 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programmable polymer-DNA hydrogels with dual input and multiscale responses†
Giovanna
Sicilia
a,
Christine
Grainger-Boultby
a,
Nora
Francini
a,
Johannes P.
Magnusson
a,
Aram O.
Saeed‡
a,
Francisco
Fernández-Trillo
b,
Sebastian G.
Spain
*a and
Cameron
Alexander
*a
aSchool of Pharmacy, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: sebastian.spain@nottingham.ac.uk; cameron.alexander@nottingham.ac.uk; Fax: +44 (0)115 951 5122; Tel: +44 (0)115 846 7678
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 4th October 2013
Abstract
Combination switchable polymer-DNA hydrogels have been synthesized to respond to both a specific oligonucleotide recognition signal and a non-specific but biorelevant environmental trigger. The hydrogels exhibit rheological properties that can be modulated through interaction with complementary DNA strands and/or reduction. Furthermore, individual and combined oligonucleotide recognition and reduction responses allow control over pore sizes in the gel, enabling programmable release and transport of objects ranging from the nano- to micro-scale.
Introduction
The structural information in nucleic acids enables encoding at the nanoscale of a variety of dynamic assemblies1–5 able to switch their conformation in response to external stimuli such as pH, small molecules (salts, ions), proteins, oligonucleotides and temperature.6–10 Among the wide range of nucleic acid based materials,11 DNA polymer hybrids and hydrogels have attracted great interest as biocompatible stimuli-responsive systems.9,12–15 In addition to the advantageous properties of hydrogels for biomedical applications,16–19 the recognition behavior, thermal stability and visco-elasticity of DNA-hybrid materials can further be tuned by changing the size and the number of branched DNA monomers.20,21 The ability of DNA hybrid networks to switch between mechanically distinct conformational states based on DNA hybridization and strand displacement mechanisms22–26 has been used for a number of label-free sensing applications with remarkable sensitivities.27 In addition to responses at the level of individual nucleic acid sequences, Lin28 and Wei29 demonstrated the hybridization triggered sol–gel transition of polyacrylamide DNA hydrogels via macroscopic rheological properties and by following the release of bioactive molecules using thrombin-binding aptamers as crosslinking agents, respectively. These studies indicate the potential for DNA-based materials to report on nanoscale phenomena via responses amplified to the macroscale.30Hydrogels crosslinked solely by DNA sequences are characteristically weak and swellable unless significant quantity of crosslinker is employed. However, on cost grounds it is important to minimize the amount of DNA in a hydrogel whilst retaining a sequence-specific response. Thus for many practical applications additional crosslinkers are needed to strengthen the gel. This strengthening must induce structural integrity without dampening the nucleic acid response. It would be advantageous therefore if a primary response in a hydrogel, e.g. to a nucleic acid sequence, could be coupled to a gel which is robust, but which also contains a secondary response mechanism that in turn can generate a structural signal and/or amplification.
Here, we describe a gel that is physically and chemically crosslinked via DNA base-pairing and disulfide bonds respectively (Scheme 1). The gel responds to DNA in a sequence specific manner while retaining macroscale integrity, and also responds to reducing conditions via disulfide bond cleavage. In addition to improving structural properties through the dual crosslinking, the resulting gels are capable of performing programmable logic operations in response to two stimuli, extending their switching capabilities from the dimensions of oligonucleotide sequences through to the microscopic scale with a view to applications in sensing and controlled release devices.
 | ||
| Scheme 1 Polymerization of acrylamide with DNA and disulfide crosslinkers results in a hydrogel that responds to a specific DNA sequence and a non-specific environmental trigger. | ||
Materials and methods
Materials
All oligonucleotides (HPLC purified, Table 1) were purchased from Biomers.net GmbH (Ulm, Germany) and used without further purification. Acrylamide (AAm, ≥98%), 3-hydroxypicolinic acid (3-HPA, ≥99%) and ammonium citrate dibasic (99%) were purchased from Fluka. N,N′-Bis(acryloyl)cystamine (BAC, 98%) and methacryloyl chloride (97%) were purchased from Alfa Aesar. Pentafluorophenol (>99%) was purchased from Fluorochem. Potassium carbonate (K2CO3, anhydrous), silica (60 Å, 35–70 μm) and sodium chloride were purchased from Fisher Scientific. N,N,N′,N′-Tetramethylethylenediamine (TEMED, 99%), ammonium persulfate (APS, 98%), water (BPC grade, DNase and RNase free), N,N-diisopropylethylamine (DIPEA, 99.5%), FITC-dextran 150 kDa, dimethyl sulfoxide (DMSO, 99.9% anhydrous), chloroform-d (99.96 atom% D), Trizma® hydrochloride, ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA), microparticles based on melamine resin, carboxylate-modified Nile Blue labeled (1 μm), FITC labeled (3 μm) and rhodamine B labeled (6 μm) were purchased from Sigma-Aldrich. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) was purchased from Thermo Scientific. Dulbecco's Phosphate Buffered Saline (DPBS, without Ca2+ and Mg2+) was purchased from Lonza Group. All other solvents were Fisher Analytical or HPLC grade. All chemicals were used as received unless otherwise stated.Analytical methods
Synthetic methods
1H NMR (400 MHz, CDCl3) δ = 6.55–6.40 (m, 1H), 5.93 (dd, J = 1.6, 1.1 Hz, 1H), 2.11 (dd, J = 1.6, 1.0 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 163.02, 141.28 (ddq, J = 251.7, 12.6, 4.2 Hz), 139.42 (dtt, J = 253.0, 13.5, 4.0 Hz), 137.89 (dtdd, J = 251.6, 13.7, 5.1, 3.1 Hz), 133.72, 129.80, 125.38, 125.36, 125.34, 18.11.
19F NMR (376 MHz, CDCl3) δ = −152.40 to −153.43 (m, 2F), −158.34 (t, J = 21.6 Hz, 1F), −161.91 to −163.23 (m, 2F).
FT-IR: ν = 1760, 1520, 1093, 996 cm−1.
| Oligo | 5′ Modification | Sequence (5′–3′) |
|---|---|---|
| A | Aminohexyl | TAACAGGATTAGCAGAGCGAGG |
| B | Aminohexyl | CCTCGCTCTGCTAATCC |
| C | CCTCGCTCTGCTAATCCTGGTA | |
| D | TTCAATCTCAACGGCTTCACCG |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :B1.
Equimolar quantities of the oligonucleotides A1 and B1 were dissolved in annealing buffer (10 mM Trizma, 50 mM NaCl and 1 mM EDTA, pH 7.5) to a final concentration of 0.93 mM. The solution was heated to 95 °C for 5 minutes then allowed to cool slowly to room temperature (approximately 50 minutes).
:B1.
Equimolar quantities of the oligonucleotides A1 and B1 were dissolved in annealing buffer (10 mM Trizma, 50 mM NaCl and 1 mM EDTA, pH 7.5) to a final concentration of 0.93 mM. The solution was heated to 95 °C for 5 minutes then allowed to cool slowly to room temperature (approximately 50 minutes).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BAC:AAm of 1:107:3357. Typically, AAm (6.72 mg, 94 μmol) and BAC (0.8 mg, 3 μmol) were dissolved in 25% ethanol solution (160.6 μL). Hybrid A1:B1 (0.35 mg, 0.03 μmol) in annealing buffer (30 μL) and TEMED (4.5 μL, 30 μmol) were added to the reaction mixture. Nitrogen was bubbled through the solution for 2 minutes The polymerization was initiated by addition of APS solution (10% w/v, 9.4 μL, 0.41 μmol) and allowed to proceed at room temperature. Gel formation occurred within 1 h.
:BAC:AAm of 1:107:3357. Typically, AAm (6.72 mg, 94 μmol) and BAC (0.8 mg, 3 μmol) were dissolved in 25% ethanol solution (160.6 μL). Hybrid A1:B1 (0.35 mg, 0.03 μmol) in annealing buffer (30 μL) and TEMED (4.5 μL, 30 μmol) were added to the reaction mixture. Nitrogen was bubbled through the solution for 2 minutes The polymerization was initiated by addition of APS solution (10% w/v, 9.4 μL, 0.41 μmol) and allowed to proceed at room temperature. Gel formation occurred within 1 h.
Rheological experiments
Dynamic rheological studies were performed on an Anton Paar MCR 302 high pressure cell rheometer fitted with a 25 mm diameter cone plate with a 50 μm gap. Samples (300 μL) were loaded onto the bottom plate using a 200 μL micropipette fitted with sterile tips. The temperature of the bottom plate was maintained at 25 °C. The upper plate was lowered slowly until the desired gap was reached.Initially, a gelation test was carried out on a solution containing AAm, BAC, hybrid A1B1, TEMED and APS (prepared as described previously) to investigate the formation of the 4% (w/v) DNA-hybrid network. The mixture was pipetted onto the bottom plate of the rheometer immediately after the addition of the free radical initiator APS.
Subsequently, the gelation test was performed on the AAm–BAC–DNA hydrogel to monitor any rheological changes over time. The gel was synthesized as described previously and loaded 16 h after its formation onto the bottom plate of the rheometer.
Finally, the gelation test was performed on the AAm–BAC–DNA hydrogel treated with TCEP·HCl in order to investigate the presence of any residual network structure after disulfide bond cleavage. TCEP·HCl (41 μmol, 10 equiv., 9 μL) was added to the gel 16 h after its formation. The sample was pipetted onto the bottom plate of the rheometer 2 h after the addition of TCEP·HCl.
Cryo-scanning electron microscopy (Cryo-SEM)
Cryo-scanning electron microscopy was performed on a JEOL JSM-6060LV scanning electron microscope fitted with an Oxford CT 1500 cryo-preparation chamber with sputter coating head and Dewar cooling apparatus. The system comprised an SEM cold stage with anti-contaminator, cryo-station with slushing chamber, temperature and cryo-trans controller units and gas flow regulators for both nitrogen and argon. All samples were loaded onto suitable stub holders with a 200 μL micropipette fitted with sterile tips. The point of the tip was cut to increase the diameter and thus minimise the shear stress applied to the gels. Samples were cryo-fixed by plunging them into sub-cooled nitrogen (nitrogen slush) close to the freezing point of nitrogen at −210 °C. The samples were then transferred in vacuo to the cold stage of the SEM cryo-preparation chamber where fracturing was performed with a fracture knife. Sublimation of ice was carried out by raising the temperature of the SEM cold stage to −90 °C. Once sublimation was observed, the samples were sputter coated with gold for 4 minutes and transferred into the SEM chamber for imaging. Images were acquired using an electron beam of 7–22 kV.For the cryo-SEM studies, AAm–BAC–DNA hydrogels were synthesized as described previously. Gels (64.2 μL each) were incubated for 16 h with oligo C (8.8 nmol, 1 equiv.), oligo D (8.8 nmol, 1 equiv.), TCEP·HCl (9.6 μmol, 10 equiv.) or water. Concentrations were such that the added volume was maintained at 6 μL. An additional hydrogel was incubated with both TCEP·HCl (9.6 μmol, 6 μL, 10 equiv.) and oligo C (8.8 nmol, 6 μL, 1 equiv.). Pore sizes were measured using the ImageJ software.31 Specifically, fifty pores were analysed for each sample considering both their short and long diameters. Pore size frequency distributions were calculated using GraphPad Prism 6.0a (GraphPad Inc.) with a bin width of 0.5 μm centered at 0.25–9.75 μm.
Release study of FITC-dextran loaded AAm–BAC–DNA hydrogels
The release profiles of FITC-dextran (150 kDa) from the 4% (w/v) AAm–BAC–DNA hydrogels were analyzed by fluorescence spectroscopy. Hydrogels (200 μL) were synthesized as described previously with the addition of 15 μg of FITC-dextran (150 kDa). Excess of FITC-dextran was removed by performing one washing step with 500 μL of DPBS buffer. Analysis of the washing buffer determined the encapsulation efficiency to be >99% in all cases.Gels were incubated with DPBS buffer (500 μL) containing either oligo C (180 μg, 28 nmol, 1 equiv.), oligo D (185 μg, 28 nmol, 1 equiv.), TCEP·HCl (8.8 mg, 30.7 μmol, 10 equiv.) or buffer alone at 37 °C. At each time point a sample (100 μL) was removed from each solution and replaced with the same volume of the appropriate incubation solution. Samples were diluted with 400 μL of DPBS buffer and transferred into a quartz fluorescence cuvette. Fluorescence measurements were performed on a Varian Cary Eclipse with the following parameters: λex = 493 nm, λem = 518 nm, excitation and emission slit widths = 2.5 nm. Release experiments were performed in triplicate (Fig. S4A†). As not all free FITC-dextran was removed at each time point fluorescence measurements were corrected using:
| Ii,corrected = Ii − (0.8 × Ii−1) |
where [FITC]sol is the concentration of FITC-dextran in the supernatant, d is the dilution factor (5 in all cases) and m is the gradient of the standard curve (7.25 in all cases). Mass release at time point i (Mi) was thus determined by:
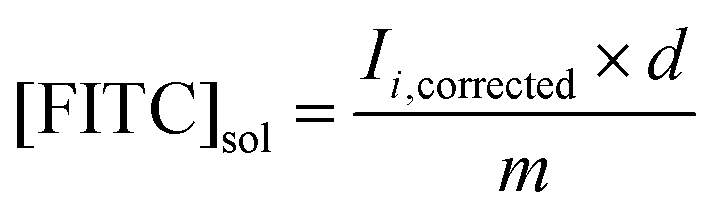
| Mi = [FITC]sol × V |
Transport of microparticles through AAm–BAC–DNA hydrogel
Particle translocation through the hydrogel was observed using an Evos f1 fluorescence microscope fitted with three light filter sets (‘cubes’) for detection of blue, green and red channels DAPI (λex = 360 nm, λem = 447 nm), GFP (λex = 470 nm, λem = 525 nm) and RFP (λex = 530 nm, λem = 593 nm). Images were taken after 30 minutes from the time of particle loading, by placing 5 μL of the hydrogel taken from the bottom part of the capillary tube between two microscope slides.
Results and discussion
The fundamental design features of the DNA hybrid gels were based on strand association and displacement sequences, and reductively-cleavable disulfide crosslinks. Oligonucleotides were designed such that the primary crosslinks were formed by two strands (A and B, Table 1) with a complementary 17-mer sequence and a 5-base overhang to enable subsequent displacement. Strand C was designed with a fully complementary sequence to A in order to break A–B links, while strand D was a non-sense sequence and thus intended as a control. In order to introduce the nucleic acid functionality into a hydrogel network, polymerizable DNA strands A1 and B1 were synthesized by reacting 5′-amino oligonucleotides A and B with pentafluorophenyl methacrylate (Fig. S1†). Conjugation was confirmed by HPLC and MALDI-TOF mass spectrometry (Fig. S2A and B†). The DNA crosslinker hybrid A1:B1 was produced by hybridization of methacrylamido strands A1 and B1 under standard conditions. Hydrogels were synthesized by copolymerization of acrylamide with N,N′-bis(acryloyl)cystamine and hybrid A1:B1 crosslinkers using ammonium persulfate and tetramethylethylenediamine as the initiating system. The formation of the crosslinked AAm–BAC–DNA hydrogels was detectable visually and confirmed by rheometry (Fig. 1A). At early timepoints (<60 minutes), the storage modulus (G′) was greater than the loss modulus (G′′) indicating the existence of a weak network prior to polymerization occurring. Although the structural details of this network could not be determined fully by rheology we have found that even small hydrophobic modifications to oligonucleotides could result in phase separation and self-assembly (data not shown) so a similar assembly process may be the cause of this weak network. At timepoints of 60–100 minutes after initiation, there was a rapid increase in, and convergence of, G′ and G′′ indicating the formation of a viscous solution with little elastic contribution as would be expected for the polymerization with minimal crosslinking. After approximately 100 minutes, G′ increased more rapidly than G′′ implying an increasing elastic contribution from crosslinked sections and gel formation as the polymerization neared completion. The double crosslinking was verified by rheological measurements in the absence/presence of the reducing agent tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl). Without TCEP·HCl the gel exhibited storage and loss moduli of 104–105 Pa (Fig. 1B, closed circles). After TCEP·HCl reduction the moduli decreased by 3 orders of magnitude to 101–102 Pa confirming the presence of a weak, secondary network (Fig. 1B, open circles). Frequency sweep tests (Fig. 1C) confirmed the decrease in moduli after treatment with TCEP·HCl. The low variation of G′ and G′′ with angular velocity (Fig. 1C, open circles) demonstrated the ability of our system to retain a stable, partially crosslinked network after the cleavage of the disulfide bonds. To gain insight into microscopic changes following the exposure to a reducing agent and/or a specific oligonucleotide sequence, gel morphology and pore structure were studied with cryo-scanning electron microscopy and pore sizes were measured from micrographs using ImageJ software (Table 2).31 Prior to freezing, gel samples were incubated in the presence/absence of oligonucleotides C and D, TCEP and water to determine the effect of each treatment on morphology.
 | ||
| Fig. 1 Rheometry measurements. Storage modulus (G′, blue points), loss modulus (G′′, red points) and tan δ (black lines). (A) In situ gelation test of the synthesis of AAm–BAC–DNA hydrogel. (B) Gelation test of AAm–BAC–DNA hydrogel before (G′ and G′′ filled circles, tan δ solid black line) and after (G′ and G′′ open circles, tan δ broken black line) reduction with TCEP·HCl. (C) Frequency sweep test of AAm–BAC–DNA hydrogel before (G′ and G′′ filled circles, tan δ solid black line) and after (G′ and G′′ open circles, tan δ broken black line) reduction with TCEP·HCl. | ||
As apparent from Fig. 2, varying pore sizes were observed in the gels after formation and after treatment with the oligonucleotides and/or reductant. The initial gel (Fig. 2A) displayed a regular structure with pore sizes of approximately 1–2 μm (mean 1.6 ± 0.49 μm). Similar structures have been observed in crosslinked polyacrylamide gels, and also in crosslinked networks formed with polyelectrolytes. Although we cannot rule out some structural changes during the cryo-SEM sample preparation, the morphologies in the initial gels were suggestive of partial phase separation of the different neutral and charged components during the polymerization and crosslinking process.32 Addition of complementary oligonucleotide C (Fig. 2B) induced a shift to a more open structure with larger pore sizes and a broader distribution (mean 3.3 ± 1.3 μm). Addition of TCEP·HCl (Fig. 2C) resulted in significant changes in morphology with formation of elongated pores and a consequent broadening of the size distribution (mean 3.1 ± 2.2 μm). When the gel was treated with both oligonucleotide C and TCEP·HCl no network structure was observed (Fig. 2D) confirming the biodegradable nature of the DNA hybrid gel. Conversely, addition of scrambled oligonucleotide D (Fig. S3A†) resulted in little change in pore size (mean 1.6 ± 0.63 μm) and a similarly small increase in pore size was observed on addition of an equivalent volume of water alone (1.6 ± 0.73 μm) implying this change was due to swelling rather than breaking of crosslinks (Fig. S3B†).
 | ||
| Fig. 2 Cryo-scanning electron micrographs and pore size frequency distributions (inset) of (A). AAm–BAC–DNA hydrogel; (B) AAm–BAC–DNA hydrogel after incubation with complementary oligonucleotide C; (C) AAm–BAC–DNA after incubation with TCEP·HCl; and (D) AAm–BAC–DNA after incubation with both oligonucleotide C and TCEP·HCl. | ||
The ability to generate matrices of different pore size through the breaking of inter-chain crosslinks suggested the AAm–BAC–DNA hydrogels might exhibit controllable release properties. Therefore, hydrogels (4% w/v in water) were synthesized in the presence of FITC-dextran (150 kDa; DH ∼ 8.5 nm), chosen as an easily detectable model of a therapeutic biopolymer. After removal of unencapsulated FITC-dextran, hydrogels were incubated at 37 °C with TCEP·HCl, oligonucleotides C and D, or buffer alone. Samples of the supernatant were analysed by fluorescence spectroscopy and cumulative release determined. As expected, release profiles (Fig. 3A) were dependent on the incubation conditions. Gels treated with TCEP rapidly liberated FITC-dextran with ∼82% of the polysaccharide detected in solution after 10 h. The level of FITC-dextran reached a maximum of ∼95% after 149 h. Treatment with complementary oligonucleotide C yielded a slower release profile with ∼43% release after 10 h followed by a gradual release to a maximum of ∼98% after 149 h. Gels incubated with scrambled oligonucleotide D, or buffer alone, displayed similar release profiles with ∼14% release after 10 h rising to ∼70% at the final time point. These data demonstrated the selectivity of AAm–BAC–DNA hydrogels as switchable release systems, and suggested the gels could be used as a matrix which generated a response upon detection of a characteristic DNA sequence.
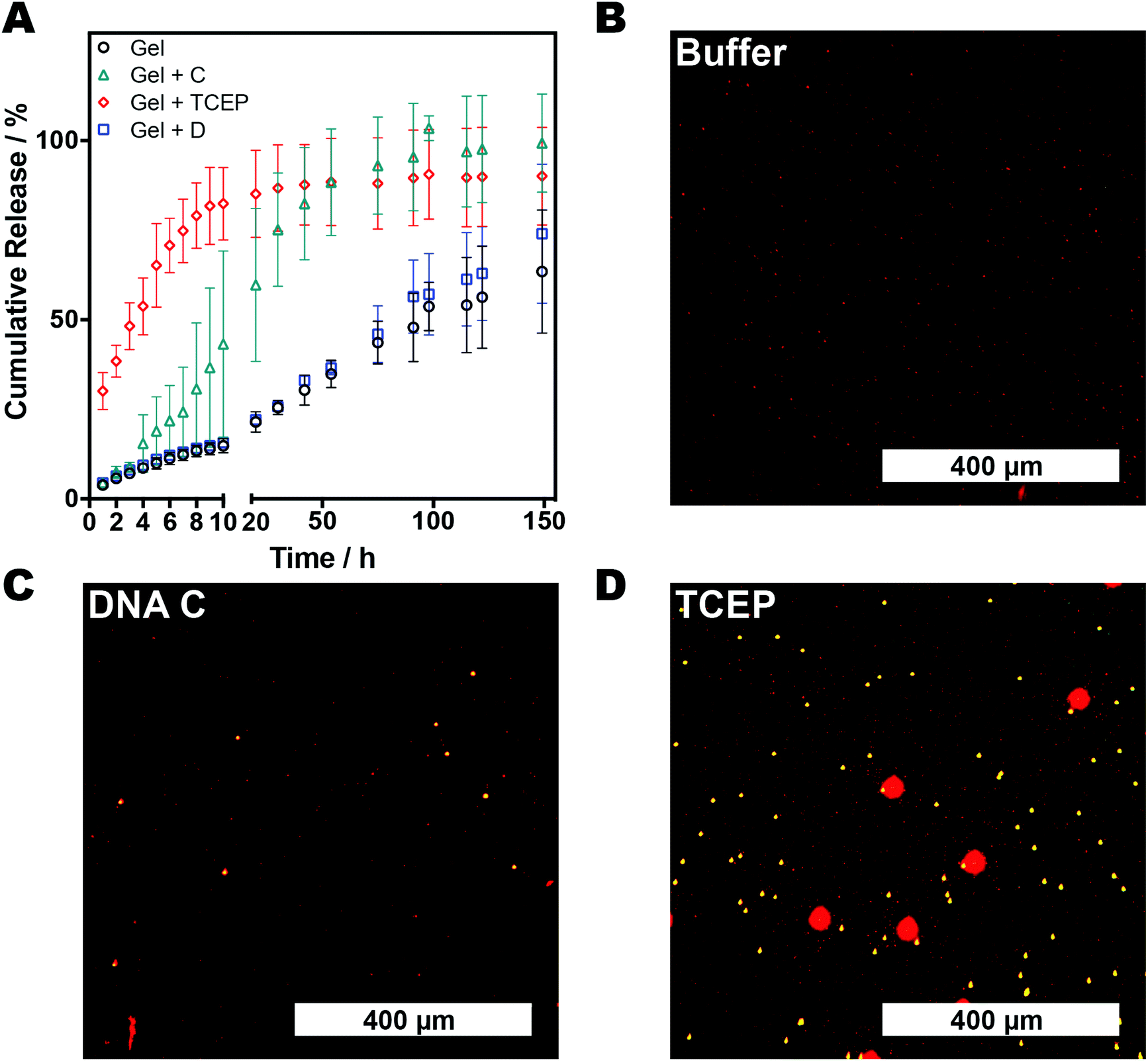 | ||
| Fig. 3 (A) Release profiles of FITC-dextran (150 kDa) from AAm–BAC–DNA hydrogels. (B–D) Fluorescence micrographs of fluorescent microparticle transport through AAm–BAC–DNA gels pre-incubated with buffer (B), oligonucleotide C (C) or TCEP·HCl (D). Gels were loaded into a capillary tube before addition of a mixture of 1, 3 and 6 μm particles to the top of the gel. After 30 minutes samples were removed from the bottom of the gel and imaged with fluorescence microscopy. | ||
We therefore set out to test the ability of the DNA hybrid network to perform a simple biosensor-type operation, in which a change in gel structure could be used to transduce the signal from a DNA sequence detection event. Fluorescent microparticles (1, 3 and 6 μm in diameter) were chosen as easily detectable ‘signals’ and their transport through gels exposed to different stimuli examined. A control AAm–BAC–DNA gel, and two analogous hydrogels treated with oligonucleotide C and TCEP respectively, were placed in capillary tubes and loaded with a mixture of microparticles. Aliquots were taken from the bottom part of the gel 30 minutes after loading. As expected based on the range of pore sizes in the matrix determined by gel response, the 1 μm Nile-Blue marked particles were able to flow across the three hydrogel matrices under all conditions (Fig. 3B–D). Addition of oligonucleotide C enabled the 3 μm particles to cross the gel (Fig. 3C), while only in the gels treated with TCEP·HCl were the 6 μm Rhodamine B marked particles able to transport (Fig. 3D). The same pattern of particle translocation was obtained with three AAm–BAC–DNA gels loaded only with one particle type (Fig. S5−S7†). In all cases, directed changes in crosslinking density of the DNA network, induced by either a specific stimulus, i.e. complementary DNA, or a non-specific trigger such as TCEP reduction, resulted in size-gated particle transport. The intention of these experiments was primarily to show that a response over the range of a few nm i.e. a DNA sequence change or breaking of disulfide crosslinks, could invoke a response detectable at a very different length scale. The movement of beads through the network yielded end-points which were easily detectable by optical microscopy, and the optical read-out thus served to amplify the chemical response of the gels at the DNA and/or disulfide crosslink regions. It is important to note that these experiments most likely relied on a kinetic resolution of changes in pore size, in that we measured transport of the beads across the gel over a relatively short period (30 minutes), rather than absolute entrapment of particles based on gel pore size. However, in principle there is no reason why specific crosslink densities in these gels based on DNA strand length and disulfide linker density could not be used for highly selective release of components based purely on size, leading to further potential uses of these hybrid gel materials.
There have been a number of elegant papers demonstrating sensor and controlled release responses from DNA nanomaterials,33–37 but there are nevertheless some practical issues that still need to be addressed with DNA hydrogels. Structural changes in a gel based on differences in DNA hybridisation states are limited by the constrained motion of nucleic acid strands bound to a network, but increasing the DNA loading to enhance a response invokes a cost penalty. It should also be stressed that not only is DNA hybridisation and strand displacement hindered when one strand is tethered to a matrix, but the diffusion of multiply-charged species in a charged network is inherently slow. This diffusion barrier to response cannot be overcome by increasing the amount of ‘responding components’ i.e. the number of strand pairing and displacement groups in a network, as indeed the greater the number of DNA crosslinks, the slower may be the observed response. Finally, while it is possible to observe and amplify nanometre changes in a gel with interferometry and other specialised instrumentation, for a number of field sensing operations, a simple UV fluorescence or basic microscopy response is preferable. Therefore, for the gels in this study we added the disulfide component to the matrix and utilised a microscale phenomenon, in this case transport of micron-size fluorescent beads, in order to have a response that could readily be detected by a cheap UV source or entry-grade microscope. As is readily apparent in Fig. 3, changes in diffusion of FITC-dextran occur following addition of a competing DNA strand, and the same strand displacement reaction can also be detected by the flow of 3 μm beads. One could therefore envisage detection of specific DNA or miRNA sequences through an initial response at the dsDNA crosslink level and amplified by relative flow of particles across a gel before and after a reduction step to accelerate flow, without the need for expensive optics.
Conclusions
In conclusion, we have shown the switchable capabilities at both the macroscopic and microscopic level of a novel dual stimuli responsive DNA hydrogel using rheological tests, cryo-SEM, fluorescence spectroscopy and microscopy. We have demonstrated that the three-dimensional network structure can be designed to undergo conformational changes involving an enlargement of the pore size in the presence of either a specific DNA sequence or a reducing agent. The significance of these data lies in the ability to amplify a nanoscale signal i.e. DNA recognition into a microscale response such as transport of fluorescent particles. In turn, this enables facile detection of molecular recognition events through pore-size change and concomitant matrix rearrangements. In addition, because these hybrid DNA gels have better mechanical properties than DNA-only systems yet retain recognition capability, they represent a promising platform for sensing, signalling and release applications.Acknowledgements
We thank the UK EPSRC (grants EP/H005625/1, EP/G042462/1, EP/H028277/1, EP/E021042/1) and the University of Nottingham for a Scholarship (GS).Notes and references
- B. Chakraborty, N. Jonoska and N. C. Seeman, Chem. Sci., 2012, 3, 168–176 RSC.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- T. Wang, D. Schiffels, S. M. Cuesta, D. K. Fygenson and N. C. Seeman, J. Am. Chem. Soc., 2012, 134, 1606–1616 CrossRef CAS PubMed.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, R. K. O'Reilly and A. J. Turberfield, J. Am. Chem. Soc., 2012, 134, 1446–1449 CrossRef CAS PubMed.
- J. Bath, S. J. Green and A. J. Turberfield, Angew. Chem., Int. Ed., 2005, 44, 4358–4361 CrossRef CAS PubMed.
- Y. H. Roh, R. C. H. Ruiz, S. Peng, J. B. Lee and D. Luo, Chem. Soc. Rev., 2011, 40, 5730–5744 RSC.
- S. Chatterjee, J. B. Lee, N. V. Valappil, D. Luo and V. M. Menon, Nanoscale, 2012, 4, 1568–1571 RSC.
- M. J. Campolongo, J. S. Kahn, W. Cheng, D. Yang, T. Gupton-Campolongo and D. Luo, J. Mater. Chem., 2011, 21, 6113–6121 RSC.
- M. Gao, K. Gawel and B. T. Stokke, Soft Matter, 2011, 7, 1741–1746 RSC.
- C. K. McLaughlin, G. D. Hamblin and H. F. Sleiman, Chem. Soc. Rev., 2011, 40, 5647–5656 RSC.
- J. Zheng, J. J. Birktoft, Y. Chen, T. Wang, R. Sha, P. E. Constantinou, S. L. Ginell, C. Mao and N. C. Seeman, Nature, 2009, 461, 74–77 CrossRef CAS PubMed.
- F. E. Alemdaroglu and A. Herrmann, Org. Biomol. Chem., 2007, 5, 1311–1320 CAS.
- J. Lee, S. M. Peng, D. Y. Yang, Y. H. Roh, H. Funabashi, N. Park, E. J. Rice, L. W. Chen, R. Long, M. M. Wu and D. Luo, Nat. Nanotechnol., 2012, 7, 816–820 CrossRef CAS PubMed.
- M. Nishikawa, Y. Mizuno, K. Mohri, N. Matsuoka, S. Rattanakiat, Y. Takahashi, H. Funabashi, D. Luo and Y. Takakura, Biomaterials, 2011, 32, 488–494 CrossRef CAS PubMed.
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797–801 CrossRef CAS PubMed.
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Delivery Rev., 2007, 59, 263 CrossRef CAS PubMed.
- J. Kopeček and J. Yang, Angew. Chem., Int. Ed., 2012, 51, 7396–7417 CrossRef PubMed.
- T. Miyata, T. Uragami and K. Nakamae, Adv. Drug Delivery Rev., 2002, 54, 79–98 CrossRef CAS PubMed.
- H. Yan, A. Nykanen, J. Ruokolainen, D. Farrar, J. E. Gough, A. Saiani and A. F. Miller, Faraday Discuss., 2008, 139, 71–84 RSC.
- Y. Murakami and M. Maeda, Biomacromolecules, 2005, 6, 2927–2929 CrossRef CAS PubMed.
- Y. Murakami and M. Maeda, Macromolecules, 2005, 38, 1535–1537 CrossRef CAS.
- G. Seelig, B. Yurke and E. Winfree, J. Am. Chem. Soc., 2006, 128, 12211–12220 CrossRef CAS PubMed.
- A. J. Turberfield, J. C. Mitchell, B. Yurke, A. P. Mills Jr., M. I. Blakey and F. C. Simmel, Phys. Rev. Lett., 2003, 90, 118102 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- H. Z. Kang, H. P. Liu, X. L. Zhang, J. L. Yan, Z. Zhu, L. Peng, H. H. Yang, Y. M. Kim and W. H. Tan, Langmuir, 2011, 27, 399–408 CrossRef CAS PubMed.
- E. J. Cheng, Y. L. Li, Z. Q. Yang, Z. X. Deng and D. S. Liu, Chem. Commun., 2011, 47, 5545–5547 RSC.
- M. J. Banholzer, J. E. Millstone, L. Qin and C. A. Mirkin, Chem. Soc. Rev., 2008, 37, 885–897 RSC.
- D. C. Lin, B. Yurke and N. A. Langrana, J. Biomech. Eng.-Trans. ASME, 2004, 126, 104–110 CrossRef.
- B. Wei, I. Cheng, K. Q. Luo and Y. Mi, Angew. Chem., Int. Ed., 2008, 47, 331–333 CrossRef CAS PubMed.
- T. Liedl, H. Dietz, B. Yurke and F. C. Simmel, Small, 2007, 3, 1688–1693 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- Z. L. Wu, T. Kurokawa, S. Liang and J. P. Gong, Macromolecules, 2010, 43, 8202–8208 CrossRef CAS.
- P. K. Lo, P. Karam, F. A. Aldaye, C. K. McLaughlin, G. D. Hamblin, G. Cosa and H. F. Sleiman, Nat. Chem., 2010, 2, 319–328 CrossRef CAS PubMed.
- F. A. Aldaye and H. F. Sleiman, J. Am. Chem. Soc., 2007, 129, 4130–4131 CrossRef CAS PubMed.
- M. Endo, Y. Yang and H. Sugiyama, Biomater. Sci., 2013, 1, 347–360 RSC.
- Y.-X. Zhao, A. Shaw, X. Zeng, E. Benson, A. M. Nystrom and B. Hogberg, ACS Nano, 2012, 6, 8684–8691 CrossRef CAS PubMed.
- X. Shen, Q. Jiang, J. Wang, L. Dai, G. Zou, Z.-G. Wang, W.-Q. Chen, W. Jiang and B. Ding, Chem. Commun., 2012, 48, 11301–11303 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed methods for synthesis, rheology and further figures depicting particle transport across gels. See DOI: 10.1039/c3bm60126a |
| ‡ Current Address: School of Pharmacy, University of East Anglia, Norwich, NR4 7TJ, UK |
| This journal is © The Royal Society of Chemistry 2014 |