Modulation of host osseointegration during bone regeneration by controlling exogenous stem cell differentiation using a material approach†
Xiaohua
Yu
*a,
Liping
Wang
b,
Zengmin
Xia
a,
Li
Chen
b,
Xi
Jiang
b,
David
Rowe
b and
Mei
Wei
*a
aDepartment of Materials Science and Engineering, University of Connecticut, Storrs, CT 06269, USA. E-mail: m.wei@ims.uconn.edu; xhyu2936@gmail.com
bDepartment of Reconstructive Sciences, School of Dental Medicine, University of Connecticut Health Center, Farmington, CT 06030, USA
First published on 23rd October 2013
Abstract
Stem cell-based tissue engineering for large bone defect healing has attracted enormous attention in regenerative medicine. However, sufficient osseointegration of the grafts combined with exogenous stem cells still remains a major challenge. Here, we developed a material approach to modulate the integration of the grafts to the host tissue when exogenous bone marrow stromal cells (BMSCs) were used as donor cells. Distinctive osseointegration of bone grafts was observed as we varied the content of hydroxyapatite (HA) in the tissue scaffolds implanted in a mouse femur model. More than 80% of new bone was formed in the first two weeks of implantation in high HA content scaffolds but host integration was poor, while only less than 5% of the new bone was formed during this time period in the No HA group but the host integration was good. Cell origin analysis leveraging the GFP reporter indicates new bone in the HA containing groups was mainly derived from donor BMSCs. In comparison, both host and donor cells were found on the new bone surface in the No HA groups which led to seamless bridging between host tissue and the scaffold. Most importantly, host integration during bone formation is closely dictated by the content of HA present in the scaffolds. Taken together, we demonstrate a material approach to modulate the osseointegration of bone grafts in the context of an exogenous stem cell-based bone healing strategy which might lead to fully functional bone tissue regeneration.
Introduction
The repair of large bone defects caused by trauma, disease, or surgical intervention still remains a major challenge in the clinic.1,2 Although autograft has been regarded as the current “gold standard” for bone defect repair, it severely suffers from a number of drawbacks such as donor site morbidity, lack of availability and the need for tissue harvest from another anatomic location.3 Allograft as an alternative approach is also limited by its own problems such as disease transmission, immunogenicity and high failure rate.4 To overcome the obstacles associated with these conventional approaches during large size bone defect healing, bone tissue engineering (BTE) combining scaffolds, stem cells and growth factors together with engineering principles offers a promising strategy to develop new bone substitutes.5–7 Recently, a stem cell-based BTE approach has attracted more and more researchers’ attention due to the new developments in stem cell biology such as pluripotent stem cells and their great potential in tissue engineering.8,9Osseointegration, the formation of direct bonding between regenerated bone and host tissue, is an indispensable requirement to achieve functional bone tissue.10,11 The osseointegration of bone grafts is of particular importance in the context of the stem cell based BTE approach since the newly generated tissues by BTE can only be functional when they are perfectly integrated with the host tissues.12 However, the integration of tissue engineered bone grafts may be very limited due to the local environment at the defect sites.13 Thus, multiple approaches have been developed to enhance the host–donor cell interactions in order to improve the integration of regenerated tissue. For example, delivery of undifferentiated mesenchymal stem cells can actively recruit stem/progenitor cells from the host into the defect zones.14 Tasso et al. found that two consecutive cell waves were recruited from host tissue and homed into the bone defects.15 Controlled release of chemotaxis cytokines such as stromal derived factor (SDF) is another approach to address this problem.16 However, leveraging a material approach to modulate the osseointegration of bone grafts has not been fully explored yet.
On the other hand, calcium phosphate (CaP)-based bioceramics, such as hydroxyapatite (HA) and tricalcium phosphate (TCP), have been used for bone tissue repair for decades due to their compositional similarity to natural bone.17 In a stem cell based approach, these bioceramics can be used as carrier material for stem cells delivery in different forms such as fillers, scaffolds and cements.18–20 Most recent reports have shown HA could actively enhance osteoprogenitor cell differentiation both in vitro and in vivo and eventually lead to osteogenesis.21–23 Yuan et al. demonstrated that porous CaP ceramic materials had equal performance in healing a critical bone defect compared to autograft or BMP-2 with excellent integration to host bone.21 Therefore, tuning physicochemical properties of biomaterials to control the differentiation of stem cells upon delivery might provide us with another alternative to modulate the osseointegration of bone grafts.
In this study, we hypothesized that the differentiation of donor bone marrow stromal cells (BMSCs) upon delivery can be controlled by a simple materials approach – incorporation of various amounts of HA into the scaffolds. Guided exogenous stem cell differentiation would then be leveraged to influence the host cell recruitment which can lead to the modulation of host integration during critical bone defect healing. To validate this hypothesis, a tissue engineering approach using a combination of both scaffold and stem cells was designed (Fig. 1): we first prepared a series of collagen–HA scaffolds with varying HA contents using a co-precipitation method developed earlier in our laboratory.24 BMSCs were isolated from transgenic mice harbouring a GFP reporter associated with osteoblasts. The impact of different materials compositions on the osseointegration of the scaffolds was evaluated using a mouse critical-size segmental bone defect model. A series of quantitative and qualitative techniques were employed to assess the bone healing at the defect sites, with particular focus on bone formation kinetics, donor cell differentiation and host cell involvement.
 | ||
| Fig. 1 Schematic of 3D scaffold fabrication and implantation. Soluble type I collagen is mixed with m-SBF and the pH of the mixture is adjusted to 6.80. The collagen–m-SBF solution is incubated at 42 °C for 24 h to allow the HA crystal growth along the collagen fibers. The collagen–HA slurry is then collected in a 35 mm dish and freeze-dried to form the 3D porous scaffold. Expanded BMSC is loaded on the scaffold before implantation. The BMSC–scaffold construct is finally implanted into a critical defect created on the femur of the mice. | ||
Experimental
Scaffold fabrication and characterization
Collagen–HA (Col–HA) scaffolds with varying HA contents were prepared by modifying the biomimetic co-precipitation method described previously.25 Briefly, type I collagen was extracted from rat tail tendon following the protocol by Rajan et al.26 Modified simulated body fluid (m-SBF) solutions with different collagen contents were then prepared as previously reported.27 To adjust the content of HA in the scaffold, the concentration of collagen in m-SBF was varied from 1.0 to 5.0 g L−1. The pH of the collagen–m-SBF solution was adjusted to 6.80 by adding 5 M NaOH dropwise with gentle stirring. The collagen–m-SBF solution was then kept stirring in a water bath at 42 °C for 24 h. After incubation, the resulting gel was centrifuged at 1.0 × 104 rpm for 15 min and washed with de-ionized water (DI water) three times after removing the supernatant. The obtained pellets were then placed in a 35 mm Petri dish and further compressed into a disc shape. Finally, the scaffolds with different HA contents were cross-linked using 2% 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) at 4 °C for 48 h and then rinsed with de-ionized water. The cross-linked pellet was treated with 0.1 M glycine aqueous solution to block non-reacted carboxyl groups. The collagen–apatite scaffolds were then rinsed with de-ionized water, and freeze-dried again. The dried scaffolds were cut into 3.5 mm × 5.0 mm × 1.0 mm pieces using a razor blade. The morphology of all scaffolds with different HA contents was examined using field emission scanning electron microscopy (FESEM, JEOL 6335F, Japan) at 5 kV after being coated with platinum. Thermogravimetry analysis (TGA, TA Instruments 2950) was used to measure the HA content in each group of scaffolds. The scaffolds were also examined by Fourier-transform infrared spectroscopy (FTIR, Nicolet Avatar 360) in the range of 4000–400 cm−1 with an average scan number of 128.Transgenic mice
To identify donor cells from host tissues, established transgenic mouse line bearing pOBCol3.6Cyan (blue) expression constructs were used as donor cell providers in this study. The pOBCol3.6GFP transgene is associated with osteoblast differentiation. The pOBCol3.6Cyan transgenic mice have been characterized previously.28 All the transgenic mice were maintained as homozygous breeders on a CD1 background.Donor cell isolation and expansion in vitro
BMSCs were derived from pOBCol3.6GFPCyan transgenic mice. Six to eight-week-old mice were sacrificed using CO2 asphyxiation. Femurs and tibias were dissected from surrounding tissues. Bone marrow was harvested by flushing bones with α-MEM medium containing 10% FBS using a 25-G needle. The cells were filtered through a 70 μm cell strainer and plated at a density of 1 × 108 cells in 100 mm culture dish. Half of the culture medium was replaced by fresh medium to remove the non-adherent cells at day 4. The cells were allowed to grow for 7–8 days in α-MEM containing 10% FBS. BMSCs at a pre-confluent stage were harvested in 0.25% trypsin–EDTA and re-suspended in α-MEM at a concentration of 1.0 × 106 cells per mL.Animal surgical procedure
All surgeries were performed under a protocol approved by the University of Connecticut Health Center Animal Care Committee. A critically-sized femoral segmental defect mouse model was used in this study. A total of 24 immunodeficient nude mice were used as the recipients and they were randomly divided into three experimental groups based on the HA content in the Col–HA scaffold (n = 8): (1) No HA group (containing 0% HA), (2) Low HA group (containing ∼25% HA) and (3) High HA group (containing ∼45% HA). Briefly, the mice were anesthetized with a ketamine–xylazine mixture (135 mg kg−1 and 15 mg kg−1). The surgical procedures are shown in Fig. S2.† An approximately 15 mm long skin incision was cut over the femur and underlying soft tissues were retracted to reveal the bone. Bilateral 3.0 mm segmental defects were created in the mid-femoral diaphysis of the mice. The limbs were stabilized by inserting a custom-made radiolucent internal fixation pin into the medullary canal of femur. To further stabilize the defect containing femur, a custom fixation plate was applied and tightened on the femur with suture. BMSCs at a dosage of 1.0 × 106 cells per scaffold were loaded into each pre-hydrated scaffold 5 min before implantation; then the scaffolds with different HA contents were wrapped along the internal pin and tied by suture. The animals were resumed to normal ambulation and behaviour after they recovered from anesthetization.Time-lapse radiological examination
New bone growth in defect sites was assessed using X-ray radiological examination. The mice were anesthetized using isoflurane and imaged using a digital microradiography unit (Faxitron X-ray LX 60, USA) 1, 14, 28, and 42 days after surgery. The X-ray photographs were taken at 30 kV and exposed for 2 s. Quantitative new bone formation was analyzed by measuring the radiopacity of host bone and new bone with an ImageJ software (National Institutes of Health, Bethesda, MD).29 Briefly, the radiopacity of the femoral defect area was measured by ImageJ, and the relative radiopacity was calculated by dividing the defect area radiopacity with host bone radiopacity.Micro-computerized tomography (micro-CT) imaging
All the mice were sacrificed at day 42 post surgery. Xylenol orange (XO) (30 mg mL−1, 90 mg kg−1), a fluorochrome chelating reagent which labels newly calcified tissue in vivo, was given to each mouse through intraperitoneal injection 24 h prior to the sacrifice. 3D structure of new bone and quantitative bone formation was assessed by 3D micro computed tomography (μCT40, Scanco Medical AG, Switzerland). Serial tomographic images were acquired at 55 kV and 145 μA, collecting 1000 projections per rotation at 300 ms integration time. Three-dimensional 16 bit greyscale images were reconstructed using standard convolution back-projection algorithms with Shepp–Logan filters. Bone volume per defect was recorded as the measurement of defect bone regeneration.Cell origin analysis
The femur explants were transferred into 30% sucrose–PBS solution and soaked overnight after IVIS imaging. The samples were subsequently embedded using a frozen specimen embedding media (Cryomatrix, Thermo-Shandon, USA). Sagittal cryosections (7 μm) of the femur were obtained with a Leica CM 3050S cryostat (Wetzlar, Germany) using a tape transfer system (Cryofilm Type 2C, Hiroshima, Japan).30 After re-hydration in PBS, the fluorescent signals in the femoral sections were examined using a Zeiss Axio Imager Z1 (Carl Zeiss, Thornwood, NY) equipped for fluorescence imaging and a computer-controlled mechanical stage as previously described.31 XO was detected with a TRITC (red) filter (Chroma Technology, Bellows Falls, VT #31002), Col3.6Cyan with a CFP (blue) filter (cat. #49001). A differential interference contrast (DIC) image was acquired at the same time as the endogenous fluorescence imaging. All images were taken in air and recorded using a Zeiss Axio digital camera. Image processing was performed using AxiaVsion 4.7 (Zeiss, Germany) and Adobe Photoshop (Adobe Systems, San Jose, CA).Histological analysis
Following detection and imaging of GFP expression, all the slides were soaked in PBS for 60 min to remove the cover slides. They were then decalcified in 15% ethylene-diamine tetraacetic acid (EDTA) overnight at room temperature. The sections of all three groups were stained with hematoxylin and eosin (H&E) after decalcification. Images of H&E staining were recorded using a Zeiss Axio Imager Z1 microscope with a RGB chromogenic filter set (Zeiss, #487933).Statistical analysis
All quantitative data were given as mean ± standard deviation. The numerical results obtained in this study were subjected to analysis of variance (ANOVA). The significance of the results was evaluated at a significance level of p < 0.05 based on the p value of comparison.Results
Scaffold characterization
It was found that the HA content of the scaffold dropped from ∼60% to about 5% as we raised the collagen concentration from 1 g L−1 to 5 g L−1 in the co-precipitation suspension (Fig. S1A†). TGA data showed that the three groups of Col–HA scaffolds used in this study have HA contents of 0%, 23%, and 45%, respectively (Fig. 2A). The FTIR spectra of the scaffolds clearly display the characteristic peaks of collagen as well as the phosphate peaks from hydroxyapatite (Fig. 2B). XRD spectra showed that the mineral phase in scaffold was poorly crystallined hydroxyapatite (Fig. S1B†). The morphology of the collagen–apatite scaffolds with different HA contents are shown in Fig. 2C. The interconnectivity of scaffolds was maintained with the change of HA contents in the scaffold. However, as the content of HA in scaffold increased, the scaffold porosity tended to decrease. The pore size of the scaffolds also decreased when the content of HA became high.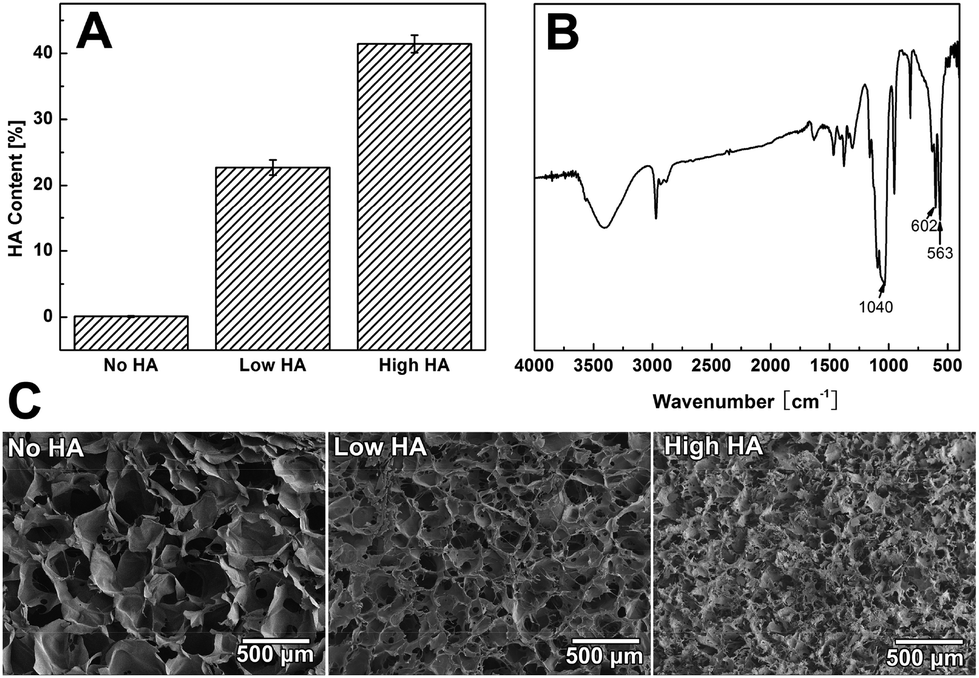 | ||
| Fig. 2 Scaffold characterization: (A) HA content in collagen–HA scaffolds was characterized by TGA. The percentage of HA in the scaffolds was controlled by varying the concentration of collagen during processing. (B) FITR spectra of collagen–HA scaffolds with different HA contents. (C) Morphology of scaffold by SEM: No HA group, Low HA group, and High HA Group. | ||
Time-lapse radiological evaluation
Time-lapse radiographs were taken at different time points for qualitative assessment of new bone formation. Bone defect without scaffold/cell was regarded as a control group and remained unbridged after 42 days (Fig. S3†). In comparison, the defect loaded with collagen scaffold (No HA) only showed some new bone formation at the two edges of the defect but was not bridged (Fig. S4†). Largely improved bone formation was observed in scaffolds with varying HA contents when they were loaded with donor BMSCs (Fig. 3): for the No HA group, small amounts of new bone originating from the cut native ends grew toward the defect zone at day 14, then the mineralized tissue became connected from both edges of the defect at day 28. The whole defect was completely bridged with mineralized tissue at day 42. In the Low HA group, newly formed bone tissue almost covered the whole defect area and no gap could be found between the new bone and the native bony tissue at day 14. The radiopaque tissue started to form a callus-like structure along the defect at day 28 and seamlessly bridged the defect at day 42. In the High HA group, a clear gap between the defect and the cutting end of the femur was observed throughout the whole implantation period, thus the defect was not bridged by this treatment (Fig. 3A, upper panel). The bone formation kinetics was studied by quantifying the radiopaque tissue in the defect area. More than 60% of bone formation took place in the first 14 days in the High HA group whereas only less than 5% of new bone formed during this period in No HA group. Bone growth in the High HA group started to slow down from day 14 and almost plateaued at day 28. Meanwhile, steady bone formation was observed in the No HA group on days 14 and 28. As for the Low HA group, the bone formation rate was always between the other two groups (Fig. 3B). However, no statistical difference was found between the groups in terms of total bone formation at day 42 (Fig. 3C).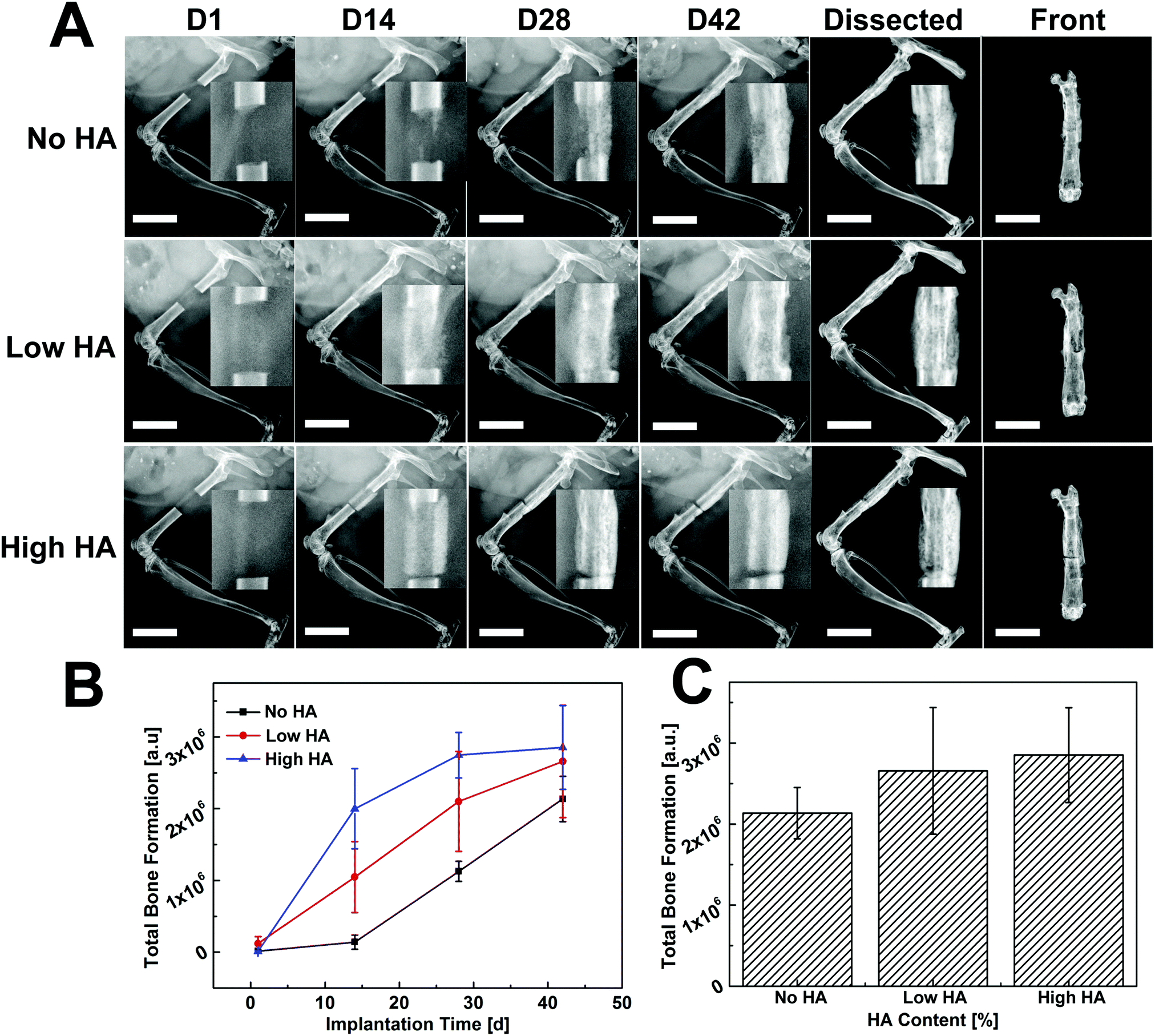 | ||
| Fig. 3 Bone formation kinetics examined by radiograph for collagen–HA scaffolds with different HA content. (A) Time lapse radiographs of segmental defects loaded with different scaffolds. New bone formation in No HA groups was initiated from host tissue while bone formation from HA-containing groups was originated from donor MSCs on the scaffolds at day 14. A gap can be observed between the defect and host bone in High HA group at day 42. Inset: high resolution radiograph of the defect sites. (B) The kinetics of bone formation through a time course of 42 days. Bone formation in HA containing groups took place much rapidly and majority of the new bone was formed within the first 28 days; new bone growth in the No HA group was delayed in the first two weeks and then accelerated between 14 and 42 days. (C) Comparison of total bone formation 42 days post-transplantation. Scale bar: 5.0 mm. | ||
Micro-CT imaging and quantification
Micro-CT images showed that the segmental defect was fully filled by newly formed bone in all groups, and substantial bone distribution differences were still observed (Fig. 4). Specifically, a gap between the new bone and native bone was shown in the HA-containing groups from all three angles of micro-CT images. Meanwhile, no gap was found at the interface between the defect and the host bone in the No HA group (Fig. 4A). The quantification of bone volume indicated that the High HA group had significantly more bone formation in the defect compared to the No HA and Low HA groups (Fig. 4B). However, the total bone volume/total volume ratio (BT/TV) did not show any statistical difference among the three groups (Fig. 4C). The micro-CT analysis elaborates that more bone formation could be achieved with high HA content scaffold but the osseointegration of the grafts was not improved.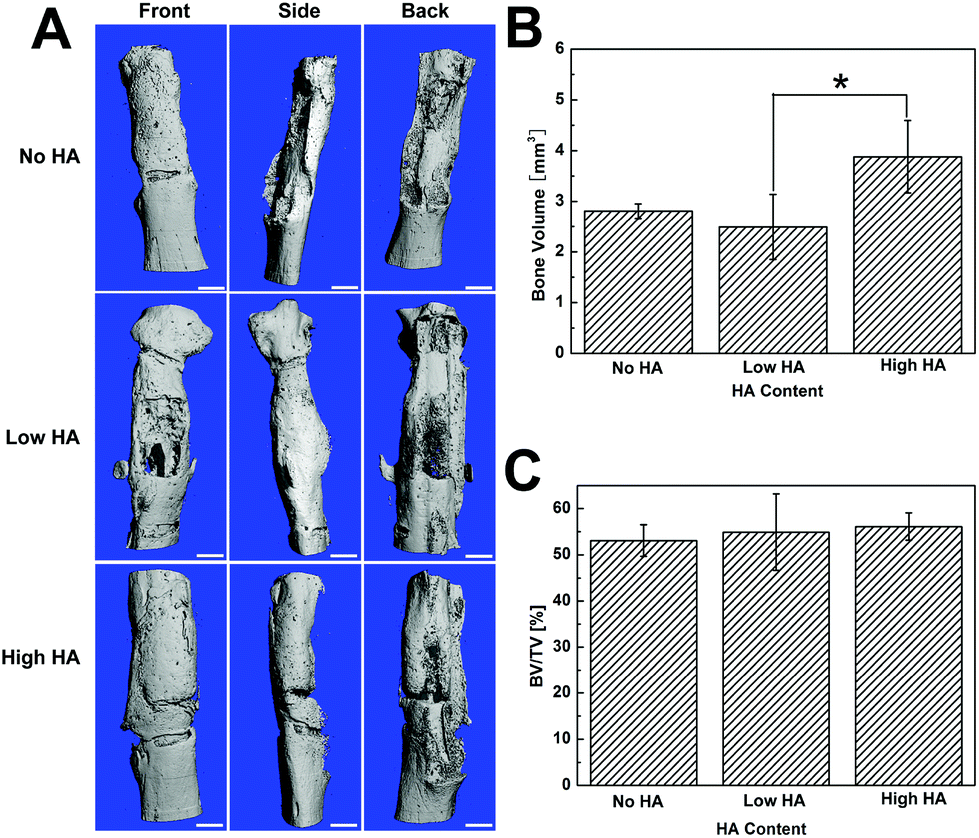 | ||
| Fig. 4 Micro-CT analysis of bone regeneration at day 42. (A) Micro-CT illustrates that all the defects were filled with new bone, but the No HA group showed better host integration when the gap between new bone and host bone was observed in HA-containing groups. (B) Quantification of regenerated total bone volume. (C) Quantification of percentage of new bone in the defect volume. (D) Local density of regenerated bone. The scale bars are 1.0 mm. | ||
Cell origin and contribution analysis at bone defect sites
The GFP reporter carried by the donor BMSCs was leveraged to analyse cell origin and contribution during bone formation. The newly formed bone in the defects was clearly displayed in all groups by DIC channel in Fig. 5 – Composite (gross view of the whole defect is shown in Fig. S5†). Seamless bridging at the defect–bone interface was only found in the No HA group (Fig. 5A). Similar gaps shown under X-ray were found again in both Low and High HA groups (Fig. 5B and C). These results indicate that the presence of HA in the scaffold had a remarkable impact on osseointegration of the scaffolds.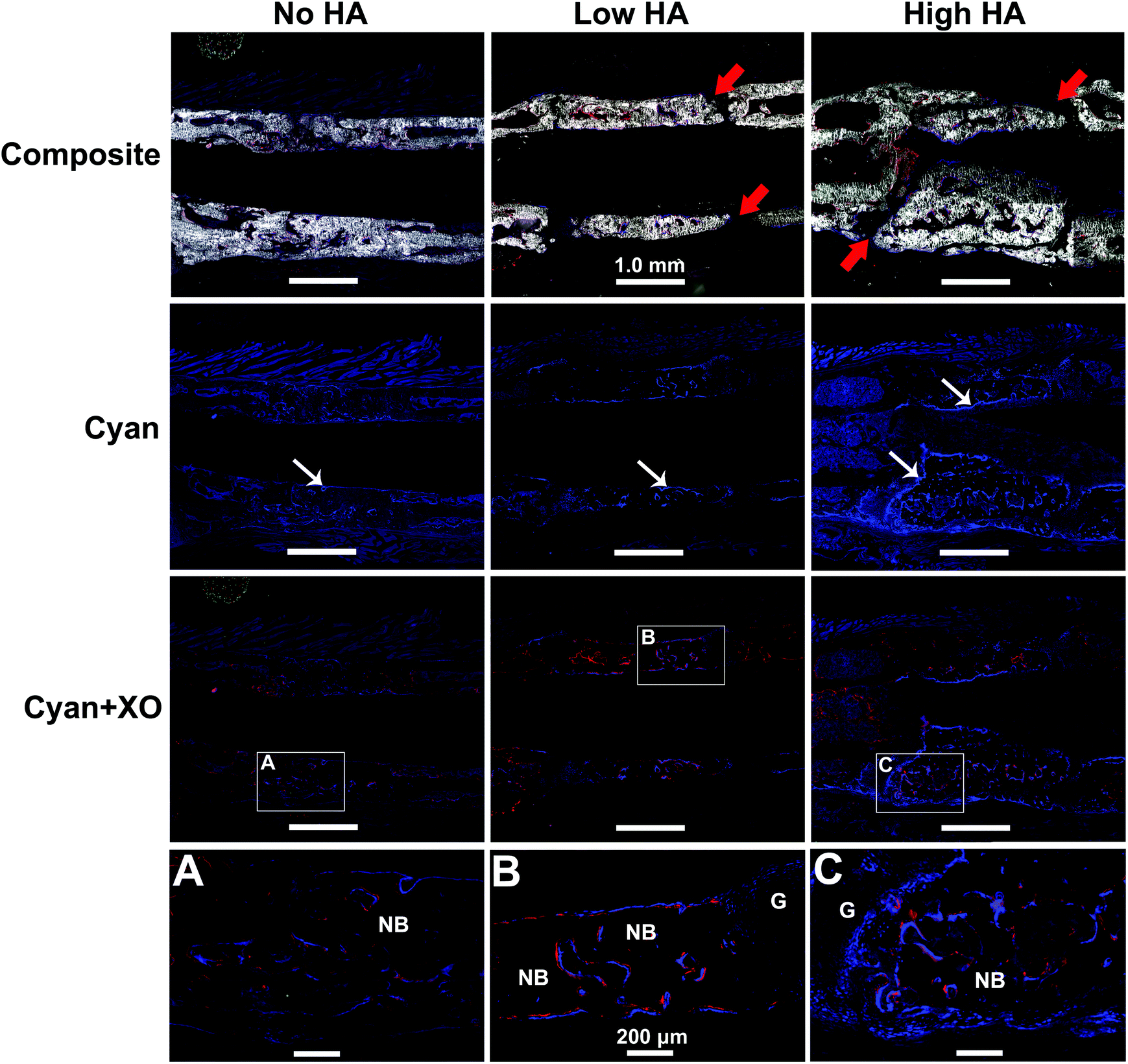 | ||
| Fig. 5 Fluorescent imaging analysis of cryosections of the implanted constructs at day 42: Composite: DIC channel combined with two fluorescence channels; Cyan: Cyan channel for blue fluorescence signal from pOBCol3.6Cyan cells, Cyan + XO: Cyan channel for blue fluorescence signal from pOBCol3.6Cyan cells plus red channel for red fluorescence signal from xylene orange. (A) Representative image of defect zone in the No HA group. (B) Representative image of defect zone in the Low HA group. (C) Representative image of defect zone in the High HA group. The new bone with the defect area was mainly derived from donor MSCs. More active Col3.6GFPCyan positive cells were found in HA-containing groups compared to the No HA group. Red arrows indicate unbridged gap; white arrows indicate donor cells; NB indicates new bone; G indicates unbridged gap. | ||
The expression of pOBCol3.6GFPCyan reporter in osteoblasts was confirmed by BMSC osteogenic differentiation in vitro (Fig. S6†) and IVIS scanning in vivo (Fig. S7†). The pOBCol3.6GFPCyan reporter was strongly expressed on the bone surface in all the tested groups by showing a bright blue signal within the defect area. However, the amount and the distribution of the reporter positive cells appeared to be remarkably distinct between tested groups (Fig. 5). The amount of positive donor cells increased as the HA content increased in the scaffolds (Fig. 5 – Cyan row). The distribution of positive pOBCol3.6GFPCyan cells in the defect was relatively sparse in the No HA group. Very little co-localization of Cyan and XO staining was found in this group (Fig. 5A). In contrast, the donor cells in the Low HA group did not only demonstrate a denser distribution, but also formed a continuous line along the new bone surface. Part of the XO staining was found to be co-localized with the Cyan signal. In the High HA group, the largest amounts of positive donor cells were found with the most intensive blue signal. The close association of Cyan and XO labelling in this group indicates that donor cells have dominated the bone formation in the defects (Fig. 5B and C).
Histological analysis
An overview of the defect femurs loaded with BMSCs/scaffold is shown in Fig. 6A–C. New bone formation can be observed in all three groups, but the osseointegration of bone grafts was closely associated with the HA content in the scaffolds which is in good agreement with radiographic and micro-CT results. In the No HA group, mineralized tissue successfully bridged the defect and the host bone, thus the border of the original defect could no longer be distinguished (Fig. 6D). No connective fibrous tissue was found at the interface of newly formed bone and host bone in this group (Fig. 6G). In the Low HA group, although the newly formed bone was well aligned with the host femur bone, two gaps at the two ends of the defect were clearly observed (Fig. 6E). These gaps were filled with large amount of fibroblast cells, which displayed a typical spindle-shaped morphology (Fig. 6H). In the third group (High HA group), several isolated bone patches were formed after 42 days of implantation. Gaps between new bone and host bone tissue were also observed in this group (Fig. 6F). However, some parts of the gaps were filled with cartilage-like tissue instead of fibrous tissue (Fig. 6I). It is also worth noting that when HA-containing scaffolds were implanted in the defects, more bone marrow was formed inside the new bone (Fig. 6F) which is consistent with our previous finding.24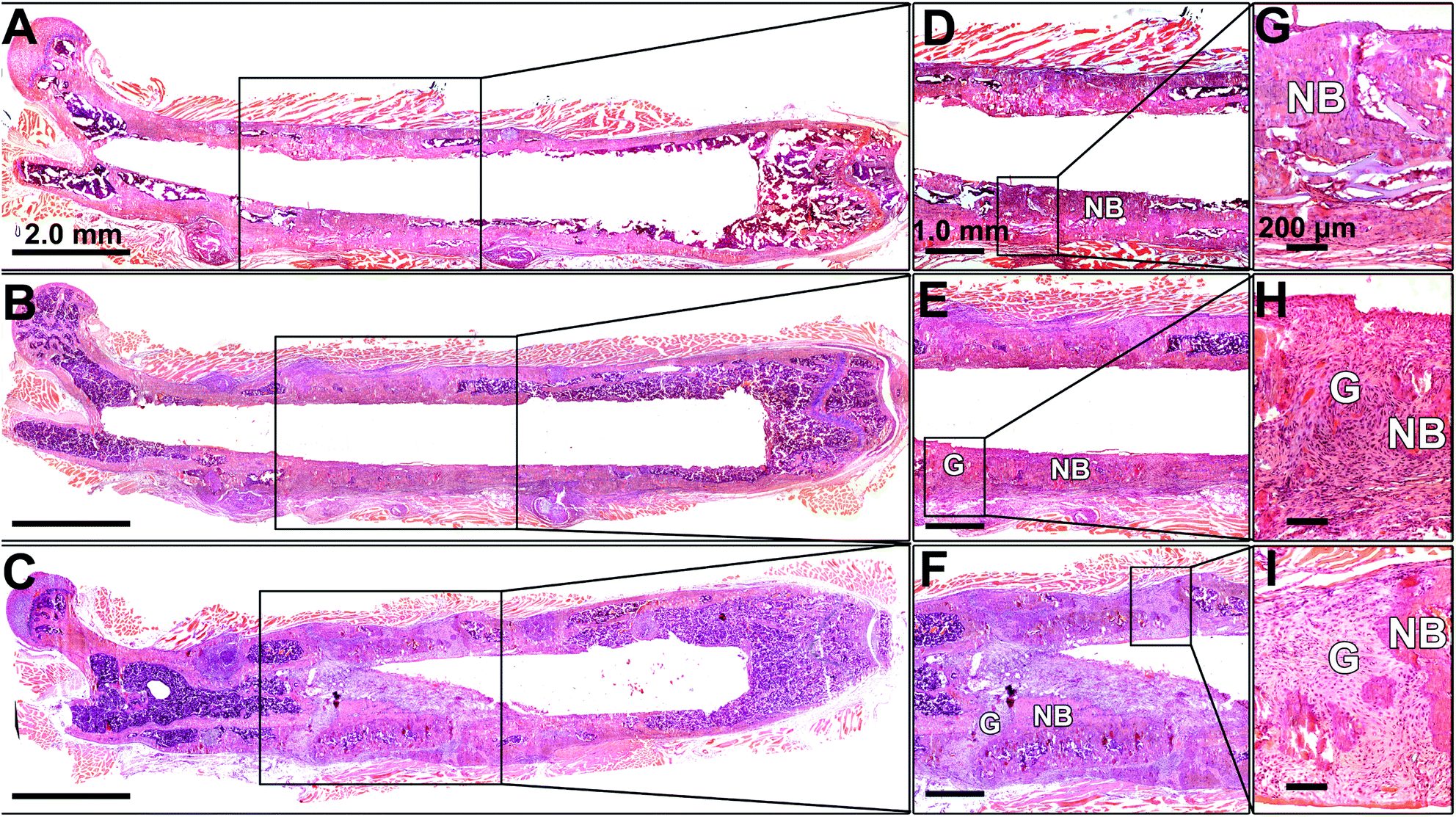 | ||
| Fig. 6 Histological photographs of H&E staining of the implanted constructs at day 42: (A) No HA group, (B) Low HA group and (C) High HA group show overview of the whole femur with segmental defect loaded with different scaffolds. (D) No HA group, (E) Low HA group and (F) High HA group show snapshots of the segmental defect zones. (G) No HA group, (H) Low HA group and (I) High HA group are representative images of high magnification photographs at the interface between new bone and host bone. NB indicates new bone; G indicates unbridged gap. | ||
The presence of HA in the scaffolds also affected the contribution of exogenous BMSCs to bone formation (Fig. 7). In the No HA group, only part of the new bone surface was lined with a layer of positive pOBCol3.6GFPCyan cells and the fluorescent intensity of the donor cells was not as strong as the HA containing groups (Fig. 7A and D). In comparison, a bright layer of donor cells was found on new bone surface in the Low and High HA groups (Fig. 7B, C, E and F). The donor cells (blue cells) were found co-localized with XO staining (red) on almost all the bone surfaces in the HA containing groups. Large amounts of donor cells can also be found in the space between the bone tissue in the HA containing groups. These histological data collectively revealed the crucial role of HA during bone defect healing in the presence of exogenous stem cells.
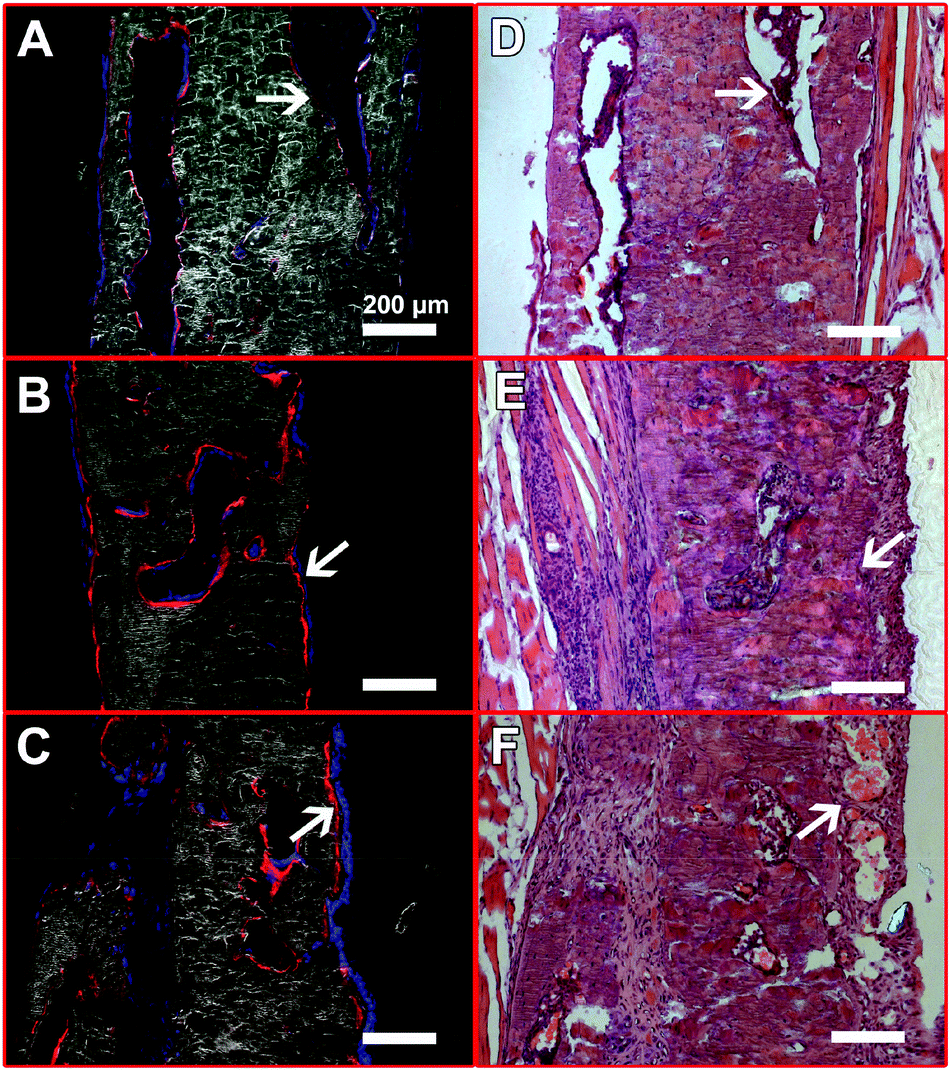 | ||
| Fig. 7 New bone formation contribution analysis by identifying donor derived cells (pOBCol3.6GFPCyan positive cells) from host cells on bone surface in the defects zone: (A) No HA, composite, (B) Low HA, composite, (C) High HA, composite, (D) No HA, H&E, (E) Low HA, H&E, (F) High HA, H&E The paired arrows with the same orientation indicate the same location on the samples. | ||
Discussion
Regeneration of functional bone tissue requires robust new bone formation associated with sufficient graft integration with the host tissue, therefore the modulation of osseointegration of bone grafts is of particular importance to the clinical success of bone regeneration. Although HA has been extensively used for bone defects repair due to its excellent biocompatibility and osteoconductivity,32–34 there have not been any systematic studies using this material to modulate the osseointegration of bone grafts in the presence of exogenous stem cells. In this study, we demonstrate that the osseointegration of bone grafts can be modulated by varying the amount of HA incorporated into the scaffolds. More importantly, the host integration of the bone grafts is strongly dictated by the composition of scaffold materials which is mainly referred to the amount of HA. Thus, the material approach demonstrated in this study may represent a particularly simple and adaptable method for efficient modulation of osseointegration.We found that new bone formation happened much earlier in the HA containing groups (Fig. 3). Furthermore, the new bone formation was derived from the donor BMSCs according to the co-localization of Cyan and XO (Fig. 5). None or little bone formation was found in the No HA group at the same time. The faster bone formation might be due to the presence of HA in these scaffolds. Although the mechanism at the cellular level for HA-mediated osteoblast differentiation is not clear yet, the observed effect may be closely related to the unique biophysical property of poorly crystalline hydroxyapatite. It has been well documented that hydroxyapatite had extremely high affinity to a diverse range of proteins and other molecules.35–37 The nano-size hydroxyapatite in the scaffold could provide large surface area for protein binding which might facilitate enrichment of signalling molecules such as endogenous growth factors on the scaffolds.38 These signalling molecules may positively affect the commitment of BMSCs into osteoblastic linage as well as their maturation.39 Besides, the soluble calcium ions released from the scaffold might also play an important role in donor stem cell differentiation. A recent study by Boer et al. identified a calcium-induced signalling cascade that could lead to hBMSCs osteogenic differentiation.40 The Col/HA scaffold was prepared by a co-precipitation technology where HA was deposited onto collagen fibers in m-SBF solution at a mild temperature, thus the HA in the scaffold was poorly crystallined and highly bio-resorbable. Our previous studies showed that this type of HA was dissolvable under a physiological environment and provided sufficient calcium ions to drive the calcium signalling pathway.27,41 Therefore, the presence of HA in the scaffolds might play an important role during exogenous stem cell differentiation and fate commitment.
The recruitment of host cells into bone defect is the key step to achieve satisfactory osseointegration of the bone grafts. Improved host participation was observed in No or Low HA groups during bone regeneration by showing better defect bridging but less donor cell involvement. We hypothesize that this phenomenon might be a combinational effect of both the exogenous cells and the composition of the scaffolds. Recent literature has shown that exogenous stem cells such as BMSCs could mobilize endogenous cells into lesions to mediate tissue repairing,42 but the host cell recruitment ability was only observed in undifferentiated stem cells.43 The BMSCs utilized in this study were only expanded briefly without exposure to any inductive factors, therefore these naive BMSCs were fully capable of recruiting host cells into the defect.43 This hypothesis is evidenced by the decreased number of donor cells present in the defects while the bone defect was maintained bridged (Fig. 7). However, the BMSCs did not induce such strong host cell ingrowth when they were encountered with high HA containing scaffold. This might be caused by rapid commitment of BMSCs into osteoprogenitor lineage which was probably triggered by the strong chemical cue from HA particles.22 As a result, less or no host cells were recruited to the defect zone and bone regeneration and poor host integration was obtained (Fig. 8). Similar mechanisms for the No HA group and Low HA group are also proposed (Fig. S8†). In the context of the current bone regeneration approach, the incorporation of HA in the scaffold can substantially accelerate the formation of new bone, but it can also impair the recruitment of host cells by exogenous BMSCs. Thus both osteogenic signals and host participating driving force should be taken into account and properly orchestrated in the design of bone tissue engineering strategies.44,45
 | ||
| Fig. 8 Proposed donor cell–host-scaffold interactions associated with new bone regeneration. (A) Early stage: upon the implantation of BMSCs loaded low HA scaffold, small amount of host progenitor cells migrate into the defect zone. (B) Medium stage: after one week, donor BMSCs start to differentiate into active osteoblast and deposit bone matrix under the regulation of HA in scaffold. Meanwhile, host derived progenitor cells also initiate bone formation towards defect area from the two ends. (C) Late stage: large amount of new bone is deposited by differentiated donor BMSCs and bridges with host derived new bone to heal the defect. The scaffold also starts to degrade associated with the remodeling of new bone at this stage. | ||
The cell fate of the exogenous BMSCs was controlled by the material composition in the scaffold, which provides a novel approach to post-transplantation stem cell fate control. Although the control of stem cell fate can be well done in vitro, there are very few approaches that could continue guiding stem cells differentiation post-transplantation.46 Therefore, most stem cells need to be pre-differentiated and matured in a culture system (e.g. bioreactor) prior to implantation.47,48 Un-differentiated BMSCs were successfully delivered and induced into osteoprogenitors, which demonstrates a typical post-transplantation cell fate control. The success of this approach is mainly attributable to the presence of HA particles in the scaffolds. The strong chemical cue provided by the HA particles might help guide the differentiation of BMSCs after they were delivered into the bone defect (Fig. 5 and 7). More differentiated donor cells were found in the defect zone where HA content in the scaffold was higher. In the case of the No HA group, the bone formation appeared to be much slower than the groups with HA incorporated, which indicates that the differentiation of BMSCs was much slower in the absence of the HA cue. The control of stem cell fate after transplantation shown in this study might be a valuable tool to extend the utilization of early stage stem cells.
Conclusions
Regeneration of functional bone tissue requires robust new bone formation associated with sufficient graft integration with the host tissue. Here, we present a material approach to modulate the osseointegration of bone grafts when exogenous BMSCs were used as donor cells. Our data show that HA particles present in the scaffold acted as a strong chemical cue to actively regulate osteogenic differentiation of exogenous BMSCs, which further affected new bone formation and host integration. High HA content in scaffolds induced fast bone formation via strong BMSCs osteogenic differentiation which also inhibited host cell participation of bone repair. In contrast, the scaffold without HA maintained BMSCs under undifferentiated status at the initial stage of implantation which led to strong host cells recruitment and better graft integration. Our study has provided a simple way of using a material approach to modulate the osseointegration of bone grafts in a stem cell-based tissue engineering strategy.Acknowledgements
The authors would like to thank the National Science Foundation (BES 0503315 and CBET-1133883) and the NIH (R21AR059962) for supporting the research.Notes and references
- J. Borrelli Jr., W. D. Prickett and W. M. Ricci, Clin. Orthop. Relat. Res., 2003, 245–254 CrossRef PubMed.
- S. Kurtz, K. Ong, E. Lau, F. Mowat and M. Halpern, J. Bone Joint Surg. Am. Vol., 2007, 89A, 780–785 CrossRef PubMed.
- G. F. Muschler, C. Nakamoto and L. G. Griffith, J. Bone Joint Surg., 2004, 86-A, 1541–1558 Search PubMed.
- D. L. Wheeler and W. F. Enneking, Clin. Orthop. Relat. Res., 2005, 36–42 CrossRef.
- G. M. Crane, S. L. Ishaug and A. G. Mikos, Nat. Med., 1995, 1, 1322–1324 CrossRef CAS.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CAS.
- D. Marolt, M. Knezevic and G. V. Novakovic, Stem Cell Res. Ther., 2010, 1, 1–10 CrossRef PubMed.
- J. Y. Yu, M. A. Vodyanik, K. Smuga-Otto, J. Antosiewicz-Bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, I. I. Slukvin and J. A. Thomson, Science, 2007, 318, 1917–1920 CrossRef CAS PubMed.
- E. M. Bueno and J. Glowacki, Nat. Rev. Rheumatol., 2009, 5, 685–697 CrossRef PubMed.
- R. Branemark, P. I. Branemark, B. Rydevik and R. R. Myers, J. Rehabil. Res. Dev., 2001, 38, 175–181 CAS.
- G. Granstrom, Oral Dis., 2007, 13, 261–269 CrossRef CAS PubMed.
- E. Kon, A. Muraglia, A. Corsi, P. Bianco, M. Marcacci, I. Martin, A. Boyde, I. Ruspantini, P. Chistolini, M. Rocca, R. Giardino, R. Cancedda and R. Quarto, J. Biomed. Mater. Res., 2000, 49, 328–337 CrossRef CAS.
- R. Cancedda, G. Bianchi, A. Derubeis and R. Quarto, Stem Cells, 2003, 21, 610–619 CrossRef CAS PubMed.
- F. Tortelli, R. Tasso, F. Loiacono and R. Cancedda, Biomaterials, 2010, 31, 242–249 CrossRef CAS PubMed.
- R. Tasso, F. Fais, D. Reverberi, F. Tortelli and R. Cancedda, Biomaterials, 2010, 31, 2121–2129 CrossRef CAS PubMed.
- A. T. Badillo, L. Zhang and K. W. Liechty, J. Pediatr. Surg., 2008, 43, 1128–1133 CrossRef PubMed.
- R. Z. LeGeros, Chem. Rev., 2008, 108, 4742–4753 CrossRef PubMed.
- Y. Zuo, F. Yang, J. G. C. Wolke, Y. Li and J. A. Jansen, Acta Biomater., 2010, 6, 1238–1247 CrossRef CAS PubMed.
- S. Deville, E. Saiz and A. P. Tomsia, Biomaterials, 2006, 27, 5480–5489 CrossRef CAS PubMed.
- A. M. P. Dupraz, J. R. de Wijn, S. A. T. v. d. Meer and K. de Groot, J. Biomed. Mater. Res., 1996, 30, 231–238 CrossRef CAS.
- H. P. Yuan, H. Fernandes, P. Habibovic, J. de Boer, A. M. C. Barradas, A. de Ruiter, W. R. Walsh, C. A. van Blitterswijk and J. D. de Bruijn, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13614–13619 CrossRef CAS PubMed.
- Y. C. Chai, S. J. Roberts, E. Desmet, G. Kerckhofs, N. van Gastel, L. Geris, G. Carmeliet, J. Schrooten and F. P. Luyten, Biomaterials, 2012, 33, 3127–3142 CrossRef CAS PubMed.
- A. M. Barradas, H. Yuan, C. A. van Blitterswijk and P. Habibovic, Eur. Cell. Mater., 2011, 21, 407–429 CAS ; discussion 429.
- X. Yu, Z. Xia, L. Wang, F. Peng, X. Jiang, J. Huang, D. Rowe and M. Wei, J. Mater. Chem., 2012, 2, 9721–9730 RSC.
- H. Qu, Z. Xia, D. A. Knecht and M. Wei, J. Am. Ceram. Soc., 2008, 91, 3211–3215 CrossRef CAS.
- N. Rajan, J. Habermehl, M. F. Cote, C. J. Doillon and D. Mantovani, Nat. Protocols, 2006, 1, 2753–2758 CAS.
- Z. Xia, X. Yu, X. Jiang, H. D. Brody, D. W. Rowe and M. Wei, Acta Biomater., 2013, 9, 7308–7319 CrossRef CAS PubMed.
- I. Kalajzic, Z. Kalajzic, M. Kaliterna, G. Gronowicz, S. H. Clark, A. C. Lichtler and D. Rowe, J. Bone Miner. Res., 2002, 17, 15–25 CrossRef CAS PubMed.
- M. D. Abràmoff, P. J. Magalhães and S. J. Ram, Biophotonics Int., 2004, 11, 36–41 Search PubMed.
- X. Jiang, Z. Kalajzic, P. Maye, A. Braut, J. Bellizzi, M. Mina and D. W. Rowe, J. Histochem. Cytochem., 2005, 53, 593–602 CrossRef CAS PubMed.
- L. Wang, Y. Liu, Z. Kalajzic, X. Jiang and D. W. Rowe, Blood, 2005, 106, 3650–3657 CrossRef CAS PubMed.
- R. Shu, R. McMullen, M. J. Baumann and L. R. McCabe, J. Biomed. Mater. Res., Part A, 2003, 67, 1196–1204 CAS.
- H. Wang, Y. Li, Y. Zuo, J. Li, S. Ma and L. Cheng, Biomaterials, 2007, 28, 3338–3348 CrossRef CAS PubMed.
- C. M. Cowan, Y. Y. Shi, O. O. Aalami, Y. F. Chou, C. Mari, R. Thomas, N. Quarto, C. H. Contag, B. Wu and M. T. Longaker, Nat. Biotechnol., 2004, 22, 560–567 CrossRef CAS PubMed.
- X. Yu, H. Qu, D. Knecht and M. Wei, J. Mater. Sci.: Mater. Med., 2009, 20, 287–294 CrossRef CAS PubMed.
- L. Jongpaiboonkit, T. Franklin-Ford and W. L. Murphy, Adv. Mater., 2009, 21, 1960–1963 CrossRef CAS.
- X. Yu and M. Wei, J. Biomed. Mater. Res., Part B, 2011, 97B, 345–354 CrossRef CAS PubMed.
- D. Suárez-González, K. Barnhart, F. Migneco, C. Flanagan, S. J. Hollister and W. L. Murphy, Biomaterials, 2012, 33, 713–721 CrossRef PubMed.
- P. Tayalia and D. J. Mooney, Adv. Mater., 2009, 21, 3269–3285 CrossRef CAS PubMed.
- A. M. C. Barradas, H. A. M. Fernandes, N. Groen, Y. C. Chai, J. Schrooten, J. van de Peppel, J. van Leeuwen, C. A. van Blitterswijk and J. de Boer, Biomaterials, 2012, 33, 3205–3215 CrossRef CAS PubMed.
- X. Yu, L. Wang, F. Peng, X. Jiang, Z. Xia, J. Huang, D. Rowe and M. Wei, J. Tissue Eng. Regen. Med., 2012 DOI:10.1002/term.1490.
- R. Tasso, A. Augello, S. Boccardo, S. Salvi, M. Carida, F. Postiglione, F. Fais, M. Truini, R. Cancedda and G. Pennesi, Tissue Eng. Part A, 2009, 15, 2203–2212 CrossRef CAS PubMed.
- J. M. Karp and G. S. L. Teol, Cell Stem Cell, 2009, 4, 206–216 CrossRef CAS PubMed.
- Y. C. Chai, A. Carlier, J. Bolander, S. J. Roberts, L. Geris, J. Schrooten, H. Van Oosterwyck and F. P. Luyten, Acta Biomater., 2012, 8, 3876–3887 CrossRef CAS PubMed.
- K. M. Dupont, K. Sharma, H. Y. Stevens, J. D. Boerckel, A. J. Garcia and R. E. Guldberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3305–3310 CrossRef CAS PubMed.
- D. Sarkar, P. K. Vemula, W. Zhao, A. Gupta, R. Karnik and J. M. Karp, Biomaterials, 2010, 31, 5266–5274 CrossRef CAS PubMed.
- S. Kumar and S. Ponnazhagan, FASEB J., 2007, 21, 3917–3927 CrossRef CAS PubMed.
- L. Meinel, R. Fajardo, S. Hofmann, R. Langer, J. Chen, B. Snyder, G. Vunjak-Novakovic and D. Kaplan, Bone, 2005, 37, 688–698 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60173k |
| This journal is © The Royal Society of Chemistry 2014 |