 93/7, 90/10 and 85/15 blends are quite close compared to electrospun PDX or 98/2 blend. This can be explained by the high packing density of fibres in those blends as observed in SEM images. Higher fibre packing density due to smaller fibre diameters and lower pore sizes led to greater flexibility. In general, the mechanical properties of the blend films decreased with increasing weight percent of PMeDX. Higher Young's modulus values were obtained for samples with up to 15 wt% PMeDX compared to PDX film but stress at break values decrease as PMeDX content increases. This is attributed to decreasing crystallinity of films as percentage of PMeDX in the blend increases. Compared to films, electrospun fibres were more flexible as shown by lower modulus values and higher elongation at break. Overall, both strain at break and peak stress values for electrospun mats increased with decreasing fibre diameter. This was in line with the work of Pai, whereby the yield stress of electrospun poly(trimethyl hexamethylene terephtalamide) was found to increase with decreasing fibre diameters.33 Wong et al.34 also found that a decrease in fibre diameter of electrospun PCL led to enhanced mechanical strength and stiffness owing to an improvement in the degree of crystallinity and molecular orientation of fibres. In a study by Stitzel et al.,35 the authors observed a decrease in modulus of blends of PCL and PLA with increasing PCL contents. Cheng et al.36 obtained higher elongation for electrospun blends of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) and poly(D,L-lactic acid) compared to the corresponding cast films. The higher elongation and lower tensile modulus values obtained with increasing PDLLA contents were explained by the decreased number of lamellae.| Blend composition PDX:PMeDX |
Electrospun mats | Solution cast films | ||||
|---|---|---|---|---|---|---|
| Strain at break (mm mm−1) | Peak stress (MPa) | Modulus (MPa) | Strain at break (mm mm−1) | Peak stress (MPa) | Modulus (MPa) | |
| 100/0 | 0.57 ± 0.11 | 4.08 ± 0.29 | 33.3 ± 2.17 | 0.07 ± 0.00050 | 3.68 ± 0.30 | 207 ± 9.11 |
| 98/2 | 0.85 ± 0.22 | 4.67 ± 0.57 | 28.5 ± 3.97 | 0.08 ± 0.00062 | 2.26 ± 0.18 | 229 ± 7.56 |
| 93/7 | 0.80 ± 0.30 | 4.54 ± 0.94 | 24.1 ± 3.35 | 0.05 ± 0.00060 | 2.60 ± 0.21 | 224 ± 7.26 |
| 90/10 | 0.83 ± 0.072 | 4.59 ± 0.61 | 23.6 ± 3.99 | 0.08 ± 0.00059 | 2.28 ± 0.16 | 227 ± 6.81 |
| 85/15 | 0.86 ± 0.063 | 5.88 ± 0.22 | 24.8 ± 2.82 | 0.11 ± 0.00014 | 0.04 ± 0.0032 | 214 ± 6.42 |
| 75/25 | — | — | — | 0.03 ± 0.00074 | 1.05 ± 0.085 | 145 ± 6.24 |
In summary, the mechanical performance of the films was affected by the overall crystallinity of the blends and therefore, the weight percent of PMeDX. On the nanoscale level, the effect of fibre properties on mechanical performance seems to be more dominant than the polymers intrinsic properties. The mechanical properties of the electrospun fibrous mats were dependent on fibre characteristics such as structural morphology and fibre diameter.
Hydrolytic degradation studies
The hydrolytic degradation of PDX/PMeDX blends was investigated in PBS solution at 37 °C. Changes in pH, mass loss and surface morphology were monitored over 3 weeks (for films) and 5 weeks (for electrospun mats). A significant drop in pH was observed for all blend films (see Fig. S5 in ESI†). This drop was more pronounced with increasing PMeDX content. Higher wt% of PMeDX lead to more amorphous zones in the blend films and hence favoured cleavage and degradation. Degradation of the poly(ester–ether)s occurs via random chain scission, resulting in chains of variable lengths as depicted in Scheme 1. The carboxylic acids formed during the process account for the sudden decrease in pH. | ||
| Scheme 1 Hydrolytic degradation mechanism highlighting formation of acid end groups (R: CH3 or H). | ||
The abrupt drop in pH noted in the case of blend films translates an autocatalytic degradation process, whereby carboxylic acids are trapped inside the polymer matrix (“cage effect”), thereby increasing local acidity37 and catalyzing dissolution of large fragments. On the other hand, if polymer degradation occurred via surface erosion, the degraded products would easily diffuse in the hydrolysis media and carboxylic acid easily neutralized, consequently leaving pH practically unchanged. The degradation is in line with bulk erosion mechanism. Contrary to blend films, no significant drop in pH was noted for electrospun mats due probably to their higher crystalline nature. This tends to support degradation via surface erosion. Surface wettability which is dependent on several factors such as fibre diameter, pore sizes/void fraction and surface roughness could also affect degradation rates. We hypothesize that the surface of the electrospun fibres is essentially constituted of PDX and that the immiscible PMeDX fraction is dispersed in the interior. Fibre diameter seemed to have a more predominant effect on degradation. Indeed, the smaller the fibre diameter, the slower the degradation. Moreover, the smaller diameter fibres showed a higher fibre packing density and hence lower porosity, which resulted in reduced water penetration and therefore slower degradation. Similar results were reported by Cui et al.,38 whereby the degree of surface roughness of the electrospun mats increased with a decrease in fibre diameters, leading to higher air entrapment between fibres, and hence decreased degradation rates.
Blend films with PMeDX contents up to 10 weight% had similar degradation profiles to PDX (Fig. 8a). Higher PMeDX contents in blend films led to significantly different hydrolytic degradation profiles. Mass loss for blend films ranges from 10 to 40% after 3 weeks for PMeDX content of 0–20%. Degradation was significantly higher as the PMeDX content increases as a result of a decrease in crystalline character. Moreover, an almost constant value of mass loss was obtained for blend films after the third week, indicating that degradation of PMeDX was almost over. Degraded blend films were further analyzed by SEM (Fig. 9). Compared to a 60/40 blend film, the presence of small particles in the SEM image of the 90/10 blend film showed that degradation was still not over at the third week. This showed that the overall crystallinity of the blend films impacted on their degradation behaviour. Mass loss profiles confirmed that blend films degraded via bulk erosion.
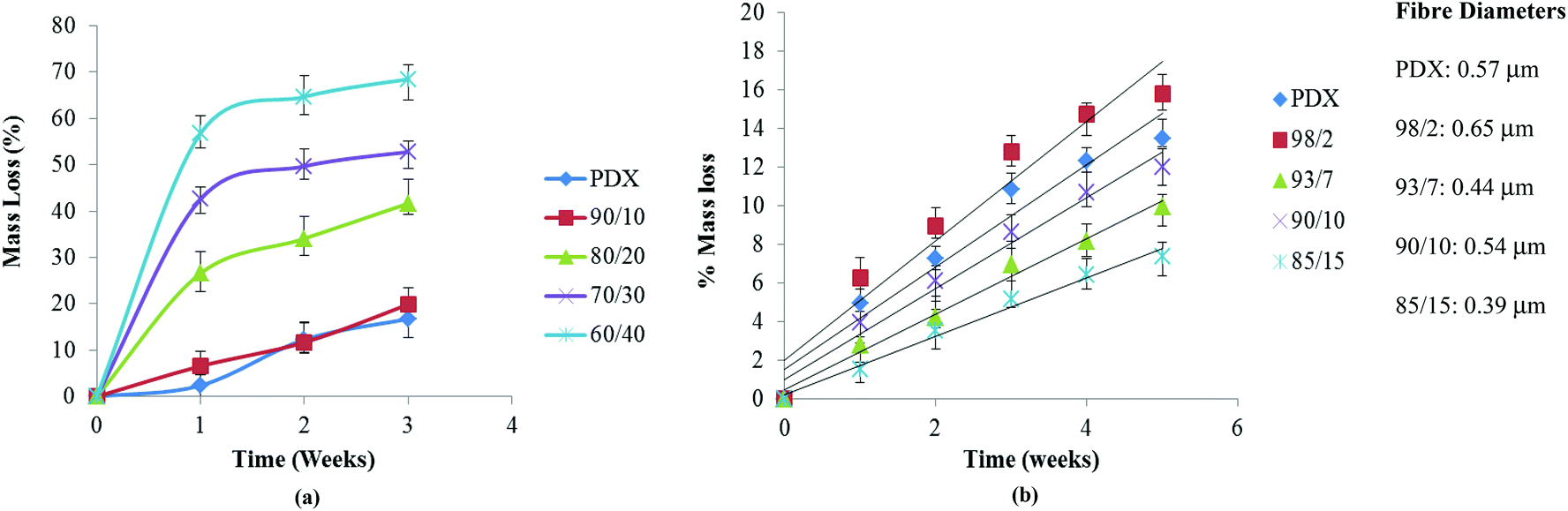 | ||
| Fig. 8 Mass loss of (a) films and (b) electrospun mats as a function of hydrolysis time in PBS at 37 °C. | ||
 | ||
| Fig. 9 SEM (500× magnification, scale bar = 50 μm) of electrospun mats of 98/2 and 85/15 at weeks 0 and 5. | ||
On the other hand, in the case of electrospun mats, the degradation trend was totally reversed with the 85/15 composition degrading at the slowest pace (Fig. 8b). Mass loss for electrospun fibres ranges from 6 to 15% after 5 weeks for PMeDX content 0–15%. Mass loss continued to increase and no constant value was reached during the degradation period investigated, confirming that fibres degraded at a much slower rate. The mass loss profile of electrospun fibres was in line with the pH evolution profile. Higher mass loss was obtained for the larger diameter 98/2 blend. At the fifth week, fibre disintegration can be observed in the electrospun 85/15 mat as degradation occurs (no fibre breaking was observed in the SEM as shown in Fig. 9). On the other hand, the morphology of the electrospun 98/2 fibre at week 5 does not resemble its original morphology at all. Indeed, visual appearance resembles a smooth polymer matrix and there is great difference in its surface morphology at weeks 0 and 5. Results obtained from this study are interesting, since they show the possibility of tuning the mechanical properties of PDX through the incorporation of PMeDX without compromising the degradation behavior as well as the thermal properties of the resulting blend fibres. The more linear mass loss profiles indicate that degradation proceeds via surface erosion rather than bulk degradation.
To sum up, mass loss profiles indicated that hydrolytic degradation of electrospun PDX/PMeDX fibres occurred via a surface erosion mechanism while in PDX/PMeDX blend films, degradation occurred via bulk erosion. Moreover, degradation of blend films showed a dependency on PMeDX content while that of fibres appears to be more affected by fibre diameter. Smaller fibre diameter mats degraded at a slower pace compared to larger diameter ones.
In vitro cell culture studies
Several groups have reported that electrospun micro/nano fibres mimic the natural ECM in the body and therefore provide a suitable matrix for cell adhesion and proliferation. Here, the effect of blending PMeDX with PDX on cell adhesion and proliferation was investigated by culturing human dermal fibroblasts on the scaffolds. SEM images revealed spindle-like shape of the attached cells with filopodia-like extension adhering to the scaffold and connecting to adjacent cells (Fig. 10). The cells intimately spread and elongate themselves on the electrospun scaffolds, forming a three-dimensional and multicellular network.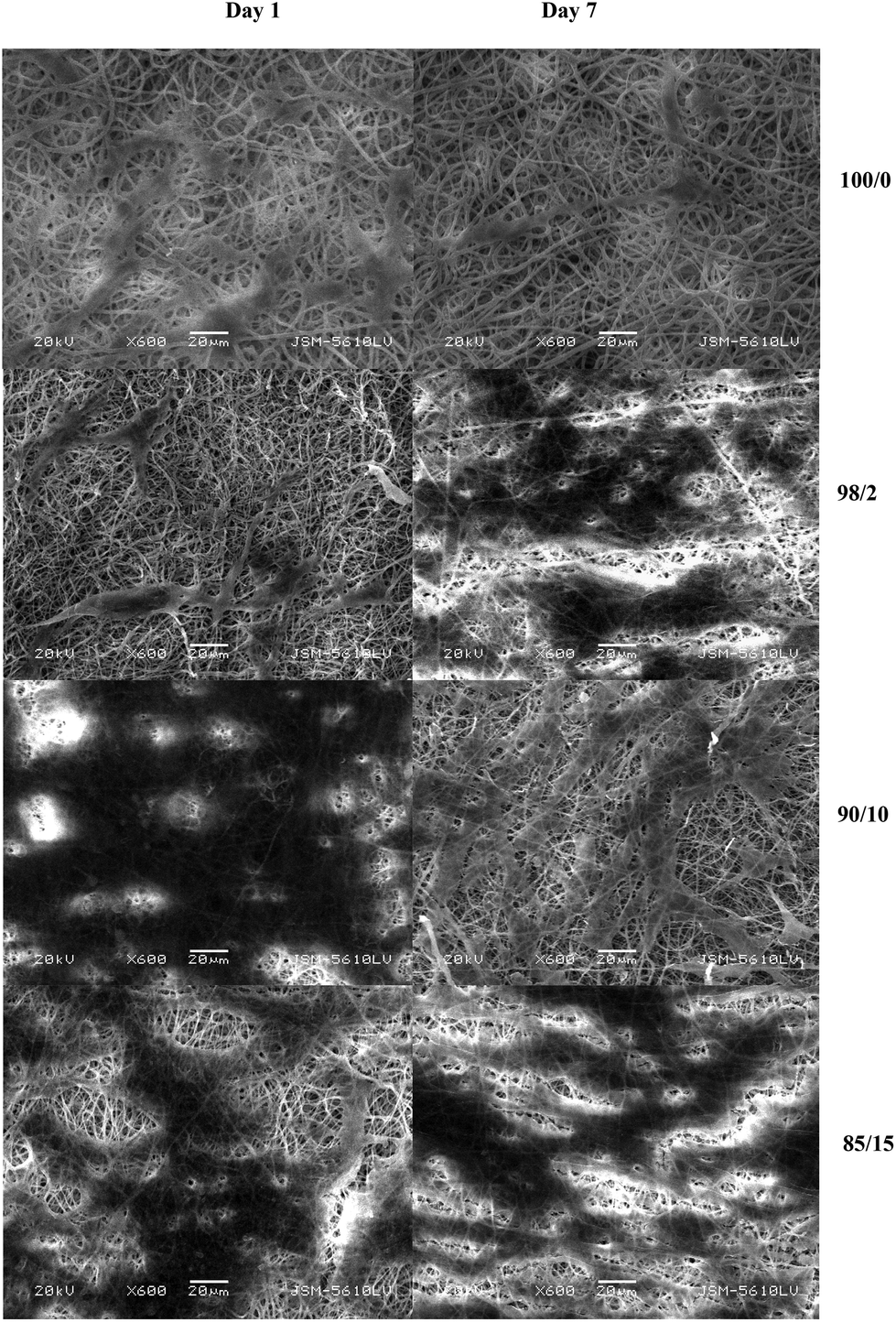 | ||
| Fig. 10 SEM images (600× magnification, scale bar = 20 μm) of cell seeded scaffolds after days 1 and 7 respectively. | ||
Cellular response is influenced by chemical properties (biomatrix composition, integrin receptor sites), topographical features (surface energy, surface morphology, surface roughness),39 and mechanical properties (elastic modulus),40,41 crystallinity42 and contact angle.43 Cell growth rates were found to be lower on crystalline substrates compared to amorphous ones42 and cell adhesion was enhanced with increasing degree of surface roughness.40 As can be observed from the SEM images in Fig. 10, there is increased cell attachment on the surface of the electrospun PDX/PMeDX scaffolds compared to electrospun PDX. An electrospun 90/10 scaffold had better cell attachment at day 1 compared to the remaining scaffolds. Cell adhesion was dependent on fibre composition. Also, at day 7, the 98/2 scaffold showed the highest cell proliferation. This could be explained by the fact that electrospun 98/2 fibre had a smaller fibre packing density compared to the remaining electrospun mats, and hence had a higher porosity, which favoured cell migration and infiltration into the nanofibrous scaffold. We have measured static water contact angles to have a better insight as to how surface wettability could affect cell attachment on the electrospun scaffolds. In contrast to what was expected, these contact angle measurements showed that the hydrophobicity of electrospun PDX/PMeDX mats decreased with increasing PMeDX contents. This suggests that PMeDX is located within the fibres and not in the topmost 1–2 nm of the surface. Several studies have shown that cell growth was favoured on less hydrophobic surfaces.43,44 This is in line with our results, whereby better HDF cell attachment was observed on the PDX/PMeDX scaffolds compared to PDX fibrous mat. However, it is noteworthy that significant increases in surface hydrophilicity may reduce cell attachment and subsequently cell proliferation.45
Fig. 11 shows the fluorescence microscopy images of HDFs cultured on scaffolds after 1 and 7 days, respectively. The cells appear as bright blue dots as indicated by red arrows in Fig. 11. Almost no cell infiltration was observed in PDX and 90/10 scaffold; the cells were attached on the scaffold surface only. A possible reason could be because the cavities (voids) formed in the electrospun PDX and 90/10 scaffolds were too small, which inhibited cell migration.46 Cell infiltration seems to be favoured in the 85/15 scaffold at day 1, but at day 7 the cell interaction is limited to a confluent layer at the scaffold surface. Since cells resided on the surface for PDX and 90/10 scaffolds, quantitative cell infiltration/migration resulted in 0% cell migration through the scaffold over 7 days. The 85/15 scaffold on day 1 was unable to be analyzed for cellular migration since the cell layer delaminated upon cryosectioning. Cellular infiltration within the nanofibrous scaffolds indicates 3D cell growth. HDF cells penetrated deeper into the 98/2 scaffold while in the case of the PDX scaffold the cells remained on the surface only. Moreover, a higher number of HDF cells penetrated the electrospun 98/2 scaffold at day 7. Cell migration analysis revealed that on average, the cells migrated up to a maximum of 45.1% (±11.8%) through the 98/2 scaffold after 7 days. While on day 1, all cells resided on the 98/2 scaffold surface (0% migration) indicating that the cells observed within the scaffold after 7 days are a result of migration and not initial cell seeding.
 | ||
| Fig. 11 Fluorescence microscopy images of HDFs on electrospun PDX/PMeDX scaffolds after 1 and 7 days. The left side of each cross section corresponds to the scaffold surface seeded with cells. Scale bar = 200 microns. | ||
In summary, better human dermal fibroblasts attachment was observed on electrospun PDX/PMeDX mats compared to electrospun PDX. Moreover, better cell infiltration was noted in the electrospun 98/2 mat possibly due to better conducive structural, mechanical and degradability properties.
Conclusions
Random electrospun mats with fibre diameters ranging from 0.39 to 0.65 μm were successfully prepared from HFIP solution of PDX and PMeDX blends. We have shown that contrary to blend films, the presence of PMeDX in the electrospun fibre does not affect PDX melting transition. We also showed that PMeDX had reduced plasticizing effect in the fibres compared to films with the possibility of weak interactions occurring between the crystalline phase of PDX and that of the PMeDX. AFM images confirmed a morphological heterogeneity, which is consistent with the proposed immiscibility of PDX and PMeDX in the electrospun fibres. The mechanical properties of the films were dependent on the overall crystallinity of the blends, but in the case of the fibres, the latter was found to be dependent on the fibre diameter and fibre packing densities. We demonstrated that electrospun fibres with PMeDX were slightly less thermally stable than PDX fibres, whereas in the case of blend films, thermal stability decreased significantly with increasing PMeDX content. Thermal degradation studies indicated that the activation energy of electrospun mats was almost twice that of films. In addition to crystallinity effects, the hydrolytic degradation of the electrospun mats was found to be highly dependent on the fibre diameter of the fibres. Electrospun mats degraded via surface erosion compared to bulk degradation for blend films. In addition, in vitro cell culture studies conducted using human dermal fibroblasts showed that the electrospun PDX/PMeDX scaffolds supported cell attachment and proliferation. Compared to electrospun PDX, a greater density of viable cells was observed on the PDX/PMeDX scaffolds, showing the potential of these electrospun poly(ester-ether) blend nanofibrous scaffold for tissue engineering applications.Acknowledgements
We thank the Tertiary Education Commission (Mauritius) for awarding a PhD scholarship to Nowsheen Goonoo. The authors acknowledge Marc Steuber and Stephan Handschuh (University of Siegen) for their contributions in performing some of the SEM measurements and the contact angle measurements, respectively. Daniel Wesner and Holger Schönherr acknowledge partial financial support from the Deutsche Forschungsgemeinschaft (DFG instrument grant no. INST 221/87-1FUGG) and the European Research Council (ERC starting grant to HS, ERC grant agreement no. 279202).Notes and references
- T. Christoforou and C. Doumanidis, J. Nanosci. Nanotechnol., 2010, 10, 6226–6233 CrossRef CAS PubMed.
- E. D. Boland, B. D. Coleman, C. P. Barnes, D. G. Simpson, G. E. Wnek and G. L. Bowlin, Acta Biomater., 2005, 1, 115–123 CrossRef PubMed.
- M. J. McClure, S. A. Sell, C. P. Barnes, W. C. Bowen and G. L. Bowlin, J. Eng. Fibers Fabr., 2008, 3, 1–10 CAS.
- S. A. Sell, M. J. McClure, C. P. Barnes, D. C. Knapp, B. H. Walpoth, D. G. Simpson and G. L. Bowlin, Biomed. Mater., 2006, 1, 72–80 CrossRef CAS PubMed.
- M. McManus, S. A. Sell, W. C. Bowen, M. Koo, D. G. Simpson and G. L. Bowlin, J. Eng. Fibers Fabr., 2008, 3, 12–22 CAS.
- V. Thomas, X. Zhang and Y. K. Vohra, Biotechnol. Bioeng., 2009, 104, 1025–1033 CrossRef CAS PubMed.
- P. A. Madurantakam, I. A. Rodriguez, C. P. Cost, R. Viswanathan, D. G. Simpson, M. J. Beckman, P. C. Moon and G. L. Bowlin, Biomaterials, 2009, 30, 5456–5464 CrossRef CAS PubMed.
- I. A. Rodriguez, P. A. Madurantakam, J. M. Mc Cool, S. A. Sell, H. Yang, P. C. Moon and G. L. Bowlin, Int. J. Biomater., 2012, 159484 Search PubMed.
- P. S. Wolfe, Y. Lochee, D. Jhurry, A. Bhaw-Luximon and G. L. Bowlin, J. Eng. Fibers Fabr., 2011, 6, 60–69 CAS.
- N. Goonoo, A. Bhaw-Luximon, I. A. Rodriguez, G. L. Bowlin and D. Jhurry, Int. J. Polym. Mat. Polym. Biomat., 2013 DOI:10.1080/00914037.2013.854224.
- N. Goonoo, A. Bhaw-Luximon, G. L. Bowlin and D. Jhurry, Polym. Int., 2013, 62, 523–533 CrossRef CAS.
- Y. Lochee, D. Jhurry, A. Bhaw-Luximon and A. Kalangos, Polym. Int., 2010, 59, 1310–1318 CrossRef CAS.
- S. Sell, C. Barnes, D. Simpson and G. Bowlin, J. Biomed. Mater. Res. A, 2008, 85, 115–126 CrossRef PubMed.
- L. H. Van Vlack, Introduction to Materials Science and Engineering, Addison Wesley, 6th edn, 1989, pp. 8–556 Search PubMed.
- W. F. Smith, Principles of Materials Science and Engineering, McGraw Hill, 2nd edn, 1990, p. 225 Search PubMed.
- B. R. Dewey, Introduction to Engineering Computing, McGraw Hill, 2nd edn, 1995, p. 69 Search PubMed.
- J. H. Han, C. Choi-Feng, D. Li and C. D. Han, Polymer, 1995, 36, 2451–2462 CrossRef CAS.
- R. D. Cardwell, L. A. Dahlgren and A. S. Golstein, J. Tissue Eng. Regen. Med., 2012 DOI:10.1002/term.1589.
- E. Saino, M. L. Focarete, C. Gualandi, E. Emanuele, A. I. Cornaglia, M. Imbriani and L. Visia, Biomacromolecules, 2011, 12, 1900–1911 CrossRef CAS PubMed.
- J. Han, C. J. Branford-White and L. M. Zhu, Carbohydr. Polym., 2010, 79, 214–218 CrossRef CAS PubMed.
- S. R. Son, N. T. Bah-Linh, H. M. Yang and B. T. Lee, Sci. Technol. Adv. Mater, 2013, 14, 015009 CrossRef.
- G. K. Shah and A. K. Gupta, Nanosystems: Phys. Chem. Math., 2013, 4(2), 288–293 Search PubMed.
- M. M. Tomadakis and T. J. Robertson, J. Chem. Phys., 2003, 119, 1741–1749 CrossRef CAS.
- G. C. Rutledge, J. L. Lowery and C. L. Pai, J. Eng. Fibers, 2009, 4, 1–13 CAS.
- K. Ishikiriyama, M. Pyda, G. Zhang, T. Forschner, J. Grebowicz and B. Wunderlich, J. Macromol. Sci. Phys. Ed. B, 1998, 37, 27–44 CrossRef.
- M. Dias, M. C. M. Antunes, A. R. Santos and M. I. Felisberti, J. Mater. Sci. Mater. Med., 2008, 19, 3535–3544 CrossRef CAS PubMed.
- M. A. Sabino, J. L. Feijoo, O. Nunez and D. Ajami, J. Mater. Sci., 2002, 37, 35–40 CrossRef CAS.
- M. A. Sabino, G. Ronca and A. J. Muller, J. Mater. Sci., 2000, 35, 5071–5084 CrossRef CAS.
- N. Goonoo, A. Bhaw-Luximon, G. L. Bowlin and D. Jhurry, Ind. Eng. Chem. Res., 2012, 51, 12031–12040 CrossRef CAS.
- B. Wunderlich, Macromolecular Physics: Crystal Nucleation, Growth and Annealing, Academic Press, New York, 1976, vol. 2 Search PubMed.
- M. G. Freire, A. R. R. Teles, R. A. S. Ferreira, L. D. Carlos, J. A. Lopes da Silva and J. A. P. Coutinho, Green Chem., 2011, 13, 3173–3180 RSC.
- A. Bianco, M. Calderone and I. Cacciotti, Mater. Sci. Eng., C, 2013, 33, 1067–1077 CrossRef CAS PubMed.
- C. L. Pai, PhD Thesis – MIT, 2011. Morphology and mechanical properties of electrospun polymeric fibers and their non-woven fabrics.
- S. C. Wong, A. Baji and S. W. Leng, Polymer, 2008, 49, 4713–4722 CrossRef CAS PubMed.
- J. Stitzel, R. Smith, D. Simpson, G. Wnek and G. L. Bowlin, Ann. Biomed. Eng., 2000, 28 Search PubMed.
- M. L. Cheng, P. Y. Chen, C. H. Lan and Y. M. Sun, Polymer, 2011, 52, 1391–1401 CrossRef CAS PubMed.
- J. N. Im, J. K. Kim, H. K. Kim, C. H. In, K. Y. Lee and W. H. Park, Polym. Degrad. Stab., 2007, 92, 667–674 CrossRef CAS PubMed.
- W. Cui, L. Xiaohong, S. Zhou and J. Weng, Polym. Degrad. Stab., 2008, 93, 731–738 CrossRef CAS PubMed.
- T. W. Chung, D. Z. Liu, S. Y. Wang and S. S. Wang, Biomaterials, 2003, 24, 4655–4661 CrossRef CAS.
- S. Nemir and J. L. West, Ann. Biomed. Eng., 2010, 38, 2–20 CrossRef PubMed.
- X. Hu, S. H. Park, E. S. Gil, X. X. Xia, A. S. Weiss and D. L. Kaplan, Biomaterials, 2011, 32, 8979–8989 CrossRef CAS PubMed.
- P. Van der Valk and A. Van Pelt, J. Biomed. Mater. Res., 1983, 17, 807–817 CrossRef CAS PubMed.
- E. Biazar, M. Heidari, A. Asefnezhad and N. Montazeri, Int. J. Nanomedicine, 2011, 631–639 CrossRef CAS PubMed.
- C. H. Kim, M. S. Khil, H. Y. Kim, H. U. Lee and K. Y. Jahng, J. Biomed. Mater. Res., 2006, 78, 283–290 CrossRef PubMed.
- M. Lampin, R. Warocquier-Clerout, C. Legris, M. Degrange and M. F. Sigot-Luizard, J. Biomed. Mater. Res., 1997, 36, 99–108 CrossRef CAS.
- Y. Zhang, H. Ouyang, C. T. Lim, S. Ramakrishna and Z. M. Huang, J. Biomed. Mater. Res. B, 2005, 72B, 156–165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60211g |
| This journal is © The Royal Society of Chemistry 2014 |