Development of a microwave-assisted digestion method for the rapid determination of chloride and fluoride in nuclear-grade boron carbide powders
M. V.
Balarama Krishna
*a,
S. V.
Rao
a,
Y.
Balaji Rao
b,
N. S.
Shenoy
c and
D.
Karunasagar
a
aNational Center for Compositional Characterization of Materials (NCCCM), Bhabha Atomic Research Centre, Department of Atomic Energy, Hyderabad, 500 062, India. E-mail: balaram@cccm.gov.in; Fax: +91 4027125463; Tel: +91 4027121365
bNuclear Fuel Complex, Department of Atomic Energy, Hyderabad, 500 062, India
cAnalytical Chemistry Division, Bhabha Atomic Research Centre, Department of Atomic Energy, Mumbai, 400 085, India
First published on 5th November 2013
Abstract
A simple and efficient microwave-assisted digestion (MAD) method for the rapid determination of chloride and fluoride in nuclear-grade boron carbide powders has been developed using a closed microwave-vessel (G30, Anton Parr) and a domestic microwave oven. Parameters optimized to get quantitative recoveries of chloride and fluoride were extractant concentration, microwave power, irradiation time and amount of the sample. The optimized MAD conditions for quantitative recovery of chloride and fluoride were obtained by irradiating ∼0.5 g of sample in 10 mL of 10% HNO3 (v/v) for about 30 s at a microwave power of 480 W. After completion of the extraction process, the sample mixture was centrifuged and then a known volume of the sample extract (clear supernatant) was transferred to another pre-cleaned tube for subsequent analyses of Cl and F using an ion-selective electrode (ISE). A pyro-hydrolysis method was used for validation of the results of the proposed MAD method. The LOD values for chloride and fluoride in conjunction with the proposed MAD were found to be 1.9 μg g−1 and 1.2 μg g−1 respectively with a relative standard deviation (RSD) of less than 10%. The studies have demonstrated that the proposed microwave-based approach constitutes a good alternative sample preparation methodology for the determination of chloride and fluoride in nuclear-grade boron carbide powder samples.
1. Introduction
Boron carbide (also known as black diamond) is highly refractory and is one of the hardest materials known, ranking third behind diamond and cubic boron nitride. Because of the combination of properties such as high melting point (2450 °C), excellent hardness (29.1 GPa), chemical inertness, low specific weight/density (2.52 g cm−3), excellent thermoelectric properties, etc., boron carbide is a suitable material for many high performance applications such as abrasives for polishing and cutting, wire drawing dies, armors for personnel and vehicle safety, as rocket propellants, for ceramic composites, etc.1–9 Furthermore, boron is a strong neutron absorber with a high neutron absorption cross-section of 3837 b for 10B for thermal neutrons while ∼2.7 b for the fast neutrons (∼100 keV). Hence, boron carbide has been used as control rods in the nuclear reactors to control neutron flux. The ability of boron carbide to absorb neutrons without forming long-lived radio-nuclides makes it attractive as a shielding material and as a neutron detector in nuclear reactors.1However, certain elements (W, Si, Ni, Fe, Al, Cl, F, etc.) cause detrimental effects in the performance of the boron carbide as a nuclear material. For example, the presence of F− and Cl− as impurities even at a trace level can accelerate the corrosion of the metallic components of the system. Most of the metallic and non-metallic impurities enter into the boron carbide matrix during the process used for conversion of boric acid to elemental boron followed by its conversion into carbide and subsequent grinding and preparation of the pellets. Therefore, depending upon the type of the nuclear application of boron carbide, specifications have been laid down for both metallic and non-metallic impurities (for chloride and fluoride specification limits have been set as 75 μg g−1 and 25 μg g−1 respectively for boron carbide).10 Hence determination of the chemical purity of boron carbide is very important towards its quality control. The present work is aimed at the determination of chloride and fluoride in boron carbide samples.
Various analytical techniques such as colorimetry,11–13 X-ray fluorescence spectrometry,14 neutron activation analysis,15 ion-chromatography16–19 and ion-selective electrode methods13 have generally been used for the determination of chloride and fluoride in a wide variety of materials. Major problems in determining chloride and fluoride in boron carbide are the hardness of the material and its high chemical resistance. Pyrohydrolysis coupled with IC is the recommended ASTM procedure for the determination of chloride and fluoride in various nuclear materials including boron carbide.20 Although this method is reliable for the quantitative determination of chloride and fluoride, it involves a large number of steps, it is time consuming and it requires high temperature for the separation through steam distillation of Cl and F from the matrix that limits the sample throughput. Thus, the determination of Cl and F in the boron carbide matrix is still considered a challenging task due to its hardness and complex nature. Thus there is a great need for the development of new analytical methodologies, which are simple, rapid, reliable and suitable for the routine analysis of trace impurities in nuclear-grade boron carbide samples.
Microwave-assisted leaching of analytes from a wide variety of solid matrices has shown tremendous potential.21–28 Owing to the advantages of rapid heating, shorter reaction times, reduction in the number of discrete sample preparation steps, lower risk of contamination and avoidance of losses of volatile analytes due to the use of closed vessels and increased sample throughput, microwave-assisted sample preparation is now a well-established and superior approach to the conventional procedures. The efficacy of microwave-based methods for sample preparation in various environmental, biological and geological applications has been reported for a wide variety of matrices (e.g., soils, sediments and biological tissues) for different purposes including total digestion for elemental analysis, extraction of selected organic compounds and in speciation analysis.29–38 In recent years, microwave-assisted extraction has attracted growing interest as it allows rapid extraction of analytes from solid matrices. A number of studies have been reported on the use of the microwave assisted approach as a sensitive sample preparation methodology.26–29 Balarama Krishna et al. have reported the use of microwave-assisted extraction for the multi-elemental analysis of sediments.39 More recently, oxidative pyrolysis combined with the microwave-assisted extraction method for the determination of metallic impurities in boron carbide powder samples has also been reported by authors from the same laboratory.40
However, simultaneous extraction of Cl and F using microwave technology from a boron carbide matrix has not been well explored. The present investigation therefore explores the feasibility of separating Cl and F together from a boron carbide matrix, employing an MAD technique by optimizing various parameters with the use of a high pressure microwave glass vessel and a domestic microwave oven and their subsequent determination using an ion-selective electrode. To the best of our knowledge, this is the first study on the use of MAD as a sample preparation methodology for the determination of chloride and fluoride in boron carbide powder samples.
2. Experimental
2.1. Instrumentation
A domestic microwave oven (Videocon, Mumbai, India) programmable for time and microwave power was used for carrying out microwave-assisted digestion (MAD) experiments throughout this work. The microwave oven has power settings from “low” through “high” which correspond to 160 through 800 W of power output. Closed microwave digestion vessels made of glass with a capacity of 30 mL obtained from Anton Parr (Model no. G30) were used for the extraction studies. The digestion vessels are designed to withstand a pressure of up to 30 bar and a temperature up to 300 °C. All the MAD experiments were carried out by keeping the microwave oven in a well ventilated fume hood to evacuate acid fumes, if any, that are generated during the extraction process.Caution: Safety guidelines regarding the work with microwave-fields in the laboratory must be observed.
A suppressor based IC system (Model no. 850, Metrohm, Switzerland) was used for the separation and determination of fluoride and chloride after the pyro-hydrolysis process. The chromatographic system equipped with a pump unit with a pulsation damper, a MSM suppressor and a conductivity detector has been employed for the analysis. A conductivity meter with full-scale sensitivity settings of the 0 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μs cm−1 single range has been employed. A six-port injection valve equipped with a 20 μL PEEK sample injection loop was used for sample introduction. The separation of chloride and fluoride was achieved on a 20 mm × 4 mm Metrosep guard column in series with a 75 mm × 4.6 mm Metrosep analytical column. All the separations were performed at room temperature under isocratic conditions. The mobile phase was a mixture of 1.3 mM Na2CO3 and 2 mM NaHCO3 solution with a flow rate of 0.6 mL min−1. The suppressor column was continuously regenerated with 50 mM H2SO4. Under the set conditions, the retention times are ∼3.7 min and ∼6.3 min for fluoride and chloride respectively.
000 μs cm−1 single range has been employed. A six-port injection valve equipped with a 20 μL PEEK sample injection loop was used for sample introduction. The separation of chloride and fluoride was achieved on a 20 mm × 4 mm Metrosep guard column in series with a 75 mm × 4.6 mm Metrosep analytical column. All the separations were performed at room temperature under isocratic conditions. The mobile phase was a mixture of 1.3 mM Na2CO3 and 2 mM NaHCO3 solution with a flow rate of 0.6 mL min−1. The suppressor column was continuously regenerated with 50 mM H2SO4. Under the set conditions, the retention times are ∼3.7 min and ∼6.3 min for fluoride and chloride respectively.
A Metrohm Ion Meter (Model no. 692, Metrohm, Switzerland) equipped with a chloride/fluoride ion-selective electrode (ISE) was used for chloride and fluoride determinations. The electrode was soaked in chloride/fluoride solution (100 μg mL−1). Before taking measurements, the electrode was thoroughly washed with high purity water and wiped dry. The sampling solutions were neutralized with dilute NaOH using a digital pH meter prior to ISE measurements.
2.2. Standards, reagents and materials
Ultra-pure water with >18 MΩ cm specific resistance produced from a Milli-Q water system (Millipore, MA, USA) was used for preparing standards and sample solutions. Sub-boiled HNO3 was prepared in-house by sub-boiling distillation in quartz stills and was used throughout this work. All containers were soaked in 20% HNO3 (v/v) and cleaned thoroughly with high purity water prior to use. Unless otherwise stated, AR grade chemicals were used for preparing reagent solutions. Chloride standard (1 mg mL−1) was prepared by dissolving an appropriate amount of NaCl (AR grade, Lab Chemie, Mumbai, India) in water and fluoride standard solutions (100 μg mL−1, Thermo Orion 940907) traceable to National Institute of Standards and Technology (NIST) reference materials were used in the preparation of the working standards by sequential dilution for analysis. The eluents of the IC system were prepared using Na2CO3 and NaHCO3 (AR grade, Thomas Baker, India) and filtered through nylon membrane filters (0.45 μm) before use.As no suitable CRM was available with us, a boron carbide powder obtained from Boron Carbide India Ltd., Mumbai, India was used as the representative material for the optimization of the MAD procedure. Boron carbide powder samples tested in this study were provided by Materials Processing Division of Bhabha Atomic Research Centre, Mumbai, India.
2.3. Microwave-assisted digestion procedure
To evaluate the optimal microwave parameters for the quantitative extraction of fluoride and chloride, 10 mL solutions of HNO3 at different concentrations were added to the representative boron carbide sample and irradiated at different microwave power and time settings. Corresponding process blanks and standards were also subjected to the general MAD procedure in order to check for possible contamination and loss of test analytes. The results obtained were compared with the ones obtained for the un-irradiated solutions. The corresponding process blank solution was utilized for the preparation of Cl and F standard solutions for ISE measurements as matrix matched standards.An accurately weighed amount (300–800 mg) of boron carbide sample, weighed to the nearest 0.1 mg, was transferred into a microwave digestion glass vessel and 10 mL extractant solution (HNO3) was added. After thorough mixing of the sample with the extractant, the vessels were closed and kept in the microwave oven and subjected to microwave irradiation for 5–40 s at a 160–800 W power. After completion of the extraction processes, the microwave vessel was allowed to cool to room temperature and the supernatant was separated from the boron carbide matrix by centrifugation for 4 min at 5000 rpm. After centrifugation, the clear supernatant was transferred to another pre-cleaned tube and then the sample extracts were brought to the volume with high purity water for subsequent analysis of chloride and fluoride by ISE as described in the next section. Each sample was processed in three replicates (n = 3) and each replicate was measured twice.
:5 was maintained for chloride measurements i.e., for a 5 mL sample solution, 1 mL of KNO3 buffer was added.
Total ionic strength adjustment buffer (TISAB III) purchased from Thermo Orion (Cat. no. 940911, Orion Research Inc., Beverly, MA, USA) was added to the samples before fluoride determinations with F-ISE. TISAB III buffer is a mixture of CDTA (trans-1,2-cyclohexylendinitrilo tetra-acetic acid), ammonium chloride, ammonium acetate (CH3COONH4) buffer and cresol red (C21H18O5S). The recommended volume ratio between TISAB III and test sample solution is 1:10. All the sample solutions were neutralized by drop-wise addition of a dilute solution of NaOH. Graduated polypropylene centrifuge tubes of 15 mL and 50 mL capacity (Abdos Labtech Pvt. Ltd, Kolkata, India) were used for preparing sample solutions throughout this work.
To check the loss of chloride and fluoride, if any, under microwave-assisted extraction conditions, a set of experiments were carried out for obtaining calibration plots with the processed standard solutions and compared with those of unprocessed aqueous standards containing known amounts of Cl and F. Similar calibration plots were obtained with optimized extractant solution into which known amounts of chloride and fluoride were spiked and subjected to the microwave extraction procedure as in the case of samples. Quantifications of Cl and F in B4C samples after applying the proposed microwave based approach are based on external calibration (in the concentration range of 0–2 μg mL−1 for fluoride and 0–10 μg mL−1 for chloride) obtained with the standards prepared in optimized extractant solution always used, to make the acidity in the standard solution used for calibration nearly the same as in the sample solution. The resultant sample solutions after suitable dilution were analysed by ISE.
The recovery of fluoride and chloride from the B4C samples after applying the proposed analytical method was calculated using the following equation:
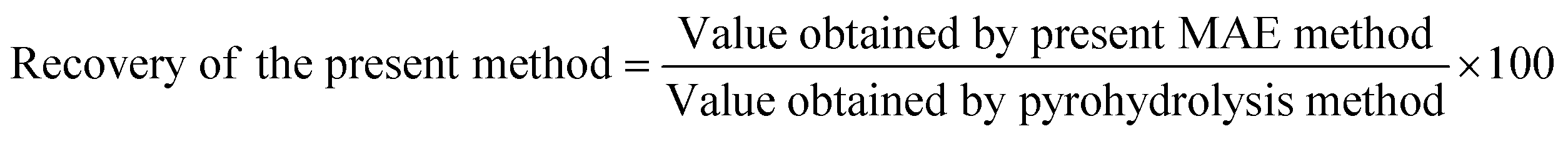
2.4. Pyrohydrolysis method
To our knowledge, no reference material of boron carbide certified for chloride and fluoride is available. Hence, the well established pyrohydrolysis method was adopted for validation of the proposed analytical method. Pyrohydrolysis combined with IC was employed for the separation and determination of F− and Cl− in boron carbide samples.A horizontal tubular furnace with a programmable temperature controller was employed for carrying out pyro-hydrolysis experiments. Initially about 1 g of the accurately weighed boron carbide sample was thoroughly mixed with approximately 2 g of U3O8 (used as an accelerator) and taken in a ceramic boat and introduced into a quartz tube loaded in the furnace. After purging through a water trap, moist oxygen gas was passed through the quartz tube from one end fitted with a PTFE cork and the evolved gas was collected in a 25 mL standard flask (collector) containing high purity water placed at the other end. The quartz tube was heated to an optimized temperature of ∼1100 °C with a heating rate of ∼20 °C min−1 and maintained at that temperature for one hour. The evolved gas was collected during the entire cycle of heating and collection was further continued for another 30 min after completion of the heating cycle for ensuring complete recovery of chloride and fluoride. The pyrohydrolysis process converts the Cl and F in the sample to HCl and HF which is carried away by the flowing moist oxygen. The process blank was prepared as described above in the absence of the boron carbide sample. At the end of the process, the trapped solutions were made up to the mark for subsequent analysis by the IC system using a sodium carbonate–bicarbonate mobile phase and conductometric detection.
3. Results and discussion
The typical major constituents of the representative material were ∼80% boron and ∼20% carbon. Since commercial CRMs are not available, the efficiency of the proposed approach was established by calculating the percentage recovery of the elements of interest using the equation shown in the Experimental section. An extractant volume of 10 mL was used throughout the study as this was found to be sufficient to ensure that the boron carbide powder is always entirely immersed in the extractant during the extraction process.3.1. Development of the microwave-assisted extraction procedure
Sample preparation is one of the most crucial steps for obtaining quality results as most analytical instruments are unable to handle solid matrices directly and therefore some form of pre-treatment is required to isolate analyte(s) of interest from the matrix. Ideally, extraction of elements of interest from solid samples must be rapid, easy and reliable. However the existing methodologies found in the literature for the separation and determination of chloride and fluoride are very tedious and time consuming. Therefore the application of MAD was considered to achieve the objective of this work.During the development of the MAD procedure, the extraction parameters such as method variables (choice of solvent and sample mass) and two instrument variables (microwave power and irradiation time) were tested to get quantitative recovery of chloride and fluoride from boron carbide samples. The first experiment for the optimization of MAD was designed to select an extractant type and its concentration for the quantitative recovery of chloride and fluoride. All the parameters were tested with a sample mass of about 0.5 g with an extractant volume of 10 mL.
The effect of the nitric acid concentration on the quantitative recovery of fluoride and chloride from the B4C matrix was studied at different HNO3 concentrations; 0 (water), 2, 5, 10, 15 and 20% (v/v) at constant microwave power (480 W), extraction time (30 s) and sample weight (∼0.5 g). In each case, the general procedure as described in the Experimental section was followed. After each extraction cycle was completed, the sample mixtures were cooled to room temperature and centrifuged. Then the clear sample extract (supernatant) was made up to the required volume with Milli-Q water for subsequent analysis by ISE.
Fig. 1 shows the recoveries obtained at different concentrations of the extractant. From the results of these studies, it was observed that the extraction efficiency of Cl and F increased from 54% to >98% by increasing the nitric acid concentration from 2–15% (v/v). A similar trend in the extraction efficiencies of both Cl and F was noticed in all the cases. The extraction efficiency reached maximum with 10 mL of 10% HNO3 and remained constant thereon and was used as an optimized extractant throughout the study. These studies proved the efficiency of using dilute solutions of HNO3 mainly when employing microwave instruments. Compared to concentrated nitric acid, diluted acid solutions generated less residue and do not require high dilution factors before analyte measurements.
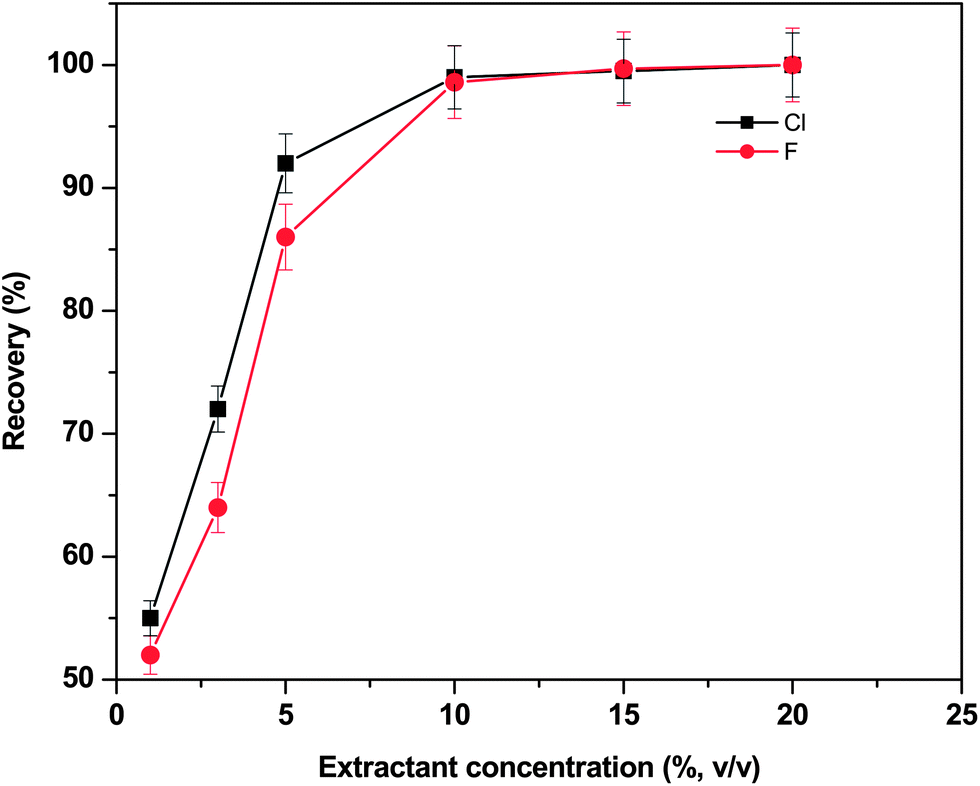 | ||
| Fig. 1 Optimization of the extractant concentration (HNO3) (v/v) for the quantitative recovery of chloride and fluoride from the boron carbide representative material; volume of extractant = 10 mL, microwave power = 480 W, microwave irradiation time = 30 s and sample mass = ∼500 mg. | ||
Extraction time is another parameter whose influence needs to be taken into account. Generally, the extraction of analytes increases by increasing the extraction time. Hence to determine the time needed to obtain a quantitative recovery, extraction studies with the representative boron carbide sample were performed for different lengths of time. Microwave irradiation times of 5, 10, 20, 30 and 40 s were evaluated keeping the concentration of the extractant (10% HNO3), microwave power (480 W) and sample weight (∼0.5 g) constant. After each extraction cycle was completed, the samples were cooled to room temperature. The sample–extractant mixture was centrifuged and the extracts were analysed by ISE for Cl and F concentrations.
From the results it was observed that the extraction efficiency of both Cl and F increased with increasing microwave irradiation time from 5–30 s and then reached a plateau. Microwave exposure for even a few seconds (10 s) was found to provide excellent yields (70–80%) for chloride and fluoride while the best recoveries (>95%) required an irradiation time of ∼30 s. The difference in the extraction behaviour of Cl and F with varying microwave power conditions and time could possibly be due to their binding nature.
Microwave power and irradiation time (i.e., extraction time) are two factors which influence each other to a great extent. In order to optimize the best combination of microwave power and irradiation time, a factorial (two factors, three levels) experimental design approach was applied and recovery of chloride and fluoride at each level of treatment was calculated. Based on the preliminary experiments, a combination of microwave power 480 W and irradiation time 30 s was chosen as a base level for the two representative materials (the upper and lower levels were obtained using a difference of ±160 W for microwave power and ±10 s for irradiation time from the base level). Corresponding extractant solutions were employed as blanks.
The recoveries obtained for chloride and fluoride when the representative boron carbide material was taken through the general microwave-assisted extraction procedure at different microwave power and irradiation settings are shown in Fig 2. These results indicate that the recovery of both chloride and fluoride increased with increasing microwave power and irradiation time while the best elemental recoveries (>98%) were obtained at different microwave power and irradiation time settings. A combination of low or moderate power with little long exposure may be a better and safer approach. Based on these aspects, a microwave power of 480 W and a microwave irradiation time of 30 s were found to be optimum for the quantitative extraction of chloride and fluoride from boron carbide samples which is advantageous for high sample throughput and hence selected for further optimization studies.
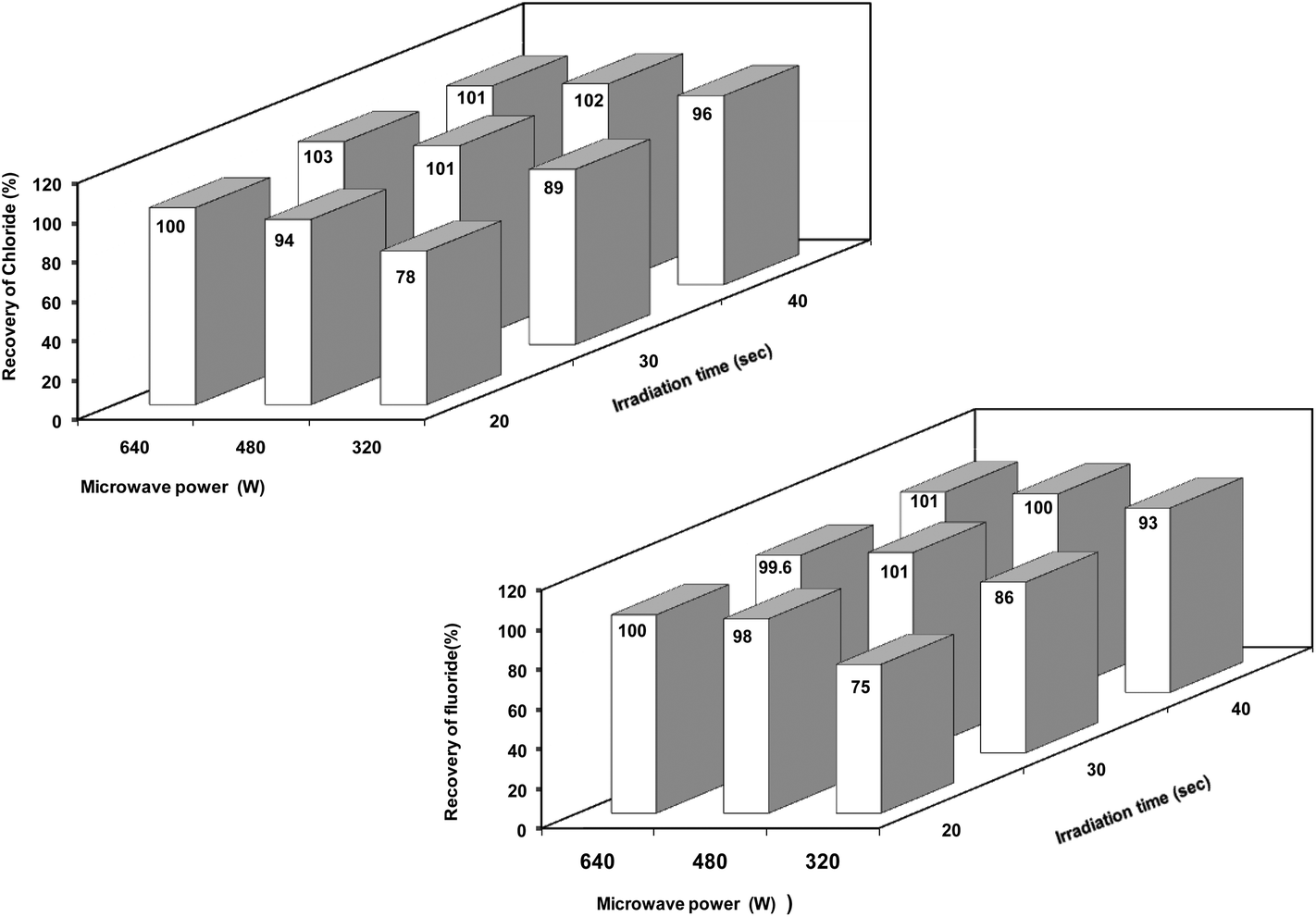 | ||
| Fig. 2 Optimization of the microwave power and irradiation time on the recovery of chloride and fluoride from the boron carbide representative sample; extractant concentration = 10% HNO3 (v/v), volume of extractant = 10 mL and sample mass = ∼500 mg. | ||
The optimized MAD method variables (composition of extractant solution and sample mass) and two instrument variables (microwave power and irradiation time) which could influence the recovery of chloride and fluoride (i.e., extraction yield) from boron carbide are listed in Table 1.
| Variable parameter | Range studied | Optimum extraction conditions |
|---|---|---|
| Instrument variables | ||
| Extractant concentration (HNO3) (%) | 2–20 | 10 |
| Sample amount (g) | 0.3–1.0 | <0.8 |
| Method variables | ||
| Microwave irradiation time (sec) | 5–40 | 30 |
| Microwave power (W) | 160–800 | 480 |
3.2. Analytical response characteristics
The effect of the acidity of extractant solution and matrix interference was checked to study the possibility of the direct determination of chloride and fluoride in final sample extracts. For this purpose, five point calibration plots were generated with pure aqueous standards and extractant solutions of 10% HNO3 (v/v) spiked with known amounts of chloride and fluoride, across a concentration range, 0 (i.e., analytical blank) to 5 μg mL−1 for the ISE method for quantification. Similar calibration plots were obtained with the same set of solutions after they were taken through the general procedure to check any loss of chloride and fluoride during the MAD process.For evaluation of the MAD procedure, the representative sample was spiked with known amounts of chloride and fluoride and taken through the MAD procedure. Similar calibration plots were also generated with standard solutions spiked into the final sample extract i.e., just before the sample analysis (after the MAD process). In this case, the analytical signal of the standard is taken as the difference between the signal obtained from spiked sample solutions and the signal of the sample solution.
The analytical curves corresponding to the aqueous standards, extractant solutions of 10% HNO3 (v/v) as well as spiked sample solutions boron carbide reference material (before and after applying MAD) have shown good linearity and satisfactory correlation coefficients (R2 > 0.995). After MW extraction the boron content was determined in the extractant solution by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) (Horiba Jobin Vyon Ultima II, France) and the concentration of boron was found to be 0.85 ± 0.04%. Hence interference studies due to boron for the determination of chloride and fluoride were also carried out by spiking a known amount of boron (using boric acid solution) in the range of 0.5–5% in the sample solutions. These studies indicated the absence of matrix-related interference for the quantification of chloride and fluoride in the samples.
The slopes of the calibration plots of standards spiked in 10% HNO3 (v/v) solution as well as in the final sample extracts are the same which demonstrates the absence of acid-related interference. Hence, the external calibration plot obtained with standards prepared in 10% HNO3 medium was always used for final quantification of chloride and fluoride.
Following the optimization of the experimental conditions of the proposed single-step microwave-assisted leaching procedure, its analytical performance was studied by examining the limit of detection (LOD), recovery of chloride and fluoride and precision (RSD). The limits of detection (LODs) (defined as the concentration equivalent to three times the standard deviation of five measurements of a blank) for chloride and fluoride in conjunction with the proposed MAD were found to be 1.9 μg g−1 and 1.2 μg g−1 respectively while the LOD value obtained by the pyrohydrolysis method was calculated to be 1.5 μg g−1 for both the tested anions.
Good agreement between the recovery values obtained with all the solutions before and after applying the MAD procedure (data not shown here) indicates no loss of chloride and fluoride during microwave irradiation as seen by the measured concentrations of the test elements.
3.3. The proposed MAD method vs. the pyrohydrolysis method
To assess the efficacy of the proposed procedure, the same boron carbide powder representative sample used in the present work was processed using the pyro-hydrolysis method, followed by analysis using IC. As seen from the results (Table 2), good agreement was obtained between the two methods for both the test elements demonstrating the efficacy of the developed method. Average recoveries of chloride and fluoride after taking through the complete MAD process were calculated based on the values obtained by the pyrohydrolysis method and were found to be in the range of 98–101%. The relative standard deviation (RSD) values were calculated by performing five microwave irradiation processes under optimum conditions on the same day and the RSD of results ranged from 3–10%.| Element | Pyro-hydrolysis method (μg g−1) | Microwave-assisted extraction (μg g−1) | Recovery (%) |
|---|---|---|---|
| Chloride | 325.4 ± 18.3 | 322.3 ± 17.7 | 99.1 |
| Fluoride | 9.2 ± 0.5 | 9.2 ± 0.4 | 100 |
An extractant concentration of 10 mL of 10% HNO3 (v/v) has been found to be sufficient for the quantitative recovery (>98%) of chloride and fluoride from boron carbide powders (up to 800 mg) and hence concentrated acids are not required. The developed procedure requires about 10 min for complete processing of one sample (including microwave irradiation, centrifugation and analysis time) making it possible to analyse as many as 40–45 samples in an 8 hour working day. The sample throughput can be further increased by using a sample holder of 10 vials. But in the case of the pyro-hydrolysis method 2–3 h of time is necessary for processing one sample which restricts the sample throughput.
Although LOD values of the MAD and pyrohydrolysis methods were comparable, the proposed extraction procedure reduces markedly the time required from >120 min to <10 min (including extraction, centrifugation and analysis) in relation to the pyrohydrolysis method. Furthermore, the proposed MAD method offers the following advantages: (1) use of diluted acids and less extractant volume; (2) quantitative recoveries under optimal conditions; (3) rapid turn-around time; (4) the possibility of performing multiple extractions (up to 10 samples with the present sample holder) resulting in increased sample throughput; (5) no need of any catalyst/accelerator.
3.4. Application to enriched boron carbide samples
To assess the efficacy and reliability, the proposed analytical method was applied to boron carbide samples of different chemical compositions for the determination of chloride and fluoride. The reliability of the proposed analytical method was checked by comparing the results with those obtained by the pyro-hydrolysis method. After leaching, the extractant solutions were appropriately diluted so that the concentrations of chloride were within the calibration range. The chloride and fluoride in boron carbide samples obtained by proposed MAD as well as pyro-hydrolysis methods are presented in Table 3. It may be seen that values were in good agreement and recoveries were found to be >98%. The impurity levels of Cl and F present in the tested samples were found to be in the range of 34–130 μg g−1 and 4–15 μg g−1 respectively. The results showed that the analysed boron carbide samples contain different levels of chloride and fluoride as impurities which may have originated either from the raw materials used to prepare the carbide or from the solutions used to remove metallic impurities from the synthesized product.| Boron carbide sample | Element | Pyro-hydrolysis method (μg g−1) | Microwave-assisted extraction method (μg g−1) |
|---|---|---|---|
| Sample 1 (boron = 76.6% and carbon = 19.8%) | Chloride | 124.4 ± 10.8 | 129.6 ± 8.2 |
| Fluoride | 7.5 ± 0.4 | 7.5 ± 0.5 | |
| Sample 2 (boron = 78.6% and carbon = 19.3%) | Chloride | 54.9 ± 4.2 | 53.7 ± 2.9 |
| Fluoride | 5.5 ± 0.4 | 5.5 ± 0.3 | |
| Sample 3 (boron = 73.7% and carbon = 20.2) | Chloride | 35.1 ± 2.2 | 34.8 ± 1.4 |
| Fluoride | 6.5 ± 0.5 | 6.5 ± 0.4 | |
| Sample 4 (boron = 74.6% and carbon = 19.4%) | Chloride | 70.4 ± 4.8 | 71.8 ± 2.3 |
| Fluoride | 8.5 ± 0.5 | 8.5 ± 0.3 | |
| Sample 5 (boron = 78.4% and carbon = 19.8%) | Chloride | 56.2 ± 3.3 | 55.4 ± 2.9 |
| Fluoride | 4.5 ± 0.5 | 4.5 ± 0.2 | |
| Sample 6 (boron = 77.6% and carbon = 19.5%) | Chloride | 35.4 ± 2.9 | 34.8 ± 1.8 |
| Fluoride | 5.5 ± 0.4 | 5.5 ± 0.3 |
4. Conclusions
A simple, fast and efficient closed microwave-assisted digestion method for the determination of chloride and fluoride in nuclear-grade boron carbide samples has been developed for the first time. After extraction with 10% HNO3 (v/v), quantification of chloride and fluoride was carried out by ISE. The optimum conditions for MAD were found to be ∼0.8 g sample, 10 mL of 10% HNO3, microwave power of >480 W and an irradiation time of 30 s. The results of the developed method were in good agreement with values obtained by the pyro-hydrolysis method with average recoveries for both chloride and fluoride in the range of 98–101%. With the optimized conditions, the proposed MAD method provided satisfactory precision and detection limits. The relative standard deviation (RSD) was better than 10% in most of the cases. Under optimal conditions, the developed method was successfully applied to the analysis of boron carbide samples of different compositions.The results obtained from the developed leaching procedure were compared with the values obtained by the pyrohydrolysis method, demonstrating that the whole process is much simpler, cost-effective and rapid. It also has inherent advantages including relatively low cost instrumentation, minimal operating expenses and ease of use, which make it attractive for the analysis of chloride and fluoride in boron carbide samples as a better alternative to existing sample preparation methods.
Acknowledgements
The authors are thankful to Dr Sunil Jai Kumar, Head, NCCCM for his constant support and encouragement.References
- K. Suri, C. Subramanian, J. K. Sonber and T. S. R. C. Murthy, Int. Mater. Rev., 2012, 55(1), 4–40 CrossRef PubMed.
- F. Thevenot and J. Euro, J. Eur. Ceram. Soc., 1990, 6, 205–225 CrossRef CAS.
- Y. Q. Li and T. Qiu, Mater. Sci. Eng., A, 2007, 444, 184–191 CrossRef PubMed.
- K. Khanra, Bull. Mater. Sci., 2007, 30(2), 93–96 CrossRef PubMed.
- D. Gosset and M. Colin, J. Nucl. Mater., 1991, 183, 161–173 CrossRef CAS.
- M. J. Pender, K. M. Forsthoefel and L. G. Sneddon, Pure Appl. Chem., 2003, 75(9), 1287–1294 CrossRef CAS.
- C. Subramanian, A. K. Suri and T. S. R. C. Murthy, Development of boron-based materials for nuclear applications, BARC News Letter, March–April 2010, Issue No. 313 Search PubMed.
- A. Jain, S. Anthonysamy, K. Ananthasivan, R. Ranganathan, V. Mittal, S. V. Narasimhan and P. R. Vasudeva Rao, Mater. Charact., 2008, 59, 890–900 CrossRef CAS PubMed.
- A. Velamakanni, K. J. Ganesh, Y. Zhu, P. J. Ferreira and R. S. Ruoff, Adv. Funct. Mater., 2008, 19, 1–8 Search PubMed.
- Specifications for nuclear-grade boron carbide powder, Annual book of ASTM standards, vol. 12.01, p. C750 Search PubMed.
- S. V. Rao, R. Singh and S. C. Chaurasia, Bhabha Atomic Research Centre (BARC) News Letter, Mumbai, India, 2002, vol. 219, pp. 6–10 Search PubMed.
- M. V. Balarama Krishna, S. V. Rao, V. S. N. Murthy and D. Karunasagar, Anal. Methods, 2012, 4, 1565–1572 RSC.
- M. K. Malde, K. Bjorrvatn and K. Julshamn, Food Chem., 2001, 73, 373–379 CrossRef CAS.
- T. Graule, A. von Bohlen, J. A. C. Broekaert, E. Grallath, R. Klockenkamper, P. Tschopel and G. Tolg, Fresenius' Z. Anal. Chem., 1989, 335, 637–642 CrossRef CAS.
- R. Verma and R. Parthasarathy, J. Radioanal. Nucl. Chem. Lett., 1996, 214, 391–397 CrossRef CAS.
- P. R. Haddad, P. N. Nesterenko and W. Buchberger, J. Chromatogr., A, 2008, 1184, 456–473 CrossRef CAS PubMed.
- S. D. Kumar, K. Venkatesh and B. Maiti, Chromatographia, 2004, 59(4), 243–245 CAS.
- S. D. Kumar, B. Maiti and P. K. Mathur, Talanta, 2001, 53, 701–705 CrossRef CAS.
- S. Jeyakumar, V. V. Raut and K. L. Ramakumar, Talanta, 2008, 76, 1246–1251 CrossRef CAS PubMed.
- Standard test methods for chemical, mass spectrometric and spectrochemical analysis of nuclear grade boron carbide, Annual Book of ASTM standards, volume 12.01:C791–04.
- Introduction to microwave sample preparation, Theory and Practice, ACS professional reference book, ed. H. M. Kingston and L. B. Jassie, American Chemical Society, Washington DC 1988 Search PubMed.
- K. J. Lamble and S. J. Hill, Analyst, 1998, 123, 103R–133R RSC.
- J. A. Nobrega, C. Pirola, L. L. Fialho, G. Rota, C. E. K. M. A. de Campos Jordao and F. Pollo, Talanta, 2012, 98, 272–276 CrossRef CAS PubMed.
- C. S. Eskilsson and E. Bjorklund, J. Chromatogr., A, 2000, 902, 227–250 CrossRef CAS.
- E. Smith and E. A. Arsenault, Talanta, 1996, 43, 1207–1268 CrossRef.
- V. Camel, Analyst, 2001, 126, 1182–1193 RSC.
- V. Camel, TrAC, Trends Anal. Chem., 2000, 19(4), 229–248 CrossRef CAS.
- V. Lopez-Avila, Crit. Rev. Anal. Chem., 1999, 29(3), 195–230 CrossRef CAS.
- L. Sanchez-Prado, C. Garcia-Jares and M. Llompart, J. Chromatogr., A, 2010, 1217, 2390–2414 CrossRef CAS PubMed.
- G. C. L. Araujo, M. H. Gonzalez, A. G. Ferreira, A. R. A. Nogueira and J. A. Nobrega, Spectrochim. Acta, Part B, 2002, 57, 2121–2132 CrossRef.
- V. Mandal, Y. Mohan and S. Hemalatha, Pharmacogn. Rev., 2007, 1, 7–18 CAS.
- S. Oztan and R.-A. During, Talanta, 2012, 99, 594–602 CrossRef PubMed.
- A. Bizzi, E. M. M. Flores, J. S. Barin, E. E. Garcia and J. A. Nobrega, Microchem. J., 2011, 99, 193–196 CrossRef PubMed.
- G. M. Mizanur Rahman and H. M. Skip Kingston, J. Anal. At. Spectrom., 2005, 20, 183–191 RSC.
- L. H. Reyes, J. L. G. Mar, A. Hernandez-Ramirez, J. M. Peralta-Hernandez, J. M. A. Barbosa and H. M. Skip Kingston, Microchim. Acta, 2011, 172, 3–14 CrossRef CAS.
- L.-F. Chang, S.-J. Jiang and A. C. Sahayam, J. Chromatogr., A, 2007, 1176, 143–148 CrossRef CAS PubMed.
- C.-S. Chiou, S.-J. Jiang and K. S. K. Danadurai, Spectrochim. Acta, Part B, 2001, 56, 1133–1142 CrossRef.
- J. L. Rodrigues, C. R. Alvarez, N. R. Farinas, J. J. Berzas Nevado, F. Barbosa Jr and R. C. R. Martin-Doimeadios, J. Anal. At. Spectrom., 2011, 26, 436–442 RSC.
- M. V. B. Krishna, K. Chandrasekharan, G. Venkateswarlu and D. Karunasagar, Anal. Methods, 2012, 4, 3290–3299 RSC.
- M. V. Balarama Krishna, G. Venkateswarlu, S. Thangavel and D. Karunasagar, Anal. Methods, 2012, 5, 1515–1523 RSC.
| This journal is © The Royal Society of Chemistry 2014 |