 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive functionalized mesoporous silica nanoparticles for drug release in response to various biological stimuli†
Xin
Chen
ab,
Xiaoyu
Cheng
ab,
Alexander H.
Soeriyadi
ab,
Sharon M.
Sagnella
ad,
Xun
Lu
ab,
Jason A.
Scott
c,
Stuart B.
Lowe
a,
Maria
Kavallaris
ad and
J. Justin
Gooding
*ab
aAustralian Centre for Nanomedicine, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: justin.gooding@unsw.edu.au
bSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia
cSchool of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia
dChildren's Cancer Institute Australia, Lowy Cancer Research Centre, UNSW Australia, Sydney, NSW 2052, Australia
First published on 23rd September 2013
Abstract
A silica-based mesoporous nanosphere (MSN) controlled-release drug delivery system has been synthesized and characterized. The system uses L-cysteine derivatized gold nanoparticles (AuNPs), bound to the MSNs using Cu2+ as a bridging ion. The AuNPs serve as removable caps that hinder the release of drug molecules inside the amino functionalized MSN mesoporous framework. The modified MSNs themselves exhibit negligible cytotoxicity to living cells, as revealed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The drug delivery system requires one of two biological stimuli to trigger drug release. These stimuli are either: low pH (pH < 5); or elevated levels of adenosine triphosphate (ATP) (concentration > 4 mM). The feasibility of biologically controlled release was demonstrated through the stimuli-induced removal of the AuNP caps over the MSN releasing the anticancer drug doxorubicin. We envisage that this MSN system could play a significant role in developing new generations of controlled-release delivery vehicles.
1. Introduction
Stimuli-responsive controlled-release systems have begun to attract attention because of their potential applications in the area of targeted drug and gene delivery.1–3 Drawing inspiration from the biological systems in which they will be applied, self-assembled carriers such as vesicles and micelles formed from organic molecules have been extensively investigated as drug delivery systems due to their biodegradability and biocompatibility.4–8 Limitations of such systems can include limited drug loading capacity and poor stability in blood after injection (leading to immediate release of encapsulated compounds upon dispersal in blood9–11). In addition, the release mechanism of many biodegradable organic molecule-based drug delivery systems rely on hydrolysis-induced erosion of the carrier structure.12–14An alternative to organic molecule based drug delivery vehicles is inorganic particulate systems such as mesoporous silica nanoparticles (MSN). Surface-functionalized MSNs have begun to be considered as potential stimuli-responsive controlled-release systems.15–17 The interest in MSNs is because they possess stable mesoporous structures, large surface areas, the ability to easily functionalize the external and/or internal area and have been shown to exhibit a “zero premature release” property. The latter is particularly useful when the drug to be delivered is toxic (e.g. antitumor drugs) or its therapeutic dosage requires precise control. Furthermore, silica-based materials are also known for their biocompatibility as they slowly degrade to orthosilicic acid which is easily carried by the blood or lymphatic system and subsequently excreted through the kidneys.18–20 This removal of Si(OH)4 from the body is sufficiently rapid so as not to induce toxic effects.21
The initial controlled drug delivery systems employing MSNs as drug carriers were mainly based upon the physical and morphological properties of the MSNs.22–24 For instance, Vallet-Regi et al. loaded ibuprofen into a series of MSNs with different pore sizes and found that the release rate of the drug was linearly dependent on the pore size.15 Such a strategy results in the released of the drug beginning immediately after administration.22–24 In some applications this immediate release results in premature and untargeted drug release that could cause undesirable side effects to normal cells and organs.25–28 Therefore, it is highly desirable to design a controlled drug delivery system that can release the loaded drug specifically in the target environment by responding to appropriate external stimuli.29 To achieve this goal, a variety of stimuli-responsive surface-functionalized, end-capped MSNs have been developed. The Lin group, Wang group and Wu group have employed quantum dots,30 gold nanoparticles,31,32 iron oxide nanoparticles,33 and polymers34 as gatekeepers to cap the pores of MSN and prevent the loaded drug from being released. The opening/closing of the gatekeepers can be controlled by various external stimuli, such as disulfide reduction,30 photochemistry35 and redox chemistry.36
The purpose of the present work is to design a MSN drug delivery system with controlled drug release triggered by biologically-relevant stimuli. Further, we intend to demonstrate a system that is responsive to two different biological stimuli that potentially could work synergistically to ensure drug release only at the target site.
Drug carriers, upon reaching the target cells, are generally internalized by endocytosis37,38 and routed to the lysosomes.39 The internalized carrier can release the drug in one of two possible ways, or both: (1) within the lysosome, followed by drug diffusion,40–42 and/or (2) in the cytosol, following carrier escape from the lysosome.43 Thus for a specific cell, the drug release rate can be expressed by the following equation:
| Rr = RLDr + RrC |
Based on these two release processes, an AuNPs-capped-MSN controlled drug delivery system was synthesized which could be triggered by two different biologically-relevant stimuli, namely pH or concentration of adenosine triphosphate (ATP). Compared with the pH value in the bloodstream, the low pH in endosomes (pH = 4.5–5.044,45) means pH becomes an effective stimulus which not only triggers the drug release, but restrict the drug release to specific organelles in the cell. Although many of the internalized carriers will release the drug within the lysosome, some of the drug delivery vehicles may escape from the lysosome before drug release.43 Hence, there is also a need for a stimulus in the cytosol to trigger drug release. Owing to ATP being predominantly present in the cytosol,46 and assuming the concentration of ATP inside cells is high enough to release the drug (concentration range of 1–10 mM47,48), ATP is considered as a second stimulus for drug release in the cytosol. Scheme 1 shows the main design features of the AuNPs-capped-MSN based drug encapsulation and release system. Ion–ligand interactions were chosen to cause capping of the MSNs by AuNPs, as these AuNP caps can be removed when the pH is lower than 5 or when ATP is present. Fluorescein was chosen as a model drug to assess the drug loading and release behavior of the AuNPs-capped-MSNs under different conditions of pH and ATP concentration. In vitro cellular cytotoxicity tests were also conducted to evaluate the biocompatibility of the system. Thereafter the performance of the AuNPs-capped-MSN in killing HeLa cells with doxorubicin was assessed.
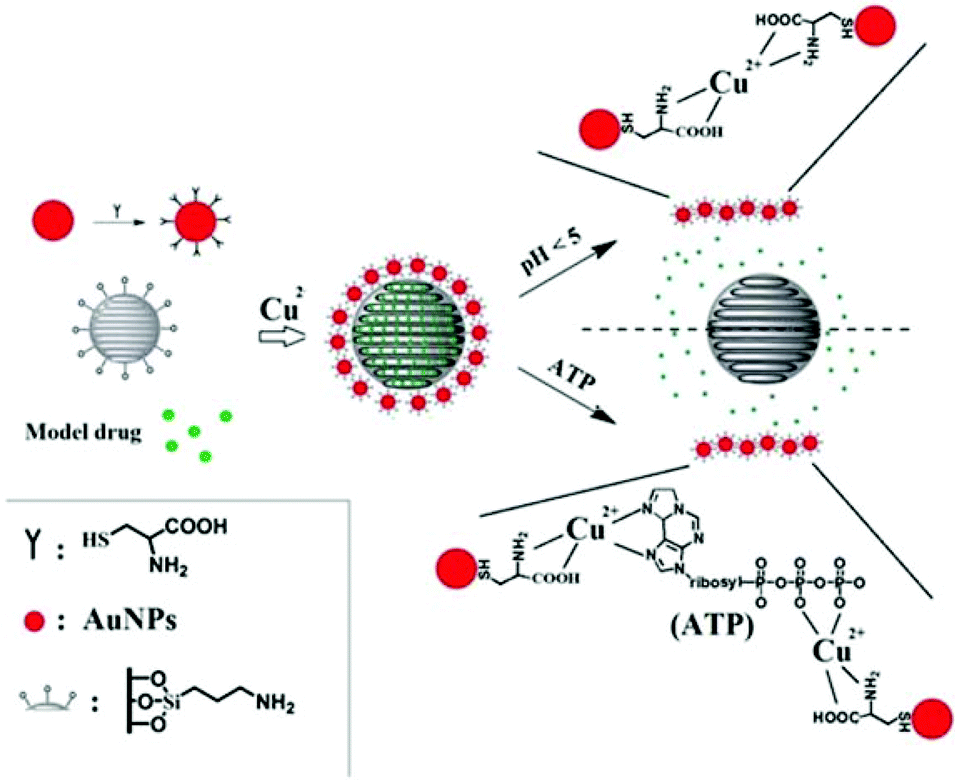 | ||
| Scheme 1 Schematic representation of the AuNPs-capped-MSN based drug delivery system. The controlled-release mechanism of the system is based on forming and breaking of ligand bond between the AuNPs caps, Cu2+ and the MSN hosts. The scheme is presented by (A) MSN functionalization, (B) AuNP functionalization, (C) AuNP capping of MSNs, (D) AuNP displacement by pH mechanism and (E) AuNP displacement by interaction with ATP. | ||
2. Materials and methods
2.1 Materials
Fluorescein sodium, cupric chloride, 3-aminopropyltriethoxysilane (APS), tetraethyl orthosilicate (TEOS), N-cetyltrimethylammonium bromide (CTAB) and ammonium hydroxide were purchased from Sigma-Aldrich (Sydney, Australia). Hydrogen tetrachloroaurate(III) trihydrate and L-cysteine were purchased from Sigma (Sydney, Australia), tri-sodium citrate (99% purity) was purchased from Ajax Chemical Co. (Sydney, Australia) and sodium borohydride (99%, Fluka). All the chemicals were analytical grade and used without further treatment.2.2 Synthesis of L-cysteine functionalized gold nanoparticles
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, strong oxidizer) and then with Milli-Q water followed by a rinsing in aqua regia (HCl–HNO3 = 3:1, highly corrosive) and then Milli-Q water. A vigorously stirred 20 mL solution containing 2.5 × 10−4 M gold chloride and 2.5 × 10−4 M trisodium citrate was heated to boil. Then 0.6 mL of ice-cold, freshly prepared 0.1 M NaBH4 solution was added to the solution. The solution turned light red immediately after addition of NaBH4 and the solution was left stirring for 5 min.
:1, strong oxidizer) and then with Milli-Q water followed by a rinsing in aqua regia (HCl–HNO3 = 3:1, highly corrosive) and then Milli-Q water. A vigorously stirred 20 mL solution containing 2.5 × 10−4 M gold chloride and 2.5 × 10−4 M trisodium citrate was heated to boil. Then 0.6 mL of ice-cold, freshly prepared 0.1 M NaBH4 solution was added to the solution. The solution turned light red immediately after addition of NaBH4 and the solution was left stirring for 5 min.
2.3 Synthesis of mesoporous silica nanoparticles (MSNs)
MSNs were prepared by a typical CTAB-templated, base-catalyzed sol–gel method.52 The pH value of 1000 mL deionized water was adjusted to approximately 11 with 52.8 mL ammonium hydroxide (29 wt% NH3 in water). The temperature was raised to 323 K, and then 1.12 g CTAB and subsequent 5.8 mL TEOS were added with rapidly stirring. After 2 h, the sample was centrifuged and washed thoroughly with distilled water and ethanol. The surfactant templates was then were removed by extraction using acidic methanol (9 mL of HCl–400 mL of methanol, 36 h) at 70 °C, which were further centrifuged, washed several times with ethanol and dried under vacuum for 20 h.2.4 Drug load and 3-aminopropyltriethoxysilane functionalized of MSN (MSN-1)
1 g MSN was dispersed in 50 mL anhydrous ethanol, and then 0.5 g fluorescein sodium or doxorubicin (Dox) was added. The mixture was stirred at room temperature for 24 h. After that, 3-aminopropyltriethoxysilane (1 mL, 50% in ethanol) was added into the suspension and stirred for another 8 h. The resultant solid was filtered, washed with ethanol, and then dried under vacuum to get MSN-1. The amount of loaded drug for MSN was determined by emission peak at 520 nm in fluorescence spectra for fluorescein sodium and 590 nm for Dox.2.5 Synthesis of gold nanoparticles capped MSN-1 (AuNPs-capped-MSNs)
According to the previous work in our group, amino and carboxyl group will form complexes with Cu2+, even though there is only 3 ppm Cu2+ in the system.51,53 Relevant research exposed that a minimum dietary value of Cu2+ for healthy growth in small mammal such as rabbits is at least 3 ppm in the diet54 and the lowest dose of Cu2+ that has been reported toxic when ingested by humans is 11 ppm.55 Thus Cu2+ was chosen to connect L-cysteine derivatized gold nanoparticles and MSN-1 to form AuNPs-capped-MSNs. Briefly, 10 mg MSN-1 was added into 20 mL above L-cysteine functionalized AuNPs (pH ∼ 7), mixed well, and stirred for 10 min. After that, CuCl2 was added into the solution with the concentration of Cu2+ of 0.1 mM (about 6 ppm) and stirred for 30 min. The resultant solid was filtered, washed with Milli-Q water and ethanol, and then dried under vacuum to get AuNPs-capped-MSNs.2.6 Stimulated drug release
In the pH triggered drug release experiment, a certain amount of fluorescein sodium loaded AuNPs-capped-MSNs powder was dispersed into 2 mL of Milli-Q water. The dispersion was transferred into a dialysis bag, and then the bag was immersed into 100 mL of PBS solution with different pHs (5, 4.5, and 4) at room temperature with magnetic stirring. An amount of 1.0 mL of solution was withdrawn at a given time interval, followed by supplying the same volume of fresh PBS solution. The amount of released drug was measured by fluorescence spectra at 520 nm.The ATP triggered in vitro drug release experiment was achieved in similar way. A certain amount of fluorescein sodium loaded AuNPs-capped-MSNs powder was dispersed in 50 mL of PBS buffer (pH 7) containing 1 mM, 5 mM and 10 mM of commercially available ATP at 25 °C. Subsequently, 2 mL of supernatant was taken periodically from the suspension at 25 °C followed by centrifugation (15000 rpm, 10 min). The release of fluorescein sodium from the pore voids to the buffer solution was determined by fluorescence emission spectroscopy (em at 520 nm).
2.7 Endocytosis and in vitro release of AuNPs-capped-MSNs in HeLa cells
HeLa cells were seeded in growth medium for 24 h prior to the experiment. After 24 h, HeLa cells are cultured in glass bottom Petri dish with growth medium containing 10% fetal bovine serum (FBS), 1% antibiotics and 0.1 mg AuNPs-capped-MSNs for different hours, then are washed with PBS for 3 times and fixed by 4% paraformaldehyde solution. After 30 min fixation, the cells are washed by PBS and subjected to fluorescent imaging.2.8 Cell uptake mechanism experiment with endocytosis inhibitor
To examine different endocytic routes of FITC MSN particle uptake, HeLa cells were incubated for 2 h in Optimem containing one of the following inhibitors: 10 μg mL−1 chlorpromazine (clathrin dependent) or 200 μM genistein (clathrin independent). Cells were then rinsed 2 times with PBS and treated with FITC MSN for 1 h prior to examination via confocal microscopy.2.9 Confocal microscopy
HeLa cells (2500 cells per dish) were plated in 35 mm cultured dishes which were pre-coated with poly-D-lysine hydrobromide for 10 minutes and left to grow for 5 days. A Zeiss LSM 780 equipped with an environmental chamber which controls the atmospheric conditions, humidity and temperature for live-cell imaging was used to acquire images with a 63× 1.3 NA water/glycerol objective.2.8 In vitro cytotoxicity (MTT method)
The experiments are performed using the HeLa cells. Before experiments, cells are cultured in 10 mL growth medium containing 10% fetal bovine serum (FBS), 1% antibiotics. Cytotoxicity assay are performed in 96-wells microtiter plates with seeding density, 10000 cells per well. Microtiter plates containing cells are pre-incubated for 24 h at 37 °C in order to allow stabilization before the addition of the test substance (AuNPs-capped-MSNs). The plates are incubated with the test substance for 24 h and 48 h respectively at 37 °C and 5% CO2. Then 5 μL MTT solution (5 mg mL−1 in PBS) is added to each well to evaluate cell viability. After 2 h at 37 °C, the solution is removed. 100 μL DMSO is added to dissolve cells. After 30 min incubation under 37 °C, the viability is measured using a microreader.
2.9 Characterization
Transmission electron microscopy (TEM) images were recorded on a Philips CM200 transmission electron microscope operated at 200 kV. For the TEM observation, samples were obtained by dropping 5 μL of solution onto carbon-coated copper grids. All the TEM images were visualized without staining. X-ray diffraction patterns were obtained in a RINT2000 vertical goniometer (Rigaku, Japan) using Cu Kα irradiation. The surface areas were calculated by the Brunauer Emmett Teller (BET) method, and the pore size distributions were calculated by the Barrett Joyner Halenda (BJH) method. The infrared (IR) spectra were measured by AVATAR 320 FT-IR using KBr pellets. The fluorescence spectra were recorded using a Varian Cary Eclipse spectrometer. Inductively coupled plasma mass spectrometry (ICP-MS) was performed using a Perkin Elmer ELAN 6100 ICP-MS. The ultraviolet-visible (UV-vis) spectra were measured with dilute aqueous solution in a 2 mm thick quartz cell using a SHIMADZU UV-2401 PC spectrophotometer. All pH value measurements were carried out on a Sartorius BECKMAN F 34 pH meter. Dynamic light scattering (DLS) and the zeta potentials were measured by a Malvern Zetasizer Nano Series running DTS software and using 4 mW He–Ne laser operating at a wavelength of 633 nm and avalanche photodiode (APD) detector. Thermogravimetric analysis was measured by PerkinElmer STA 6000 simultaneous thermal analyzer from 25 °C to 800 °C at heating rate of 10 °C min−1.3. Results and discussion
3.1 Synthesis and characterization AuNP-capped-MSNs
As depicted in Scheme 1, the AuNP-capped-MSNs require the synthesis of cysteine functionalized AuNPs and the functionalized MSNs. AuNPs was synthesized with average size of 5 nm based on DLS and TEM measurements (Fig. S1 and S2†). The preparation of 3-aminopropyltriethoxysilane (APTES) functionalized MSN (MSN-1) was first investigated by FTIR spectroscopy. As can be seen from Fig. S3,† the asymmetric vibration of Si–OH at 957 cm−1 disappeared and a new in-plane bending vibration of N–H from the NH2 groups was observed at 1560 cm−1 after attachment of APTES, which demonstrates that OH groups are replaced by NH2 group on the surface of MSN. The structure of MSN-1 was then further characterized by TEM. As shown in the TEM images (Fig. 1a), the prepared MSNs were uniform spherical nanoparticles with a mean diameter of approximately 100 nm. The MSN that has been synthesized here is of type MCM-41, which is defined by its uniform two-dimensional (2D) hexagonal p6m mesopores. This type of MSN is chosen due to its simplicity in synthesis and is one of the most popular choices of MSN for drug delivery applications.56 This highly ordered mesoporous network with a hexagonal array could be clearly seen from the TEM images and was also confirmed by XRD measurements (Fig. S4†). The three well-resolved diffraction peaks observed in the XRD pattern of MSN, assigned as (100), (110) and (200) planes, are consistent with the characteristic diffraction pattern of MCM-41 type MSN.57,58 | ||
| Fig. 1 Transmission electron micrographs of bare MSN (a), AuNPs-capped-MSN at pH 5 (b) and pH 4 (c) after 20 h. Arrow indicates aggregated gold nanoparticles. Samples were obtained by dropping 5 μL of PBS solution with different pH containing 0.1 mg mL−1 AuNPs-capped-MSN or MSN onto carbon-coated copper grids, and then dried for 10 h in air. | ||
The successful immobilization of AuNPs on the MSN matrix is confirmed by various analytical and microscopic methods. As shown in Fig. S4a,† the immobilization of the surface functionalized AuNPs to the MSN material reduced the intensity of the powder X-ray diffraction (XRD) peaks. Such a reduction of scattering contrast between the pores and the framework of the MCM-41 materials due to the pore-filling effect has been reported previously in the literature.59–64 Compared with the d100 value of the MSN material, a small increase in the d100 value of AuNPs-capped-MSN was observed. The decrease of the d100 values may be attributed to the ligand linkage induced pore-filling effect between the AuNPs and the mesoporous silica matrix.30 Fig. S4b† shows the high-angle XRD diffraction patterns of the AuNPs-capped-MSN within the 2θ range of 30°–90°. As can be seen from this figure, four typical reflections of Au atoms are observed that can be attributed to the diffraction of (111), (200), (220) and (311) lattice planes of the AuNPs attached to the mesoporous silica.59,63
TEM investigations of the AuNPs-capped-MSNs also provided direct evidence of the AuNPs distribution on the functionalized MSN material (Fig. 1b). Individual AuNPs are clearly visible on the outside of the MSN. Furthermore, scanning electron microscopy (SEM) was also used to investigate the AuNP coverage on the MSNs because of the dramatic increase of conductivity that results from AuNP attachment onto the MSN particles. As shown in Fig. S5,† comparing before and after attachment of AuNPs, the image of as-prepared MSNs has no obvious features present. On the other hand, after modification with AuNPs, a clear image of MSNs with a large number of bright dots was observed on the SEM micrograph.
Next, the total surface areas and the average pore diameters of MSN and AuNPs-capped-MSNs were analyzed by N2 adsorption/desorption measurements. As shown in Fig. S6a,† the BET isotherms of the unmodified MSN exhibited the characteristic type IV N2 adsorption/desorption patterns according to the IUPAC classification, with well-defined steps at relative pressures P/P0 of 0.4–0.8, which indicated that the material possesses both uniform mesoporous channels and narrow pore size distribution.65 After AuNPs capping, the N2 adsorption/desorption isotherms of the AuNPs-capped-MSNs displayed a relatively flat isotherm, which is quite different from that of the pure MSNs. The specific surface area of the MSN was calculated to be 1100 m2 g−1 (surface BET) and the cumulative pore volume was found to be 1.75 cm3 g−1. After AuNPs capping, the values of calculated specific surface area and cumulative pore volume decreased to about 200 m2 g−1 and 0.6 cm3 g−1, respectively. Furthermore, the average pore size decreased from 3.9 nm for the unmodified MSNs to lower than 2.0 nm for AuNPs-capped-MSNs (Fig. S6b†). All these results are in agreement and suggest that the majority of the pores have been capped by AuNPs.
The L-cysteine functionalized AuNPs and MSN-NH2 mixed system in the presence and absence of Cu2+ have been investigated to understand the interaction between AuNPs and MSN-NH2. As can be seen in Fig. S7,† a very high density of AuNPs were found on the surface of MSN for the mixed system when Cu2+ is present. However, the surface coverage of AuNPs is extremely low for the case when Cu2+ is absent, which shows the ion–ligand interaction is the main interaction to attach AuNPs to MSN instead of electrostatic interaction. More direct evidence was presented by the IR spectrum (Fig. S8 and Table S1†). As can be seen from the IR results, a lowering of the νCOO(s) band and an increase of the νCOO(as) band are observed after coordination of Cu2+, due to the generation of the Cu–O ligand and partial reconstruction of C–O double bond after forming the ligand. Moreover, the δNH band disappeared in the IR spectra of the complexes compared to the position of these bands in the IR spectrum of MSN-NH2 and L-cysteine functionalized AuNPs, which means the forming of ion–ligand between –NH2 and Cu2+. All these observations suggest that the coordination of the L-cysteine through –NH2 and –COO– group in a bidentate fashion.
3.2 Stimuli responsive drug release
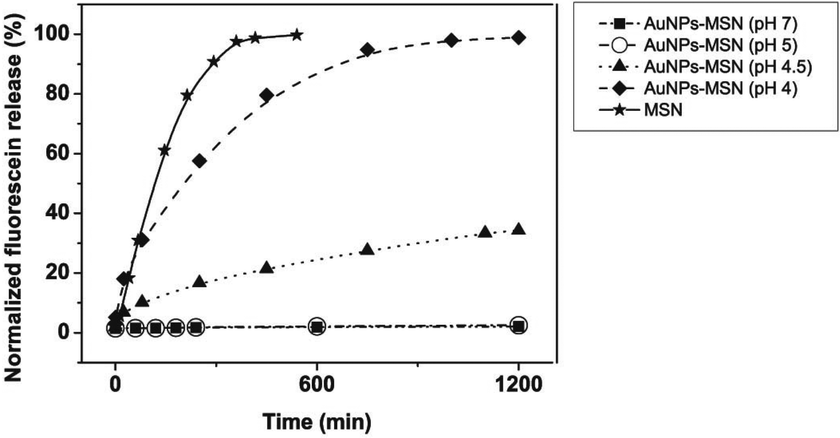 | ||
| Fig. 2 pH-dependent release kinetics of fluorescein sodium loaded MSNs comparing to the free release from unmodified MSN. The figure is represented by ■ (pH 7 for AuNPs-capped-MSN), ○ (pH 5 for AuNPs-capped-MSN), ▲ (pH 4.5 for AuNPs-capped-MSN), ◆ (pH 4 for AuNPs-capped-MSN), ★ (pH 7 for MSN). The drug release percentage (normalized fluorescein release) is calculated by the weight of released drug in PBS solution divided by the initial weight of drug in MSN. | ||
The observed pH-dependency of drug release can be understood by considering the ζ-potential of the L-cysteine modified AuNPs (Fig. 3). At pH > 5, the L-cysteine AuNPs are negatively charged and tend to be electrostatically attracted to the protonated amino groups on the MSN surface. The addition of Cu2+ ions allows for a network of AuNPs to form around the MSN due to metal chelation by free carboxy and amino groups from both AuNPs and MSN.51,52 With the decrease of pH below 5, more of the L-cysteine on the surface of the AuNPs is protonated creating electrostatic repulsion between L-cysteine modified AuNPs and NH3+ groups on the modified MSN. This results in dissociation between AuNPs and MSNs so that the AuNPs caps are removed and the drug is released. ζ-Potential measurement of both L-cysteine modified AuNPs and AuNPs-capped-MSN at pH 4 indicates that they are both positively charged, giving rise to electrostatic repulsion. It is also important to pinpoint that the stability constant log β of Cu2+–NH2 and Cu2+-L-cysteine is 3.4 and 13.72 respectively.66,67 The weak interaction between Cu2+ and NH2 group provides a potential breaking point for the removal of the AuNPs caps. Direct evidence of the removal of AuNPs was observed by TEM and SEM images. As can be seen from Fig. 1c and Fig. S5c,† most of the AuNPs were removed from the surface of MSN when the system pH reaches 4. The removed AuNPs do not distribute well but appear to be aggregated from observation based on TEM. DLS analysis of the AuNPs-capped-MSN solution at pH 4 provided the further evidence that the aggregation occurs in solution, and not from the drying process during the preparation of TEM or SEM samples. As can be seen from Fig. S11,† in DLS measurements at pH of 4, besides the sharp peak of MSN at 150 nm, a broad peak distribution from 4 nm to 150 nm is also evident in the size distribution which is attributed to aggregates of AuNPs released from the surface of the MSNs.
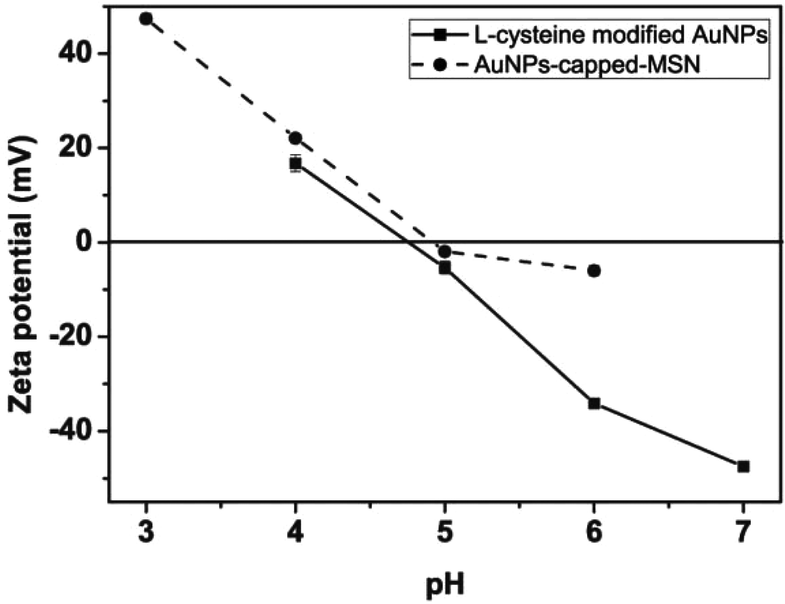 | ||
| Fig. 3 pH dependence of the zeta potentials of L-cysteine modified AuNPs (●) and AuNPs-capped-MSN (■). | ||
TGA analysis of the system (Fig. S12†) showed that the percentage of model drug that could be incorporated in the system is 28% (w/w). That is, in the fully-loaded system, for every 1000 mg of drug loaded AuNPs-capped-MSN, there is 280 mg of drug incorporated. It is also of interest to investigate the dose of Cu2+ delivered in this system. Inductively coupled plasma mass spectrometry (ICP-MS) was used to investigate the content of Cu2+ at different parts of the AuNPs-capped-MSN during the drug delivery process (free Cu2+ in aqueous solution, combined with MSN or combined with AuNPs) at pH 7 and pH 4 (Table S2†). As can be seen from Table S2,† the total amount of Cu2+ in this system is 3.91 mg per 1000 g AuNPs-capped-MSN. With removal of the particles, the remaining solution is shown to have only trace amounts of Cu2+ (0.06 ppm) indicating that the majority of Cu2+ still remains associated with the AuNPs or the MSN.
From the TGA and ICP-MS data, it is possible to calculate the ratio of MSN:drug:Cu is 1:0.39:3.9 × 10−6. According to the literature, typical weekly doses for the common anti-cancer drug, doxorubicin, is around 20 mg,68,69 which means the weekly dose of the AuNPs-capped-MSNs is less than 51.2 mg, and the weekly dose of Cu2+ is less than 2 × 10−4 mg, which is 0.00095% of daily recommended intake of copper for adult males.70
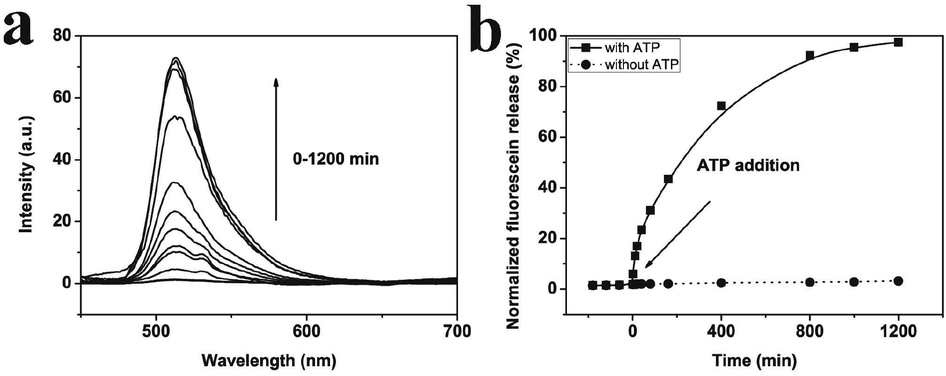 | ||
| Fig. 4 (a) Time-dependent fluorescence spectrum of fluorescein sodium solution in PBS by adding adenosine triphosphate (ATP). (b) The percentage of the fluorescein sodium released from AuNPs-capped-MSN versus the incubation time in PBS with (■) and without (●) 10 mM ATP. | ||
 | ||
| Fig. 5 Transmission electron micrographs of AuNPs-capped-MSN after adding 10 mM adenosine triphosphate (ATP) for 20 h. Arrow indicates aggregated gold nanoparticles. Samples were obtained by dropping 5 μL of PBS solution containing 0.1 mg mL−1 AuNPs-capped-MSN and 10 mM ATP onto carbon-coated copper grids, and then dried for 10 h in air. | ||
3.3 Cellular internalization and delivery studies
Having demonstrated stimuli-responsive behavior of the AuNPs-capped-MSN drug delivery system, cellular uptake experiments were performed in order to confirm a possible therapeutic application of these nanoparticles as drug carriers. In this case, experiments employing HeLa cells were conducted. As can be seen from Fig. 6b, green fluorescence was observed in the cytoplasm of HeLa cells after 2 h incubation with fluorescein sodium loaded AuNPs-capped-MSNs (50 μg mL−1). The inhomogeneous fluorescence distribution suggests that the AuNPs-capped-MSNs were taken up by endosomes. After incubation for 6 hours, as can be seen from Fig. 6c, green fluorescence homogeneously filled the whole cells and a small amount of green fluorescence appeared in the cell nucleus (setting the background as 0, the relative fluorescence intensity of cell nucleus and cytoplasm is 55 and 122, respectively). It has previously been shown that MSNs with size larger than 50 nm cannot enter the cell nucleus,72 which leads us to believe that the green fluorescence in the cell nucleus is due to free fluorescein sodium released from AuNPs-capped-MSNs. This result suggests that the fluorescein sodium molecules were released from the MSN intracellularly. The control samples without any nanoparticles do not exhibit extensive fluorescence.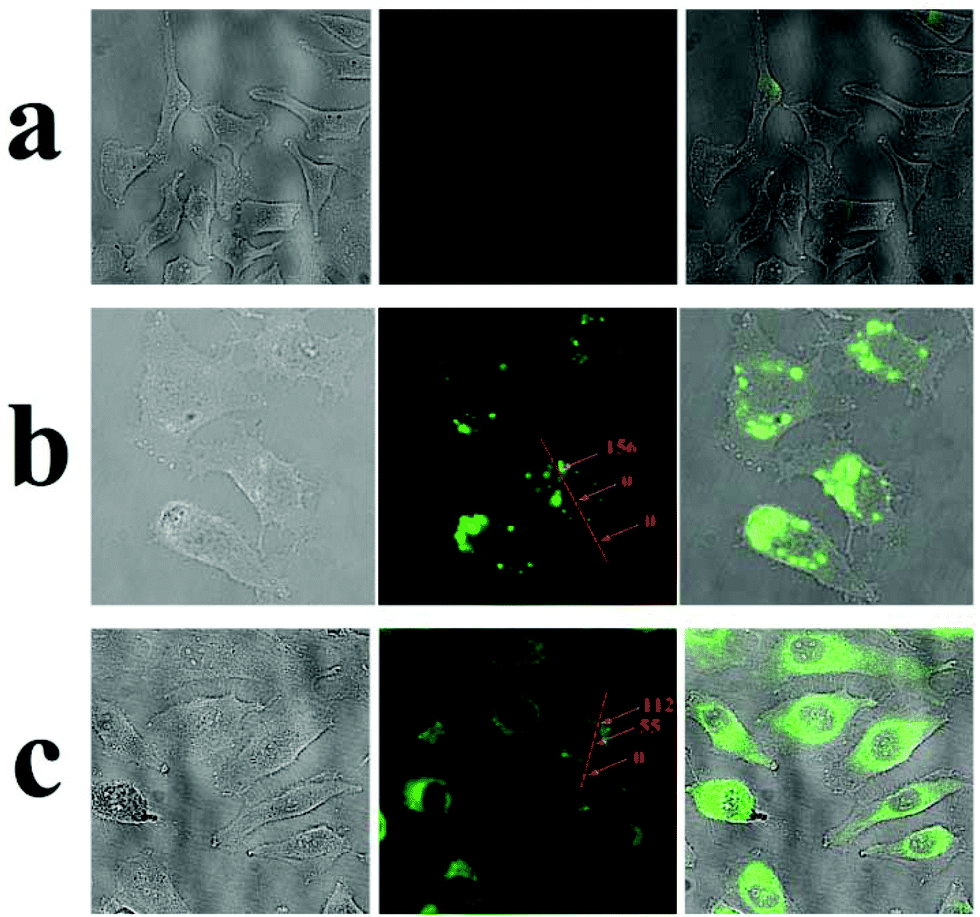 | ||
| Fig. 6 Optical microscope images (left), fluorescent images (middle) and overlay images (right) of HeLa cells before (a) and after incubation with fluorescein sodium loaded AuNPs-capped-MSN for 2 h (b) and 6 h (c). The insert numbers are the relative fluorescence intensity (arbitrary units) at different parts of the cells. | ||
3.4 In vitro cell uptake and cytotoxicity
The in vitro cell cytotoxicity of MSNs, AuNPs, and AuNPs-capped-MSNs was tested on HeLa cells using the MTT assay. It can be seen from Fig. 7 that the AuNPs-capped-MSNs showed no obvious cytotoxic effects on the HeLa cells at 6.25–100 μg mL−1 after incubation for 24 h and 48 h. At AuNPs-capped-MSN concentrations as high as 100 μg mL−1, the cell viability was about 80% even after incubation for 48 h. These results demonstrate that the AuNPs-capped-MSNs are well-tolerated, even at higher concentrations. Other groups have reported that the concentration of MSNs required for effective drug delivery to kill cancer cells is lower than 10 μg mL−1.65 Therefore, the AuNPs-capped-MSN system is suitable for use as the drug carriers in controlled drug delivery systems.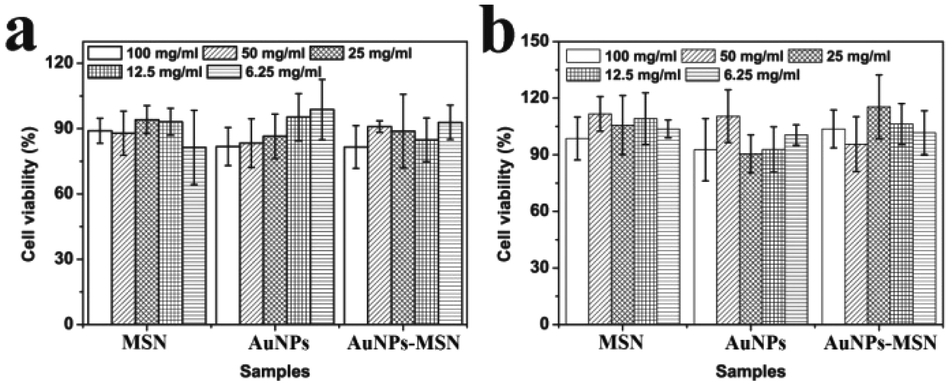 | ||
| Fig. 7 Cell viability of HeLa cells exposed to different concentrations of MSNs, AuNPs and AuNPs-capped-MSNs, as determined by the MTT assay. Materials at the indicated concentrations were incubated with cells for 24 hours (a) and 48 hours (b). For each sample, columns represent 100 mg mL−1, 50 mg mL−1, 25 mg mL−1, 12.5 mg mL−1, 6.25 mg mL−1 respectively from left to right. | ||
To determine the specific endocytic pathway of MSNs uptake, endocytosis inhibition studies were carried out in which common chemical inhibitors of clathrin-dependent (CDE) and clathrin-independent (CIE) endocytosis were employed. Chlorpromazine, an inhibitor of CDE, is a cationic amphiphilic molecule which translocates clathrin and its related proteins to intracellular vesicles thus suppressing the formation of clathrin coated pits.73 Caveolae-mediated endocytosis was inhibited by the tyrosine-kinase inhibitor genistein.74
Inhibitor specificity for the CDE and CIE inhibitors was established in HeLa cells by their ability to block uptake of molecules known to be internalized via the specific endocytic pathway with minimal toxic effects. In the case of CDE, uptake of human transferrin receptor (hTf), known to be taken up via CDE, was effectively blocked via treatment with chlorpromazine (Fig. S15†) with little change in cellular morphology and minimal cell death. Lactosylceramide (LacCer) uptake, which is known to be taken up via CIE,75,76 was effectively inhibited via tyrosine kinase inhibitor genistein (Fig. S16†). Once the specificity and effectiveness of these particular inhibitors was established, HeLa cells were first exposed to one of the above mentioned CDE or CIE inhibitors followed by incubation with FITC labelled MSNs and examined via confocal microscopy (Fig. 8). Clathrin dependent endocytosis appears as the dominant uptake mechanism for the MSN's with little to no particles apparent in the cytoplasm of cells treated with chlorpromazine. Furthermore, genistein had little to no effect on uptake. Clathrin dependant uptake has been shown in previous studies to dominate in the uptake of certain MSN formulations.77–79 Hao et al. demonstrated that particle shape and aspect ratio had a major effect on the uptake route of MSNs, with CDE dominating the uptake of spherical MSN, while CIE routes dominated the uptake of MSN with larger aspect ratios in HeLa cells.78
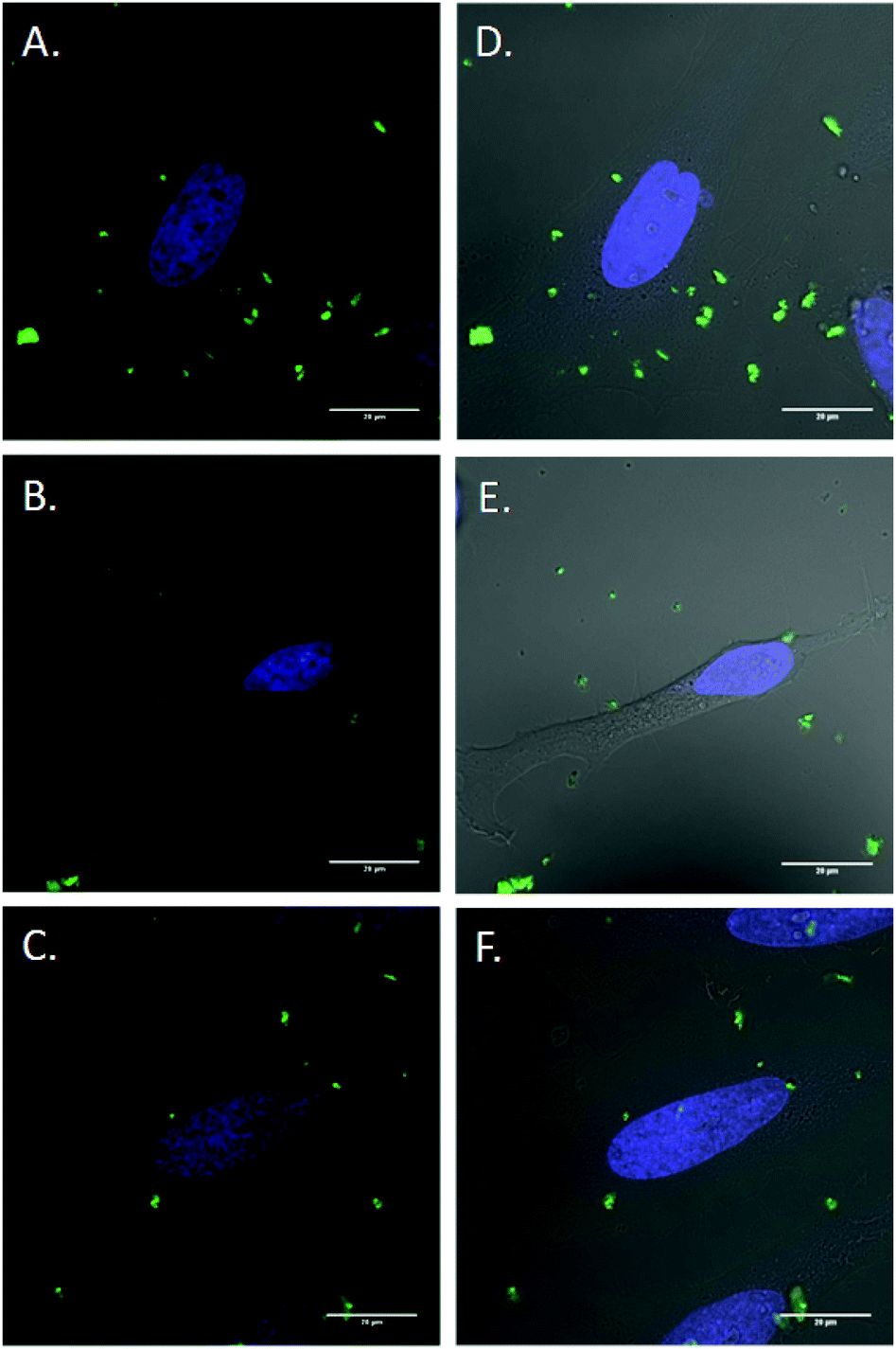 | ||
| Fig. 8 Endocytosis inhibition of FITC labeled MSNs. Cells were incubated for 2 h in either (A,D) Optimem or (B,E) Optimem containing 10 mg mL−1 chlorpromazine or (C,F) Optimem containing 200 mM genistein. Cells were rinsed with PBS and then incubated with FITC labeled MSNs for 1 h prior to imaging. Green: FITC labelled MSNs, blue: Hoescht 33342 live nuclear stain. | ||
The in vitro release of an anticancer drug from the AuNPs-capped-MSNs was also performed by using Dox. Fig. S17† shows the Dox release behavior of AuNPs-capped-MSNs in PBS buffers as a function of pH (7 and 4) at room temperature. The pH-triggered release of Dox was monitored by fluorescence emission spectroscopy at 590 nm. As can be seen from the figure, there is almost no Dox release after 20 h under pH 7. However, the cumulative release of Dox reached up to 87% after 20 h at pH 4 indicating that the low pH could effectively trigger the Dox release. The ATP triggered Dox release from the MSNs was monitored in the presence of 10 mM ATP at pH 7. As seen in Fig. S18,† the emission intensity of Dox begins to increase almost immediately following the addition of ATP, indicating rapid release. The emission intensity asymptotically approaches its maximum value after approximately 1200 min while prior to the addition of ATP, the emission intensity of Dox shows negligible increase, indicating that the Dox remains trapped in the pores of the silica particles. Moreover, HeLa cells were used to investigate the in vitro drug release in biological system. As can be seen from Fig. S19,† the Dox loaded MSN shows high toxicity to the HeLa cells after 24 h and 48 h, which means the Dox was successfully released from the uptaken MSN and killed HeLa cells. Moreover, HeLa cells show much lower viability after 48 h, which suggests the sustained release ability of the AuNPs-capped-MSN for long time treatment. These results present the successful in vitro release of real anticancer drug from our AuNPs-capped-MSNs.
The comparison between free Dox and Dox loaded AuNP-capped-MSNs were also performed. As can been seen from Fig. S20,† the free Dox and Dox loaded MSN all show toxicity to HeLa cells after 24 h incubation. The free Dox killed the cells more rapidly, but the effect does not be continued for long time periods and it did not have stimuli targeting mechanism that the particles possess.
4. Conclusions
A mesoporous silica-based system for loading and releasing drug molecules was developed using a capping concept where the release is controlled by the removal of AuNP caps that block the pores of the MSNs. The capping is achieved using NH2-group modified MSNs, Cu2+ ions and L-cysteine functionalized gold nanoparticles. Both pH and ATP concentration can trigger the release of the caps and hence delivery of the drug. In relation to the pH-controlled delivery, the release of the drug is inhibited at pH above 5, whereas there is rapid release of the guest molecule from the mesoporous silica host at pH below 5. The pH-controlled “opening” mechanism is associated with the switch in the ζ-potential of the cysteine-coated AuNPs from negative to positive at pH < 5. At the same time the use of Cu2+ ligands also opens up the possibility of ATP as a suitable stimulus for release procedures. This is related to the disruption of the capping by formation of ion–ligand bond between Cu2+ and ATP. Under both stimuli, the AuNPs remain associated with Cu2+ after leaving the MSN surface, allowing the release of the entrapped drugs and minimal release of Cu2+. Both low pH and ATP biologically-relevant stimuli are appealing methods of releasing entrapped guest molecules within either the lysosome (low pH) and/or the cytosol (contains high concentration of ATP) respectively. The incorporation of both pH and ATP responses provides multiple routes for intracellular drug release and represents a novel strategy for maximizing drug delivery. The modular nature of the construct means that the system has wide applicability for controlled delivery purposes and is demonstrated here for the delivery of the anticancer drug doxorubicin to cancer cells under stimuli responsive conditions.Acknowledgements
The authors thank the Australian Research Council (DP110902183) and the National Health and Medical Research Council (APP1024723) and the University of New South Wales for financial support for different aspects of this work.Notes and references
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- A. B. Descalzo, R. Martinez-Manez, R. Sancenon, K. Hoffmann and K. Rurack, Angew. Chem., Int. Ed., 2006, 45, 5924–5948 CrossRef CAS PubMed.
- S. Angelos, M. Liong, E. Choi and J. I. Zink, Chem. Eng. J., 2008, 137, 4–13 CrossRef CAS.
- X. L. Hu, S. Liu, Y. B. Huang, X. S. Chen and X. B. Jing, Biomacromolecules, 2010, 11, 2094–2102 CrossRef CAS PubMed.
- M. L. Gou, K. Men, H. S. Shi, M. L. Xiang, J. A. Zhang, J. Song, J. L. Long, Y. Wan, F. Luo, X. Zhao and Z. Y. Qian, Nanoscale, 2011, 3, 1558–1567 RSC.
- B. S. Kim, S. W. Park and P. T. Hammond, ACS Nano, 2008, 2, 386–392 CrossRef CAS PubMed.
- W. N. E. vanDijkWolthuis, J. A. M. Hoogeboom, M. J. vanSteenbergen, S. K. Y. Tsang and W. E. Hennink, Macromolecules, 1997, 30, 4639–4645 CrossRef CAS.
- Z. H. Gan, T. F. Jim, M. Li, Z. Yuer, S. G. Wang and C. Wu, Macromolecules, 1999, 32, 590–594 CrossRef CAS.
- S. Radin, P. Ducheyne, T. Kamplain and B. H. Tan, J. Biomed. Mater. Res., 2001, 57, 313–320 CrossRef CAS PubMed.
- W. Aughenbaugh, S. Radin and P. Ducheyne, J. Biomed. Mater. Res., 2001, 57, 321–326 CrossRef CAS PubMed.
- P. Kortesuo, M. Ahola, M. Kangas, I. Kangasniemi, A. Yli-Urpo and J. Kiesvaara, Int. J. Pharm., 2000, 200, 223–229 CrossRef CAS PubMed.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- R. Langer, Acc. Chem. Res., 1993, 26, 537–542 CrossRef CAS.
- Y. X. Li and T. Kissel, J. Controlled Release, 1993, 27, 247–257 CrossRef CAS.
- M. Vallet-Regi, A. Ramila, R. P. del Real and J. Perez-Pariente, Chem. Mater., 2001, 13, 308–311 CrossRef CAS.
- B. Munoz, A. Ramila, J. Perez-Pariente, I. Diaz and M. Vallet-Regi, Chem. Mater., 2003, 15, 500–503 CrossRef CAS.
- A. Ramila, B. Munoz, J. Perez-Pariente and M. Vallet-Regi, J. Sol-Gel Sci. Technol., 2003, 26, 1199–1202 CrossRef CAS.
- P. Kortesuo, M. Ahola, S. Karlsson, I. Kangasniemi, J. Kiesvaara and A. Yli-Urpo, J. Biomed. Mater. Res., 1999, 44, 162–167 CrossRef CAS PubMed.
- P. Kortesuo, M. Ahola, M. Kangas, A. Yli-Urpo, J. Kiesvaara and M. Marvola, Int. J. Pharm., 2001, 221, 107–114 CrossRef CAS PubMed.
- W. Lai, P. Ducheyne and J. Garino, Removal pathway of silicon released from bioactive glass granules in vivo, World Scientific, New York, 1998 Search PubMed.
- W. Lai, J. Garino and P. Ducheyne, Biomaterials, 2002, 23, 213–217 CrossRef CAS PubMed.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed.
- T. Azais, C. Tourne-Peteilh, F. Aussenac, N. Baccile, C. Coelho, J. M. Devoisselle and F. Babonneau, Chem. Mater., 2006, 18, 6382–6390 CrossRef.
- J. Fan, C. Z. Yu, T. Gao, J. Lei, B. Z. Tian, L. M. Wang, Q. Luo, B. Tu, W. Z. Zhou and D. Y. Zhao, Angew. Chem., Int. Ed., 2003, 42, 3146–3150 CrossRef CAS PubMed.
- C. Q. Huang, X. X. Zhang, J. M. Ramil, S. Rikka, L. Kim, Y. Lee, N. A. Gude, P. A. Thistlethwaite, M. A. Sussman, R. A. Gottlieb and A. B. Gustafsson, Circulation, 2010, 121, 675-U698 CrossRef PubMed.
- E. B. Keeffe, Acute liver failure, Lang Medical Books/McGraw-Hill, New York, 2003 Search PubMed.
- H. F. Galley, J. Roy. Coll. Surg. Edinb., 2000, 45, 44–50 CAS.
- R. D. Keidan, J. Fanning, R. A. Gatenby and J. L. Weese, Dis. Colon Rectum, 1989, 32, 206–209 CrossRef CAS PubMed.
- Y. N. Zhao, B. G. Trewyn, I. I. Slowing and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398–8400 CrossRef CAS PubMed.
- C. Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- F. Torney, B. G. Trewyn, V. S. Y. Lin and K. Wang, Nat. Nanotechnol., 2007, 2, 295–300 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 238 Search PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS PubMed.
- D. R. Radu, C. Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216–13217 CrossRef CAS PubMed.
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350–353 CrossRef CAS PubMed.
- R. Hernandez, H. R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS PubMed.
- A. M. Kaufmann and J. P. Krise, J. Pharm. Sci., 2007, 96, 729–746 CrossRef CAS PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
- Q. H. Sun, M. Radosz and Y. Q. Shen, J. Controlled Release, 2012, 164, 156–169 CrossRef CAS PubMed.
- X. J. Huang, Y. Xiao and M. D. Lang, J. Colloid Interface Sci., 2011, 364, 92–99 CrossRef CAS PubMed.
- E. S. Lee, K. Na and Y. H. Bae, J. Controlled Release, 2005, 103, 405–418 CrossRef CAS PubMed.
- J. Chen, X. Z. Qiu, J. Ouyang, J. M. Kong, W. Zhong and M. M. Q. Xing, Biomacromolecules, 2011, 12, 3601–3611 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- R. M. Steinman, I. S. Mellman, W. A. Muller and Z. A. Cohn, J. Cell Biol., 1983, 96, 1–27 CrossRef CAS PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- D. L. Cook, L. S. Satin, M. L. J. Ashford and C. N. Hales, Diabetes, 1988, 37, 495–498 CrossRef CAS PubMed.
- J. G. Fitz, Trans. Am. Clin. Climatol. Assoc., 2007, 199–208 Search PubMed.
- I. Beis and E. A. Newsholme, Biochem. J., 1975, 152, 23–32 CrossRef CAS PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- J. Dyne, Y. S. Lin, L. M. H. Lai, J. Z. Ginges, E. Luais, J. R. Peterson, I. Y. Goon, R. Amal and J. J. Gooding, ChemPhysChem, 2010, 11, 2807–2813 CrossRef CAS PubMed.
- W. R. Yang, J. J. Gooding, Z. C. He, Q. Li and G. N. Chen, J. Nanosci. Nanotechnol., 2007, 7, 712–716 CAS.
- X. Chen, X. Y. Cheng and J. J. Gooding, Analyst, 2012, 137, 2338–2343 RSC.
- W. R. Yang, D. B. Hibbert and J. J. Gooding, J. Electroanal. Chem., 2001, 516, 10–16 CrossRef CAS.
- C. E. Hunt and W. W. Carlton, J. Nutr., 1965, 87, 385–393 CAS.
- Registry of toxic effects of chemical substances, ed. D. V. Sweet, National Institute for Occupational Safety and Health, Cincinnati, Ohio, 1997 Search PubMed.
- P. P. Yang, S. L. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- F. Carniato, C. Bisio, G. Paul, G. Gatti, L. Bertinetti, S. Coluccia and L. Marchese, J. Mater. Chem., 2010, 20, 5504–5509 RSC.
- K. Miyasaka, A. V. Neimark and O. Terasaki, J. Phys. Chem. C, 2009, 113, 791–794 CAS.
- E. Aznar, M. D. Marcos, R. Martinez-Manez, F. Sancenon, J. Soto, P. Amoros and C. Guillem, J. Am. Chem. Soc., 2009, 131, 6833–6843 CrossRef CAS PubMed.
- B. Marler, U. Oberhagemann, S. Vortmann and H. Gies, Microporous Mater., 1996, 6, 375–383 CrossRef CAS.
- H. Winkler, A. Birkner, V. Hagen, I. Wolf, R. Schmechel, H. von Seggern and R. A. Fischer, Adv. Mater., 1999, 11, 1444–1448 CrossRef CAS.
- W. H. Zhang, J. L. Shi, L. Z. Wang and D. S. Yan, Chem. Mater., 2000, 12, 1408–1413 CrossRef CAS.
- W. H. Zhang, J. L. Shi, H. R. Chen, Z. L. Hua and D. S. Yan, Chem. Mater., 2001, 13, 648–654 CrossRef CAS.
- M. M. Chili, V. S. R. R. Pullabhotla and N. Revaprasadu, Mater. Lett., 2011, 65, 2844–2847 CrossRef CAS.
- L. Yuan, Q. Q. Tang, D. Yang, J. Z. Zhang, F. Y. Zhang and J. H. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CAS.
- L. Ilcheva and J. Bjerrum, Acta Chem., Scand. Ser. A, 1976, 30, 343–350 CrossRef.
- C. J. Hawkins and D. D. Perrin, Inorg. Chem., 1963, 2, 839–843 CrossRef CAS.
- W. Scheithauer, C. Zielinksi and H. Ludwig, Weekly low dose doxorubicin monotherapy in metastatic breast cancer resistant to previous hormonal and cytostatic treatment, Breast Cancer Res. Treat., 1985, 6, 89–93 CrossRef CAS PubMed.
- P. G. Rose, Pegylated Liposomal Doxorubicin: Optimizing the Dosing Schedule in Ovarian Cancer, Oncologist, 2005, 10, 205–214 CrossRef CAS PubMed.
- Anon, Recommended Dietary Allowances, National Academy Press, Washington DC, 1989 Search PubMed.
- A. D. Silva, A. L. R. Merce, A. S. Mangrich, C. A. T. Souto and J. Felcman, Polyhedron, 2006, 25, 1319–1326 CrossRef.
- L. M. Pan, Q. J. He, J. N. Liu, Y. Chen, M. Ma, L. L. Zhang and J. L. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed.
- L. H. Wang, K. G. Rothberg and R. G. W. Anderson, J. Cell Biol., 1993, 123, 1107–1117 CrossRef CAS PubMed.
- I. R. Nabi and P. U. Le, J. Cell Biol., 2003, 161, 673–677 CrossRef CAS PubMed.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharmacol. Rev., 2006, 58, 32–45 CrossRef CAS PubMed.
- M. Fisichella, H. Dabboue, S. Bhattacharyya, G. Lelong, M. L. Saboungi, F. Warmont and P. Midoux, J. Nanosci. Nanotechnol., 2010, 10(4), 2314–2324 CrossRef CAS PubMed.
- N. J. Hao, L. L. Li, Q. Zhang, X. L. Huang, X. W. Meng, Y. Q. Zhang, D. Chen, F. Q. Tang and L. F. Li, Microporous Mesoporous Mater., 2012, 162, 14–23 CrossRef CAS.
- D. M. Huang, Y. Hung, B. S. Ko, S. C. Hsu, W. H. Chen, C. L. Chien, C. P. Tsai, C. T. Kuo, J. C. Kang, C. S. Yang, C. Y. Mou and Y. C. Chen, FASEB J., 2005, 19(12), 2014–2016 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures associated with this article can be found in the online version. See DOI: 10.1039/c3bm60148j |
| This journal is © The Royal Society of Chemistry 2014 |