Bio-based hyperbranched poly(ester amide)–MWCNT nanocomposites: multimodalities at the biointerface†
Sujata
Pramanik
a,
Rocktotpal
Konwarh
a,
Nilakshi
Barua
b,
Alak K.
Buragohain
b and
Niranjan
Karak
*a
aDepartment of Chemical Sciences, Tezpur University, Tezpur-784028, India. E-mail: karakniranjan@yahoo.com
bDepartment of Molecular Biology and Biotechnology, Tezpur University, Tezpur-784028, India
First published on 27th September 2013
Abstract
There has been a growing interest in the use of nanomaterials featuring potent biocompatibility and biodegradability together with the added facet of antibacterial activity, particularly against drug-resistant bacterial species. Addressing these three features at the biointerface, we report the fabrication of multimodal bio-based hyperbranched poly(ester amide) (HBPEA)–microwave functionalized multiwalled carbon nanotube (f-MWCNT) nanocomposites by incorporation of various weight percentages (1, 2.5, and 5 wt%) of the f-MWCNTs into HBPEA by using an ex situ polymerization technique. Fourier transform infrared spectroscopy confirmed the structural changes upon interaction of the f-MWCNTs with HBPEA. The formation of thermosetting nanocomposites resulted in an acceptable improvement of the desired properties including their mechanical properties (∼170%), instrumental for providing mechanical integrity in cultured cells. The nanocomposite films were found to be biocompatible substrates for the in vitro adhesion and proliferation of peripheral blood mononuclear cells (PBMC) with enhanced cell viability correlating with the increase of the f-MWCNT content. The antibacterial results, monitored by a CFU count and the protein concentration, demonstrated that the prepared nanocomposites were more toxic towards Gram positive bacteria and Mycobacterium smegmatis than the Gram negative ones. The damage of bacterial cells upon interaction with the nanocomposites was validated by UV-visible spectroscopy and a SEM study. The antibacterial and biocompatibility studies suggested that these microporous nanocomposite films (3D interconnected porous structures with pore diameters of 5–105 μm and a porosity of 39.90%) possess concurrent long-term lethal activity against the bacterial cells and biocompatibility with PBMC. Thus, the prepared nanocomposites may find potential bio-medical applications, particularly as antimicrobial dressing materials for infected burn wounds.
1. Introduction
The steep augmentation in deadly infections associated with infected wounds including burns has posed a serious nuisance in the realm of medical sciences. The prevalence of community-associated and hospital-acquired infections has increased abruptly in the last 20 years, particularly with the methicillin-resistant Staphylococcus aureus (MRSA) and the drug resistant Mycobacterium smegmatis and so on, accompanied by a rise in antibiotic-resistant strains.1,2 These multi-drug resistant opportunistic bacteria, ubiquitous in distribution, are recognized as deadly pathogens which require a foreign body such as sutures or burn wounds to invade tissues. The loss of skin, availability of bacterial nutrients in the burn locale and destruction of the vascular system near the burned region lead to immune-suppression, thereby making burn wounds susceptible to infection.3 Such bacterial colonization near the periphery of the injury region causes sepsis.4 The prevailing scenario of the infection susceptibility of burn patients by drug-resistant bacterial species has caused a global need for antibacterial dressing materials. Thus, a major drive to search for suitable antimicrobial materials is underway.In this vein, a nanotechnology-based approach can be effective in addressing the aforementioned challenges. Amidst the genre of nanomaterials, polyaniline (PAni) nanofibers5 and multiwalled carbon nanotubes (MWCNTs)6 have carved a unique niche of their own as potent antibacterial materials in the biomedical field. The functionalization of MWCNTs via covalent and non-covalent approaches is an apt strategy to address the practical challenges of the chemical inertness of the graphitic network, poor dispersion, interfacial bonding with others, and so on.7 The microwave (MW) assisted functionalization of MWCNTs, covalently with poly(glycidyl methacrylate) (PGMA) and non-covalently with PAni nanofibers in a single pot, proved to be an efficient technique amongst all others, particularly in the context of green chemistry.7 Thus, in the present study, MW-assisted PGMA and PAni nanofiber functionalized MWCNTs (f-MWCNT) will be employed as efficient antibacterial materials. The effects of functionalization and synergism of MWCNTs and PAni nanofibers as the single nanohybrid in the antibacterial efficacy against a spectrum of bacterial species will be delved into. Further, the potential toxicity of these nanomaterials implies an urgent need for the development of biocompatible nanomaterials.
However, the rapid advancement of nanotechnology in the realm of bio-medical science over the past few decades has resulted in growing concern regarding the biosafety of these nanomaterials to human health, particularly the immunologic toxicity. The conventional antibacterial materials including antibiotics, metal ions, and quaternary ammonium compounds used in the domain of bioscience suffer from limitations such as short-term efficacy, a poor release profile together with cumulative toxicity, and subsequently, microbe resistance.8 The unison of nanotechnology and polymer science may be imperative in amalgamating the biocompatibility together with antibacterial efficacy to address the above problems. The inclusion of a small wt% of f-MWCNTs into a biocompatible polymeric matrix holds potential in reducing the cytotoxicity of the nanotubes. The blood compatibility is one of the key problems that limit the applications of these materials in blood-contacting environments such as in the cardiovascular system.9 The peripheral blood mononuclear cells (PBMC) are of immunological importance, and particularly help in the healing of wounds and in controlling the response of the host matrix towards a foreign body.10 The response of the prepared nanocomposites towards the same is dictated by the adhesion and proliferation of PBMC onto the host matrix, making the interaction imperative in evaluating the biocompatibility. The rationale behind the adherence and proliferation of PBMC is for the practical utility of the nanocomposites as antibacterial dressing materials for infected burn wounds. The lipid protein layer (LPC) produced in the skin after the burning accident inhibits the immune response and the production of interleukin 2 (IL2), a cytokine.11 PBMCs stimulate the release of IL2, essential for the growth of IL2-dependent lymphocytes in the periphery of the infected burn wound region.12 Thus, the current focus remains on scrutinizing the compatibility of the f-MWCNTs with the polymer along with its interaction with the PBMC for their potent application as antibacterial dressing materials for infected burn wounds.
Again, the biodegradability of the polymeric materials is of paramount importance because this provides long term biocompatibility as well as leaving room for tissue regeneration.13 In addition to the above, no post-healing treatment is required in the context of the biodegradable dressing material. Thus, there has been a paradigm shift from non-degradable to biodegradable polymeric materials for applications in the arena of bio-medical science. This is attested to by the latter's inherent amenability for tailorable degradation kinetics, which is useful for various applications.14 In this milieu, bio-based hyperbranched poly(ester amide) (HBPEA) is an important proposition as the global scientific community thrusts to opt for renewable bioresources endowing green credentials.15 The unison of ester and amide moieties in the hyperbranched architecture bequeaths the resin with the synergism of high functionality, adequate mechanical strength and flexibility together with improving the biocompatibility and biodegradability of the same.16 It is pertinent to mention that non-edible castor oil containing more than 90–95% ricinoleic acid is a good candidate for the preparation of HBPEA.17
Thus, the judicious inclusion of the antibacterial f-MWCNTs into a bio-based HBPEA matrix constitutes an interesting strategy for the fabrication of novel antibacterial, biocompatible and biodegradable materials. A perusal of the literature has shown that the preparation of bio-based HBPEA–MWCNT nanocomposites having antimicrobial properties, biocompatibility and biodegradability under the biological conditions of stimulated human body fluid has never been attempted, which encouraged us to initiate the present study. Thus, in continuation of our exploration of f-MWCNTs,7 we herein wish to report the fabrication of castor oil based HBPEA–f-MWCNT nanocomposites. The response of these nanocomposites towards a number of bacteria including drug resistant bacterial species was investigated. The UV absorption at 260 nm and SEM studies were done to gain insight into the antibacterial action of the nanocomposites. Further, the potential of the same to support the attachment and proliferation of PBMC, and the biodegradability in simulated body fluid was also delved into.
2. Materials and methods
2.1 Materials
Castor oil (Sigma Aldrich, India), diethanol amine (Merck, India), phthalic anhydride (Merck, India) and isophthalic acid (Sisco Research Laboratory Pvt. Ltd, India) were used after being dried under vacuum at 50 °C overnight. Maleic anhydride (Merck, Germany) was used after being dried in a vacuum desiccator for 2 days. The MWCNTs (purity ∼95 wt%, external diameter of about 10–15 nm) were purchased from Hanwha Nanotech Corp., Korea. Glycidyl methacrylate (GMA), 2,2′-azobis-(isobutyronitrile) (AIBN), ammonium peroxydisulfate (APS) and hydrochloric acid (HCl) (36.46 g mol−1, 11.6 N) were obtained from Merck, India, and used as received. Aniline (Merck, India) was purified by vacuum distillation in the presence of zinc dust (SD Fine-Chem Limited, India, atomic weight 65.37 g mol−1) and stored at 4 °C prior to use. The solvents such as benzene, methanol, tetrahydrofuran (THF), dimethyl acetamide (DMAc) and dimethyl sulfoxide (DMSO) were obtained from Merck, India, and distilled before use. The bisphenol-A-based epoxy resin (Araldite GY 250) (epoxy equivalent: 180–190 g per equiv. and density: 1.16 g cc−1 at 25 °C) and poly(amido amine) hardener ((HY 840) Hindustan Ciba Geigy Ltd, India) were used as received. Histopaque (density 1.077 g mL−1) was obtained from Sigma Aldrich. Sodium citrate was obtained from Merck, India, and phosphate buffer saline from HIMEDIA. All other chemicals and bacterial strains used in the biological study were procured from the Department of Molecular Biology and Biotechnology, Tezpur University.2.2 Characterization of the nanocomposites
The interaction of the f-MWCNT and HBPEA matrix was confirmed by Fourier transform infrared (FTIR) spectroscopy using a Nicolet Impact-410 FTIR spectrophotometer. The spectral studies were conducted in transmission mode within the wavenumber range of 500–4000 cm−1 using KBr pellets. The prepared nanocomposites were exposed to MW irradiation using a Catalyst Scientific microwave system at 30% power of 210 W at 60–80 °C for 2 min in pulsed mode. The crystallinity of the prepared nanocomposites was evaluated by a Rigaku Miniflex X-ray diffraction (XRD) system using CuKα radiation with a wavelength (λ) of 1.5406 Å in the range of 2–70°. The dispersion of the f-MWCNT in the HBPEA matrix was done using a standard sonotrode of 3 mm tip-diameter in a Hielscher UP200S high intensity ultrasonic processor at a 60% amplitude and 0.5 cycles. The thermal stability of the nanocomposites was determined by thermogravimetric analysis (TGA) using a Shimazdu TGA 50 thermal analyzer at a nitrogen flow rate of 30 mL min−1 and at the heating rate of 10 °C min−1 within the temperature range of 25 to 700 °C. The mechanical performance of the cured thermosets (as per the ASTM D 412-51 T)18 was determined using a Zwick Z010 universal testing machine (UTM) with a load cell of 10 kN and at a 40 mm min−1 jaw separation speed. The results were averaged over three replicates. The gloss, scratch hardness and impact resistance of the cured films were measured as per the standard methods.19 The absorbance for the MTT assay was measured by ultraviolet (UV)-visible spectroscopy using a UV-1700 PharmaSpec spectrophotometer. The surface morphology of the thermosets was studied by high-resolution transmission electron microscopy (HRTEM) using a JEOL 2100X electron microscope at an operating voltage of 200 kV after casting on carbon coated copper grids. The adhesion of the PBMC onto the thermosets and the antibacterial interaction of the used bacterial strains with the nanocomposites was assessed by scanning electron microscopy (SEM) using a JEOL JSM-6390LV microscope after platinum coating on their surface.3. Results and discussion
3.1 Fabrication of the nanocomposites
A solution technique was adopted to fabricate the HBPEA–f-MWCNT nanocomposites. Firstly, the MWCNTs were functionalized with PGMA and PAni nanofibers, wherein the functionalization occurred at the defects – the prime sites present on the nanotube edges and on the sidewalls.20 The epoxy groups and benzenoid-quinoid moieties generated by the MW-assisted functionalization inferred interfacial adhesion and wettability of the same with the HBPEA. The interaction of the MWCNTs with the PGMA and PAni nanofibers dictated the dispersion of the debundled nanotubes in the HBPEA matrix. The preparative layout of the HBPEA–f-MWCNT nanocomposites is shown in Scheme 1. | ||
| Scheme 1 Preparation of the bio-based HBPEA–f-MWCNT nanocomposites by the synergism of ultrasonication and MW irradiation. | ||
3.2 Possible mechanism of the formation of the nanocomposites
The ultrasonic waves as well as the MW irradiation played vital roles in the formation of the HBPEA–f-MWCNT nanocomposites with a preferred directionality, as evident from the HRTEM study. Pulsed ultrasound was reported to be effective in the exfoliation and dispersion of MWCNT agglomerates in the HBPEA matrix.21 The effects of the ultrasound are associated with the rapid collapse of cavitation bubbles on a microsecond time scale, creating microscopic domains of high temperature and pressure, which opened up the non-covalently wrapped PAni nanofibers from the nanotube surface. The formation of microjets of energy or hot spots caused enhanced exfoliation of the f-MWCNTs,22 thereby allowing a greater surface area of the same to interact with the HBPEA chains. Dictated by previous reports on the active MW absorption by the MWCNTs,23 the prepared nanocomposites were exposed to MW radiation for 2 min (divided into six cycles, each cycle containing an irradiation of 10 s succeeded with an interval of 10 s) to assist in the dispersion of the f-MWCNTs in the HBPEA matrix. The strong MW absorption is ascribed to the presence of free π-electrons moving out of the graphitic framework of the f-MWCNTs with a simultaneous volume expansion of the nanotubes.24 The MW absorption by the MWCNTs at a relatively low temperature caused a thermal expansion of the nanotube layers, thereby facilitating the formation of interfacial interactions between the MWCNTs and the polymer matrix.24 The MW irradiation induced the localized heating of the f-MWCNTs at the nanotube–HBPEA interface such that the nanotubes aligned in a preferred direction within the matrix.23 The alignment of the f-MWCNTs in the HBPEA is supported by the morphological study and the presence of long-range order by the XRD study (ESI† and Table 1). Upon direct MW irradiation, the static temperature gradients resulted in well oriented and aligned nanotubic arrays in the HBPEA matrix.| Physico-mechanical property | HBPEA | HBPEAM1 | HBPEAM2.5 | HBPEAM5 |
|---|---|---|---|---|
| a The maximum limit of the instrument is 100 cm. | ||||
| Curing time (h) | 10 | 1.5 | 1.2 | 1 |
| Gel fraction (%) | 77.2 | 78.4 | 79.7 | 80.2 |
| Scratch hardness (kg) | 8.5 | 9.5 | 10 | >10 |
| Impact resistancea (cm) | >100 | >100 | >100 | >100 |
| Tensile strength (MPa) | 7.2 | 9.5 | 12.7 | 16.7 |
| Elongation at break (%) | 88.1 | 83.3 | 77.5 | 70.3 |
| Domain length (Å) | — | 9.7 | 12.4 | 16.2 |
3.3 Morphology of the nanocomposites
The stable and uniform dispersion of the f-MWCNTs in the HBPEAM5, as evident from the HRTEM study, is instrumental in comprehending the enhancement of the structural and thermal properties of the nanocomposites. The HRTEM image (Fig. 1a) shows that the pristine MWCNTs have an average external diameter of ∼15 nm. A representative HRTEM micrograph (Fig. 1b) shows the anchoring of a PGMA layer onto a MWCNT (having a diameter of ∼35 nm). A reconstruction of the wall-to-wall distance of f-MWCNTs was observed in the MW treated nanocomposites as compared to the untreated f-MWCNTs. The wall-to-wall distance of the untreated f-MWCNTs was ∼0.37 nm, which increased to ∼0.4 nm in the case of MW irradiated f-MWCNTs dispersed in the HBPEA nanocomposite (inset Fig. 1b).24 This change in the intertubular distance of the f-MWCNTs is indicative of the high temperature and pressure encountered by the nanotubes during MW irradiation. The dispersion of the PAni nanofibers in the nanocomposite was observed in Fig. 1c. The MW irradiation suffices for the appearance of a furrowed-like architecture of the nanocomposite (Fig. 1d). This is further confirmed by the presence of long range order in the XRD study (Table 1). | ||
| Fig. 1 High-resolution TEM images of (a) a pristine MWCNT, (b) a f-MWCNT based nanocomposite with the inset of an enlarged wall-to-wall distance of the f-MWCNT, (c) a dispersion of PAni nanofibers in HBPEAM5, and (d) a dispersion scenario of f-MWCNTs in HBPEAM5. | ||
3.4 Performance of the nanocomposites
The performance of the epoxy-poly(amido amine) cured thermosetting nanocomposites effectively changed with the incorporation of f-MWCNTs. Table 1 lists properties such as the curing time, gel fraction, scratch hardness, impact resistance, gloss, tensile strength and elongation at break of the nanocomposites. It is evident from Table 1 that the curing time of the epoxy-poly(amido amine) cured HBPEA and its nanocomposites baked at 150 °C decreased significantly with the increase of the f-MWCNT content. This was attributed to the presence of the N-atom of the benzenoid ring and the oxirane ring (adhered to the f-MWCNTs) which aided the crosslinking reaction of HBPEA. In other words, the covalent bonding between the epoxide groups adhered to the f-MWCNTs and –OH groups present in the periphery of the HBPEA backbone resulted in a strong interfacial adhesion between the nanotubes and the polymer matrix.25 The preferred orientation of the f-MWCNTs in the HBPEA matrix, as evident from the HRTEM study, also helped in decreasing the curing time. The increment in the gloss with the f-MWCNT content in the nanocomposites indicated that the cured thermosets possessed a good dimensional stability and smooth surface morphology. The increment in scratch hardness of the nanocomposites with the f-MWCNT content was attributed to the enhanced strength of the nano-reinforcing functionalized nanotubes and flexibility of the HBPEA chains. The high impact resistance of the thermosets reflected optimum crosslinking and flexibility of the long hydrocarbon chains of the fatty acid part of the HBPEA. The pristine HBPEA thermoset possessed a tensile strength of 7.2 MPa which increased from 9.5 to 16.2 MPa with the incorporation of f-MWCNTs from 1 to 5 wt%. The increment in the magnitude of the tensile strength of the HBPEA nanocomposites to ∼170% as compared to the pristine one further supports the presence of strong f-MWCNT–HBPEA interfacial interactions. The interactions between f-MWCNTs and HBPEA, as evident from the FTIR study, enhanced the dispersion of the f-MWCNTs in the matrix and hence led to the formation of dense crosslinked structures, which hindered the mobility of the HBPEA chains, thereby improving the mechanical properties.26 In other words, the incorporation of the f-MWCNTs in the HBPEA imparted a nano-reinforcing effect, which is responsible for providing mechanical integrity to the PBMC. The restricted mobility of the long fatty acid chains due to the incorporation of f-MWCNTs justified the decrease (20.2%) in the elongation at break of the nanocomposites. The minimal decrement in the elongation at break also reflects sufficient flexibility of the nanocomposite films for being suitable for a number of applications including wound dressing materials.The thermal stability and degradation pattern of the HBPEA and its nanocomposites were assessed by thermogravimetry (Fig. 2). The HBPEA thermoset exhibited a two-step degradation profile, with an initial and final degradation at around 277 °C (weight loss due to ester groups) and 521 °C (weight loss due to amide groups), respectively.15 The HBPEA nanocomposites also presented a two-step degradation pattern with a dose-dependent increment of thermal stability. The increase (277–325 °C) of the initial degradation temperature of the nanocomposites with increasing f-MWCNT content is attributed to the nano-mechanical interlocking of the f-MWCNTs within the HBPEA matrix via the adherence of functionalities onto the nanotube surface. An increment in the weight residue of the nanocomposites (∼2–3% as compared to the HBPEA) observed at 800 °C reflects the presence of thermo-stable f-MWCNTs.
 | ||
| Fig. 2 Thermogravimetric analysis of HBPEA, HBPEAM1, HBPEAM2.5 and HBPEAM5. | ||
3.5 Antibacterial activity of the nanocomposites
The effect of the incorporation of f-MWCNTs in the HBPEA on bacterial growth was assessed by inoculating the nanocomposite films with the test strains. A substantial reduction in the attachment of the Gram positive and acid fast bacterial species was observed as assessed through enumeration of the colony forming units (CFU) (Fig. 3). The nanocomposites exhibited pronounced antibacterial efficacy against B. subtilis (Fig. 3a) and S. aureus (Fig. 3b), as compared to E. coli (Fig. 3c) and K. pneumonia (Fig. 3d). A significantly lower number of bacterial colonies was observed on the nanocomposites, implying the superior antibacterial activity of the same over the pristine HBPEA. It can thus be inferred from the above observations that the nanocomposites were more effective against Gram positive bacteria as compared to the Gram negative ones, showing differential interaction of the same with the surface moieties of the two different bacterial strains. The basic architectural differences between the cell wall structures in these bacterial strains may play a key role in the determination of their antibacterial activities.27 The M. smegmatis mc2 155 (Fig. 3e) (conventionally available, antibiotic ampicillin resistant) and M. smegmatis ATCC14468 (Fig. 3f) exhibited low colonization onto the nanocomposites as compared to the pristine HBPEA (Fig. 4a).
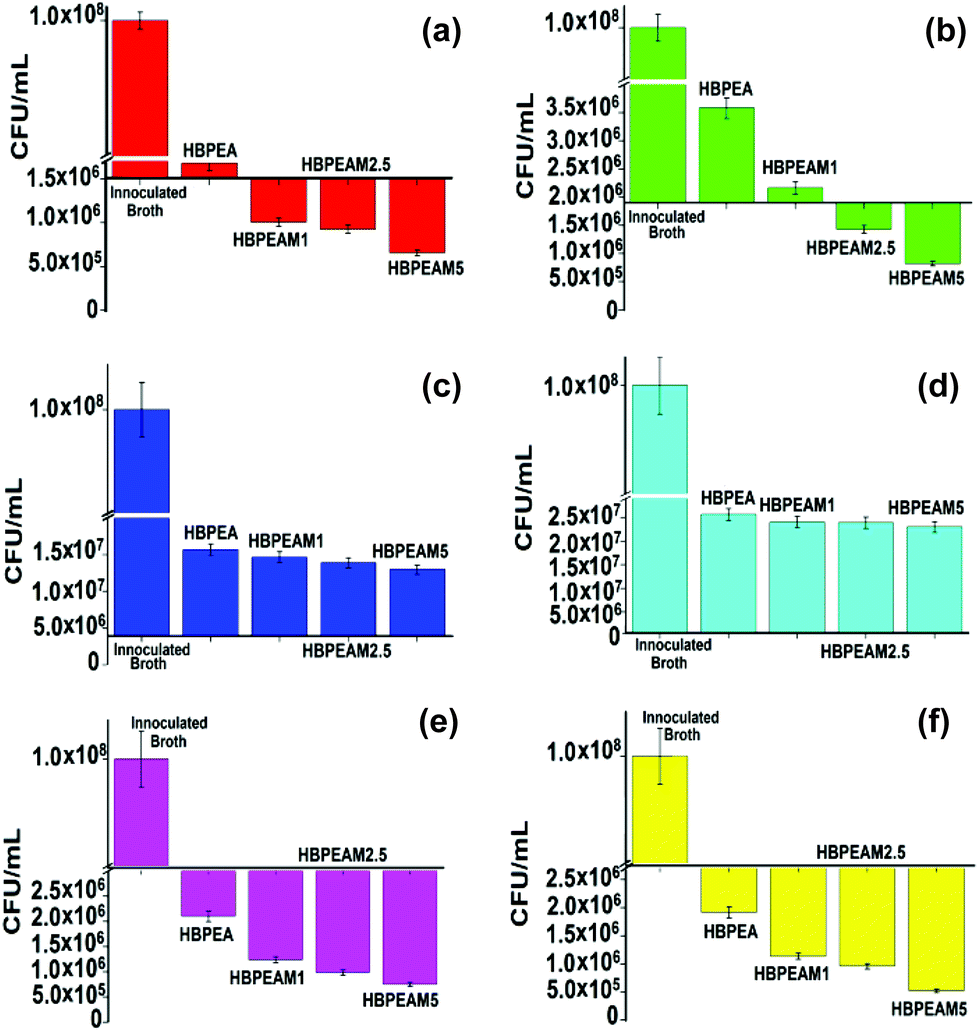 | ||
| Fig. 3 Antimicrobial activity of HBPEA, HBPEAM1, HBPEAM2.5 and HBPEAM5 towards (a) B. subtilis, (b) S. aureus, (c) E. coli, (d) K. pneumonia, (e) M. smegmatis mc2 155, and (f) M. smegmatis ATCC14468. The CFU results are compared with the control sample of inoculated broth. | ||
 | ||
| Fig. 4 Comparative antibacterial study in terms of (a) colony-forming units enumerated from, and (b) protein adsorbed on HBPEA, HBPEAM1, HBPEAM2.5 and HBPEAM5 towards B. subtilis (colored in red), S. aureus (colored in green), E. coli (colored in blue), K. pneumonia (colored in cyan), M. smegmatis mc2 155 (colored in purple) and M. smegmatis ATCC14468 (colored in yellow). | ||
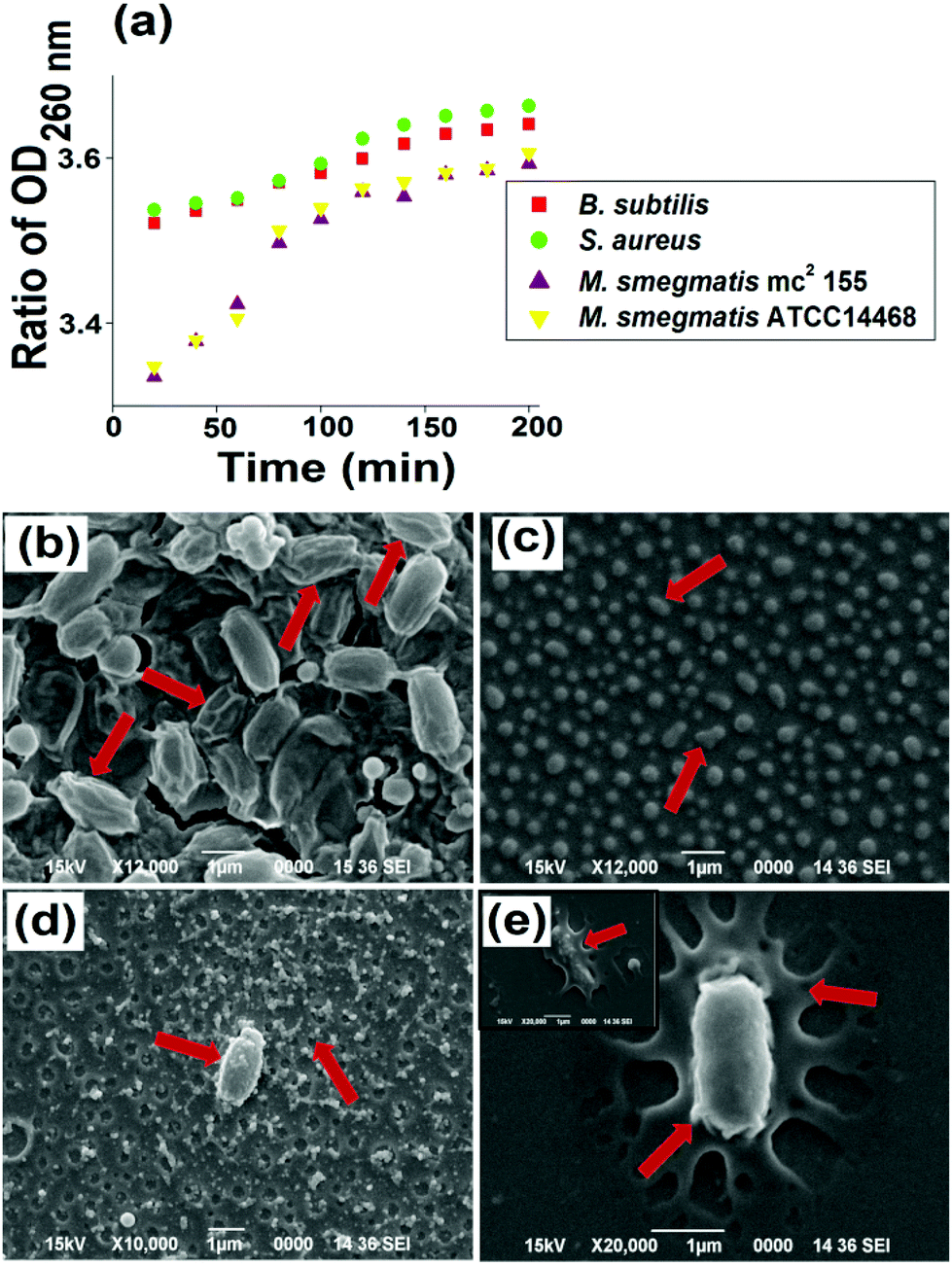 | ||
| Fig. 5 (a) Release of 260 nm absorbing materials from Gram positive and acid fast positive bacterial strains. SEM images of (b) B. subtilis, (c) S. aureus, (d) M. smegmatis mc2 155, (e) M. smegmatis ATCC14468 upon interaction with HBPEAM5. | ||
Further, the SEM study showed a distinct difference in cellular morphology of the bacterial cells after treatment with HBPEAM5. The B. subtilis are elongated and evenly shaped cells, which became corrugated with a ruptured cell surface upon interaction with the tested material (Fig. 5b). The cell wrinkling and shortening together with the formation of craters were also observed. Upon interaction with the nanocomposite, the deformation and disintegration of the cells were observed from round and proliferating cells with intact and well-defined membranes of the control S. aureus (Fig. 5c). The M. smegmatis bacterial strains have a uniform cylindrical-shape morphology. These cells exhibited cell lesions together with the formation of mycothiol for survival under hostile oxidative stress conditions, upon interaction with the nanocomposite (Fig. 5d). In other words, the cells ruptured, becoming flattened together with compromising their cellular integrity while remaining embedded in the mycothiol (Fig. 5e).31 The different morphological observations of the two tested bacterial strains indicated different strain susceptibility towards the nanocomposite. Thus, on the basis of the above study, it can be inferred that there exists a positive correlation of the loss of cellular integrity and the release of cytoplasmic constituents from the bacterial cells. These results confirmed the interaction between the nanocomposite and the bacterial cells. This is due to the fact that the cells are damaged upon interaction with the nanocomposite, which is clearly seen from the SEM micrographs. The nano-biointeraction is further supported by the UV-visible results, as the peak at 260 nm confirmed the release of DNA and RNA from the bacterial cytoplasm (absorbance at 260 nm).
3.6 In vitro biocompatibility of the nanocomposites
 | ||
| Fig. 6 Representative pictures of cell viability using the trypan blue exclusion assay after 48 h of incubation of PBMC with (a) control, (b) HBPEA, (c) HBPEAM1, (d) HBPEAM2.5, (e) HBPEAM5. (f) % Cell viability. | ||
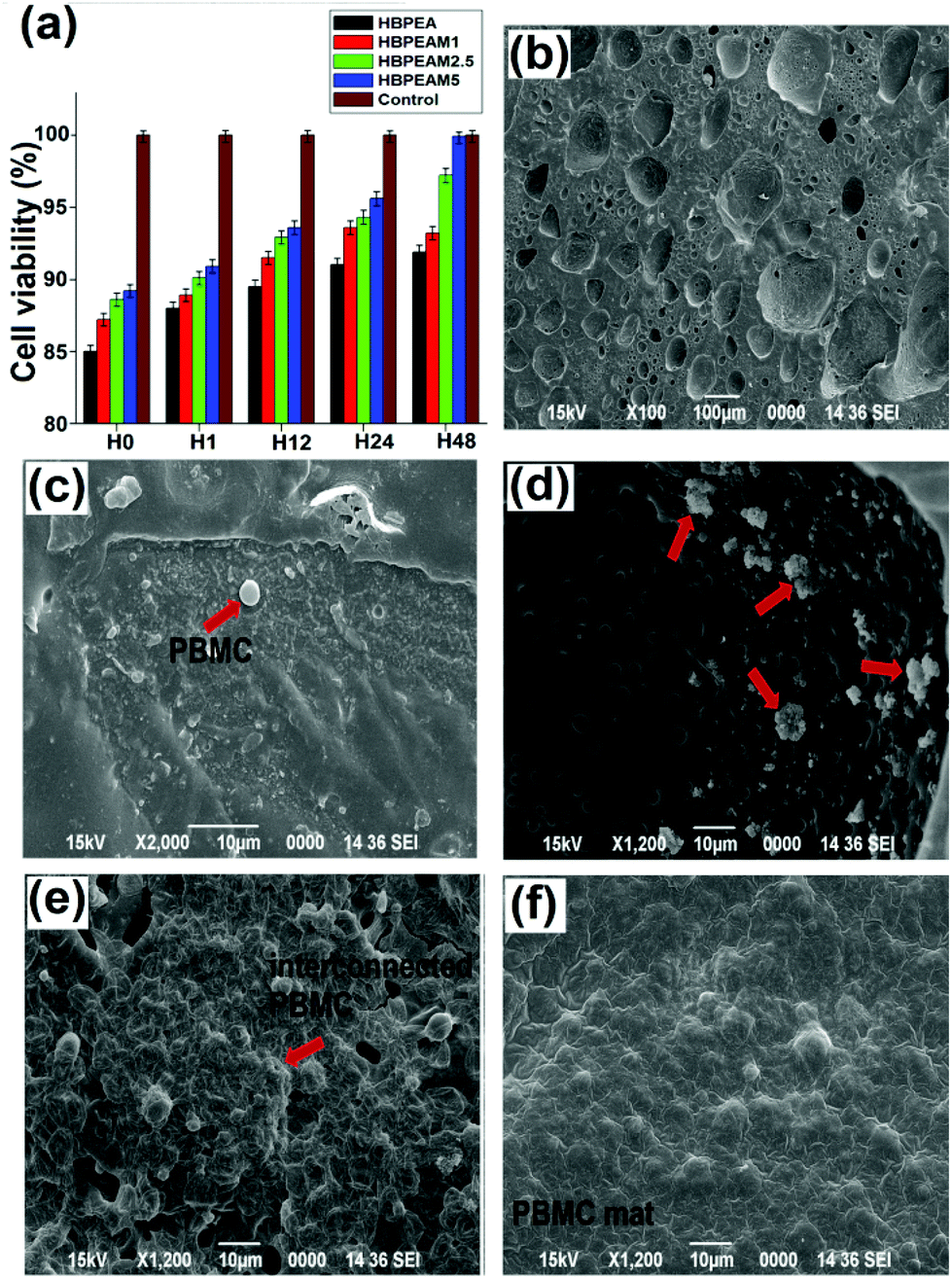 | ||
| Fig. 7 (a) The cell viability of PBMC in terms of absorbance at 540 nm at different time intervals of 0, 1, 12, 24 and 48 h. SEM images showing the adhesion and proliferation of PBMC after seeding on the surfaces of (b) HBPEA, (c) HBPEAM1, (d) HBPEAM2.5, and (e) HBPEAM5. | ||
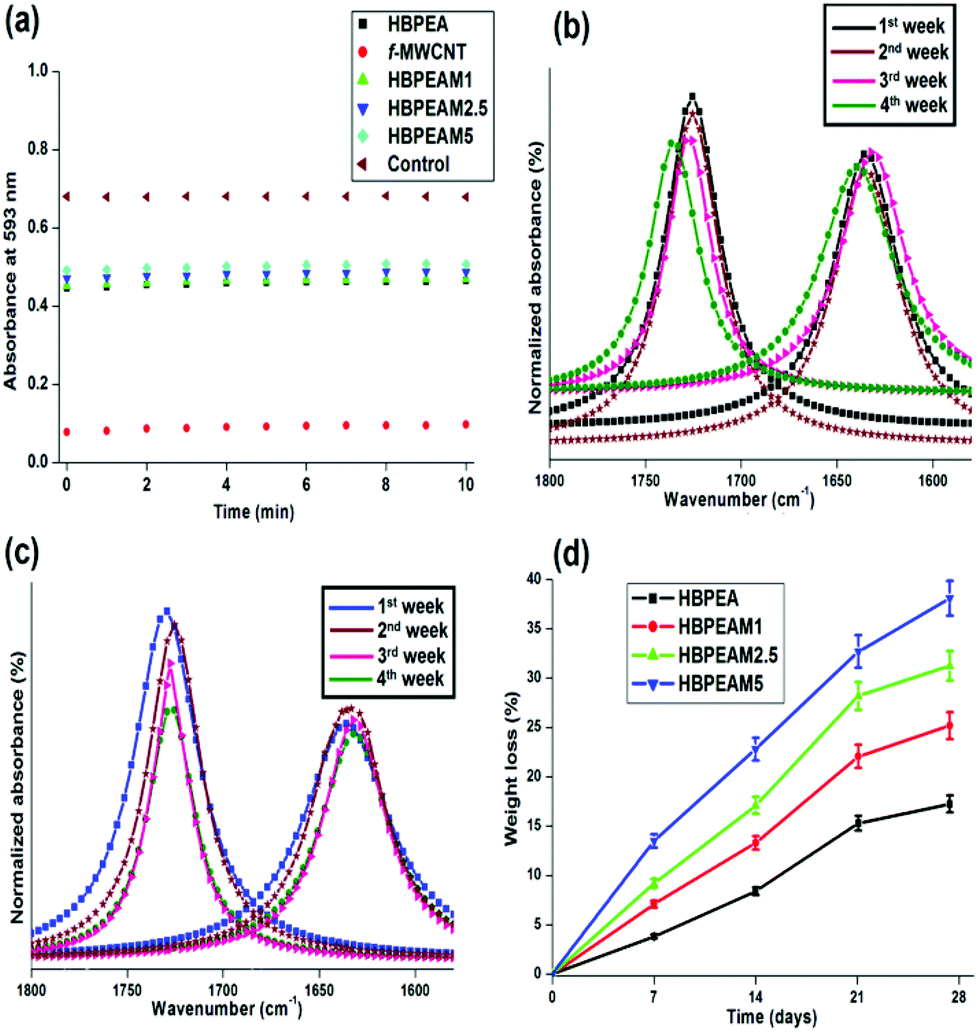 | ||
| Fig. 8 (a) ROS reaction kinetics, and deconvoluted Iester/Iamide FTIR peaks of (b) HBPEA, and (c) HBPEAM5. (d) Weight loss profile of HBPEA and its nanocomposites. | ||
Thus, the in vitro cell response studies showed that the adhesion and proliferation rate of the PBMC increased with increasing f-MWCNT content, without having any hemolysis activity or without inducing any production of ROS. In this context, both the 3D porous interconnected network structure and the presence of f-MWCNTs proved to be the prime factors in promoting cell growth.8
3.7 In vitro biodegradation of the nanocomposites
The in vitro biodegradation of pristine MWCNTs and HBPEA–f-MWCNT nanocomposites was studied by subjecting the films to the degrading action of a lipase–phosphate buffer solution, which mimics the pseudo-chemical environment of simulated animal body fluid.42 The biodegradation of the pristine and nanocomposite films occurred as an extracellular process in PBS catalyzed by lipase via an enzymatic oxidative mechanism. Lipase, interfacially activated at the water–polymer interface, assists in the enzymatic hydrolysis of the ester moieties present in the polymeric backbone.43 The enzyme contains buried catalytic sites and their activation occurs as a result of a conformational change induced upon binding to the polymer substrate. In aqueous media, the α-helical lid covers the active site of the enzyme and blocks its access to the substrate. Upon contact with the hydrophobic polymeric substrate, the lid rolls back and the active site becomes accessible, and thus, the enzyme gets in its active conformation.43,44 Firstly, the inclusion of water into the polymer matrix occur via attack of the polar groups, followed by bond scission catalyzed by the enzyme. Thus, the bulk degradation takes place by the uptake of water, followed by surface erosion at the interfacial region between the polymeric surfaces and the aqueous media. The increase in the f-MWCNT content in the HBPEA matrix directly correlates with the increase in the degradability of the fabricated films.The FTIR spectroscopic analysis of the ester to amide ratio (Iester/Iamide) of the nanocomposite films before and after biodegradation provided a mechanistic understanding of its enzymatic degradation. The scission kinetics of the ester bond of the biodegraded films was investigated from the FTIR spectrum. The pristine HBPEA exhibited an absorption peak at around 1730 cm−1, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching. The integrated intensity of Iester/Iamide was analyzed by fitting with a Lorentzian profile. The enzymatic hydrolysis of ester moieties of the pristine MWCNTs (Fig. 8b) and the HBPEA nanocomposites (Fig. 8c) by lipase is evident from the decrement in integrated intensity of Iester/Iamide (Table 2) for the biodegraded films.
O stretching. The integrated intensity of Iester/Iamide was analyzed by fitting with a Lorentzian profile. The enzymatic hydrolysis of ester moieties of the pristine MWCNTs (Fig. 8b) and the HBPEA nanocomposites (Fig. 8c) by lipase is evident from the decrement in integrated intensity of Iester/Iamide (Table 2) for the biodegraded films.
| Sample | I ester/Iamide | |||
|---|---|---|---|---|
| 1st week | 2nd week | 3rd week | 4th week | |
| HBPEA | 1.12 | 1.05 | 0.92 | 0.89 |
| HBPEAM5 | 1.11 | 0.89 | 0.75 | 0.61 |
The degradation by a surface erosion mechanism, characterized by the loss of material from the surface, is evident from the mass loss profile of the films (Fig. 8d). With the increase in f-MWCNT content, a considerable decrement in the mass of the films (17.09, 25.04, 31.16 and 38.19% mass loss for HBPEAM1, HBPEAM2.5 and HBPEAM5, respectively) was observed. Thus, the hydrolysis of the ester bonds in HBPEA and its nanocomposites has been postulated as the mechanism of its biodegradation.15 Further, the observed dose-dependent biodegradation is attributed to the increasing f-MWCNT content. The increment of the degradation of the nanocomposites with the f-MWCNT content is attributed to the presence of polar moieties on the f-MWCNTs, such as epoxy, carbonyl and PAni nanofibers. These groups aided in the enhancement of the wettability and hydrolytic degradation of the nanocomposites by sorption and penetration of water into the polymer matrix. The penetrated water molecules promoted the accessibility of the polymer chains towards lipase, thereby enhancing chain scission, which hence resulted in good biodegradation.
However, upon degradation of the nanocomposites in due course of time, the f-MWCNTs may be released from the polymer matrix, although the rate of degradation slows down with time. The fate of the released MWCNTs in the body yields concern regarding their cytotoxicity. In such a situation, the nanotubes are catalytically biodegraded by the radical intermediates generated inside the biofluids in the body, such as myeloperoxidase and hypochlorite, resulting in the release of carbon dioxide without generating any inflammatory response.45
3.8 Antibacterial efficacy of the biodegraded nanocomposites
The long-term antibacterial potency of the biodegraded films was investigated by inoculating the biodegraded nanocomposites with the tested bacterial strains and calculating the CFU count. It was clearly evident from Fig. 9 that there is a slight increment in the CFU count with time on HBPEA. The increase in the CFU count on the film is illustrative of the enhancement of the bacterial adhesion onto HBPEA with time. This may be attributed to the increase in oligomeric products with degradation time. These low-molecular-weight HBPEAs may increase the accessibility of the film towards bacteria by serving as the carbon source for their growth. However, the antibacterial activity of the degraded nanocomposites was fairly constant with degradation time. In other words, the inhibition of bacterial growth on the biodegraded nanocomposites was similar to that of the non-biodegraded ones (Fig. 9). This is attributed to the presence of f-MWCNTs – the prime factor for antibacterial activity in the degraded nanocomposites. Also the in vitro biodegradation study of the HBPEAM5 (the maximum biodegraded nanocomposite) showed that it required four weeks to degrade 38.19 wt% with a gradual decrease in the rate of degradation with exposure time. Moreover, the above degradation accounted for the polar moieties such as esters. The nanotubes remained intact for longer (more than four weeks). Thus, these results indicate that the nanocomposite retained the antibacterial efficacy for a longer time period. | ||
| Fig. 9 Antimicrobial activity (CFU count) of HBPEA, HBPEAM1, HBPEAM2.5 and HBPEAM5 towards (a) B. subtilis, (b) S. aureus, (c) M. smegmatis mc2 155, and (d) M. smegmatis ATCC14468. | ||
4. Conclusions
The present study highlighted the fabrication of multifunctional bio-based HBPEA–f-MWCNT nanocomposites. The antibacterial nanocomposites triggered the release of a significant amount of cytoplasmic constituents of Gram positive bacteria and M. smegmatis. The mitochondrial enzyme activity assay established the biocompatibility of the nanocomposites with PBMC, further confirmed by the significant PBMC attachment and proliferation in response to a varied f-MWCNT content of the nanocomposites and exposure time. The time-course dependent biodegradation of the nanocomposites augmented in line with the increase in the f-MWCNT content. Thus, the study opens up the promising application of bio-based HBPEA–f-MWCNT nanocomposites as antibacterial dressing materials for infected burn wounds, where a decrease in the bacterial colonization onto the wound surface is favorable for the reduction in the infection rate of burn wounds. However, before the practical utilization of these microporous nanocomposites as wound dressing materials for burn infections, further in vivo studies need to be performed. The authors are happy to announce that the study of the prepared nanocomposites as prospective scaffold materials for facilitating the proliferation of epithelial cells, an integral part of the wound healing process, is in progress, and the preliminary results are very encouraging.Acknowledgements
The authors express their gratitude and thanks to the research project assistant given by DRL, India, through the grant no. DRL/1047/TC, dated 2nd March, 2011, SAP (UGC), India, through the grant no. F.3-30/2009(SAP-II) and FIST program-2009 (DST), India, through the grant no. SR/FST/CSI-203/209/1 dated 06.05.2010. Mr Joston P. Nongkynrih and SAIF, NEHU, Shillong are gratefully acknowledged for TEM imaging. R. Konwarh acknowledges the receipt of his senior research fellowship from the Department of Biotechnology, Government of India. N. Barua thankfully acknowledges the receipt of a senior research fellowship from the CSIR, Government of India.References
- L. Broxmeyer, D. Sosnowska, E. Miltner, O. Chacon, D. Wagner, J. McGarvey, R. G. Barletta and L. E. Bermudez, J. Infect. Dis., 2002, 186, 1155–1160 CrossRef PubMed.
- B. R. Copp and A. N. Pearce, Nat. Prod. Rep., 2007, 24, 278–297 RSC.
- T. Dai, Y. Y. Huang, S. K. Sharma, J. T. Hashmi, B. D. Kurup and M. R. Hamblin, Recent Pat. Anti-Infect. Drug Discovery, 2010, 5, 124–151 CrossRef CAS.
- J. P. Barret and D. N. Herndon, Plast. Reconstr. Surg., 2003, 111, 744–750 CrossRef PubMed.
- S. Kang, M. Herzberg, D. F. Rodrigues and M. Elimelech, Langmuir, 2008, 24, 6409–6413 CrossRef CAS PubMed.
- M. R. N. Gizdavic, J. R. Bennett, S. Swift, A. J. Easteal and M. Ambrose, Acta Biomater., 2011, 7, 4204–4209 CrossRef PubMed.
- S. Pramanik, R. Konwarh, R. C. Deka, L. Aidew, N. Barua, A. K. Buragohain, D. Mohanta and N. Karak, Carbon, 2013, 55, 34–43 CrossRef CAS PubMed.
- S. C. M. Fernandes, P. Sadocco, A. Alonso-Varona, T. Palomares, A. Eceiza, A. J. D. Silvestre, I. Mondragon and C. S. R. Freire, ACS Appl. Mater. Interfaces, 2013, 5, 3290–3297 CAS.
- E. K. Yim, I. C. Liao and K. W. Leong, Tissue Eng., 2007, 13, 423–433 CrossRef CAS PubMed.
- R. Abe, S. C. Donnelly, T. Peng, R. Bucala and C. N. Metz, J. Immunol., 2001, 166, 7556–7562 CAS.
- B. G. Sparkes, Vaccine, 1993, 11, 504–510 CrossRef CAS.
- B. G. Sparkes, Burns, 1991, 17, 128–135 CrossRef CAS.
- B. Das, P. Chattapadhyay, M. Mandal, B. Voit and N. Karak, Macromol. Biosci., 2012, 1, 126–139 Search PubMed.
- P. A. Gunatillake and R. Adhikari, Eur. Cells Mater., 2003, 5, 1–16 CAS.
- S. Pramanik, R. Konwarh, K. Sagar, B. K. Konwar and N. Karak, Prog. Org. Coat., 2013, 76, 689–697 CrossRef CAS PubMed.
- M. Deng, J. Wu, C. A. Reinhart-King and C. C. Chu, Biomacromolecules, 2009, 10, 3037–3047 CrossRef CAS PubMed.
- S. Pramanik, K. Sagar, B. K. Konwar and N. Karak, Prog. Org. Coat., 2012, 75, 569–578 CrossRef CAS PubMed.
- D. G. Nair, B. G. Fry, P. Alewood, P. P. Kumar and R. M. Kini, Biochem. J., 2007, 402, 93–104 CrossRef CAS PubMed.
- Oil and Colour Chemist's Association of Australia, Surface Coatings, Chapman & Hall, Methuen, New York, 1st edn, 1986, vol. 1, p. 139 Search PubMed.
- M. A. Hamon, H. Hu, P. Bhowmik, S. Niyogi, B. Zhao, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2001, 347, 8–12 CrossRef CAS.
- F. H. Gojny, M. H. G. Wichmann, U. Kopke, B. Fiedler and K. Schulte, Compos. Sci. Technol., 2004, 64, 2363–2371 CrossRef CAS PubMed.
- R. Konwarh, S. Pramanik, D. Kalita, C. L. Mahanta and N. Karak, Ultrason. Sonochem., 2012, 19, 292–299 CrossRef CAS PubMed.
- J. Chang, G. Liang, A. Gu, S. Cai and L. Yuan, Carbon, 2012, 50, 689–698 CrossRef CAS PubMed.
- H. Hu, Z. Zhao, Q. Zhou, Y. Gogotsi and J. Qiu, Carbon, 2012, 50, 3267–3273 CrossRef CAS PubMed.
- S. Pramanik, J. Hazarika, A. Kumar and N. Karak, Ind. Eng. Chem. Res., 2013, 52, 5700–5707 CrossRef CAS.
- W. Yuan and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2012, 4, 2065–2073 CAS.
- R. Konwarh, B. Gogoi, R. Philip, M. A. Laskar and N. Karak, Colloids Surf., B, 2011, 84, 338–345 CrossRef CAS PubMed.
- C. Sun, D. K. Hunt, R. B. Clark, D. Lofland, W. J. O. Brien, L. Plamondon and X. Y. Xiao, J. Med. Chem., 2011, 54, 3704–3731 CrossRef CAS PubMed.
- S. Liu, L. Wei, L. Hao, N. Fang, M. W. Chang, R. Xu, Y. Yang and Y. Chen, ACS Nano, 2009, 3, 3891–3902 CrossRef CAS PubMed.
- S. Liu, L. Wei, L. Hao, N. Fang, M. W. Chang, R. Xu, Y. Yang and Y. Chen, ACS Nano, 2009, 3, 3891–3902 CrossRef CAS PubMed.
- C. C. Miller, M. Rawat, T. Johnson and Y. A. Gay, Antimicrob. Agents Chemother., 2007, 51, 3364–3366 CrossRef CAS PubMed.
- X. Sun, L. Zhang, Z. Cao, Y. Deng, L. Liu, H. Fong and Y. Sun, ACS Appl. Mater. Interfaces, 2010, 2, 952–956 CAS.
- R. Rajaraman, R. A. Fox, V. G. Vethamany, L. A. Fernandez and J. M. Macsweene, Exp. Cell Res., 1977, 107, 179–190 CrossRef CAS.
- A. A. Shvedova, E. R. Kisin, D. Porter, P. Schulte, V. E. Kagan, B. Fadeel and V. Castranova, Pharmacol. Ther., 2009, 121, 192–204 CrossRef CAS PubMed.
- E. G. R. Fernandes, V. Zucolotto and A. A. A. D. Queiroz, J. Macromol. Sci., Part A: Pure Appl. Chem., 2010, 47, 1203–1207 CrossRef CAS.
- W. Lew, J. J. Oppenheim and K. Matsushima, J. Immunol., 1988, 140, 1895–1902 CAS.
- S. Hofmanna, H. Hagenmüllera, A. M. Kocha, R. Müllerb, G. V. Novakovicc, D. L. Kaplane, H. P. Merklea and L. Meinel, Biomaterials, 2007, 28, 1152–1162 CrossRef PubMed.
- S. Huang and D. E. Ingber, Exp. Cell Res., 2000, 261, 91–103 CrossRef CAS PubMed.
- Z. Liu, X. Dong, L. Song, H. Zhang, L. Liu, D. Zhu, C. Song and X. Leng, J. Biomed. Mater. Res., Part A, 2013 DOI:10.1002/jbm.a.34729.
- N. R. Jacobsen, G. Pojana, P. White, P. Møller, C. A. Cohn, K. S. Korsholm, U. Vogel, A. Marcomini, S. Loft and H. Wallin, Environ. Mol. Mutagen., 2008, 49, 476–487 CrossRef CAS PubMed.
- I. F. F. Benzie and J. J. Strain, Anal. Biochem., 1996, 239, 70–76 CrossRef CAS PubMed.
- R. R. Mitry, R. D. Hughes, M. M. Aw, C. Terry, G. Mieli-Vergani, R. Girlanda, P. Muiesan, M. Rela, N. D. Heaton and A. Dhawan, Cell Transplant., 2003, 12, 69–74 CrossRef.
- A. M. Brzozowski, U. Derewenda, Z. S. Derewenda, G. G. Dodson, D. M. Lawson, J. P. Turkenburg, F. Bjorkling, B. H. Jensen, S. A. Patkar and L. Thim, Nature, 1991, 351, 491–494 CrossRef CAS PubMed.
- N. K. Singh, B. D. Purkayastha, J. K. Roy, R. M. Banik, M. Yashpal, G. Singh, S. Malik and P. Maiti, ACS Appl. Mater. Interfaces, 2010, 2, 69–81 CAS.
- V. E. Kagan, N. V. Konduru, W. Feng, B. t. Allen, J. Conroy, Y. Volkov, I. I. Vlasova, N. A. Belikova, N. Yanamala, A. Kapralov, Y. Y. Tyurina, J. Shi, E. R. Kisin, A. R. Murray, J. Franks, D. Stolz, P. Gou, j. K. Seetharaman, B. Fadeel, A. Star and A. A. Shvedova, Nat. Nanotechnol., 2010, 5, 354–359 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60170f |
| This journal is © The Royal Society of Chemistry 2014 |