Focussed ion beam serial sectioning and imaging of monolithic materials for 3D reconstruction and morphological parameter evaluation†
Mercedes
Vázquez
*ab,
David
Moore
b,
Xiaoyun
He
a,
Aymen
Ben Azouz
ab,
Ekaterina
Nesterenko
a,
Pavel
Nesterenko
c,
Brett
Paull
c and
Dermot
Brabazon
ab
aIrish Separation Science Cluster, National Centre for Sensor Research, Dublin City University, Glasnevin, Dublin 9, Ireland. E-mail: mercedes.vazquez@dcu.ie; Fax: +353 1 700 8021; Tel: +353 1 700 7602
bAdvanced Processing Technology Research Centre, School of Mechanical Engineering, Dublin City University, Glasnevin, Dublin 9, Ireland
cAustralian Centre for Research on Separation Science, University of Tasmania, Hobart, Tasmania, Australia
First published on 17th October 2013
Abstract
A new characterisation method, based on the utilisation of focussed ion beam-scanning electron microscopy (FIB-SEM), has been employed for the evaluation of morphological parameters in porous monolithic materials. Sample FIB serial sectioning, SEM imaging and image processing techniques were used to extract the pore boundaries and reconstruct the 3D porous structure of carbon and silica-based monoliths. Since silica is a non-conducting material, a commercial silica monolith modified with activated carbon was employed instead to minimise the charge build-up during FIB sectioning. This work therefore presents a novel methodology that can be successfully employed for 3D reconstruction of porous monolithic materials which are or can be made conductive through surface or bulk modification. Furthermore, the 3D reconstructions were used for calculation of the monolith macroporosity, which was in good agreement with the porosity values obtained by mercury intrusion porosimetry (MIP).
1. Introduction
A significant body of research within the field of analytical chemistry over the last decade has been focused on the development and application of novel porous monolithic materials. These have found widespread application within chromatography,1 solid-phase extraction (SPE),2 spectroscopy3 and electrochemical analysis.4 This research effort has resulted in the successful commercialisation of a number of monolithic separation columns for high-performance liquid chromatography based on porous organopolymer and silica monolithic stationary phases. These monolithic materials possess unique and advantageous properties for flow-through applications, such as high porosity, well developed surface area and hydraulic permeability, and can be readily modified to provide a wide range of surface chemistries, making them attractive for an extensive variety of analytical purposes. In chromatographic applications, the high permeability of monolithic columns allows pumping of the mobile phase at relatively high flow rates, leading to faster separations at lower back-pressures compared to conventional packed columns.Recently, porous carbon monoliths have attracted considerable attention due not only to their unique physicochemical properties, such as chemical inertness, thermal stability and electrical conductivity, but also due to the availability of template-based preparation methods5,6 that provide a means to precisely control pore size and pore structures in the resulting carbon monolith.
Indeed, the performance of monolithic materials is significantly affected by morphological parameters such as the pore size, surface area and the total porosity. Common analytical techniques used for morphological characterisation of monolithic materials include transmission electron microscopy (TEM), scanning electron microscopy (SEM), nitrogen adsorption and mercury intrusion porosimetry (MIP).
MIP provides information on the porosity, pore size distribution and specific surface area of porous materials containing pores with sizes in the range 3.5 nm to 500 μm (i.e. mesopores and macropores).7 Nitrogen adsorption measurements are commonly used for calculation of the specific surface area (SBET) via the Brunauer–Emmett–Teller equation, as well as for the determination of the micro- and mesoporosity.8 However, calculations of the total pore volume by both techniques exclude closed pores (pores isolated from the material surface) and assume an ideal pore shape, typically spherical.7,8 In addition, both MIP and nitrogen adsorption techniques are unsuitable for analysis of monoliths prepared in capillaries with internal diameters (ID) in the micrometer range due to insufficient sample amount (minimum amount of sample required for analysis is approximately 35 mg), or rather complex sample preparation. Therefore, considering that the morphology of porous monolithic materials prepared in bulk (macroscopic scale) can significantly differ from those prepared in a much smaller, confined space,9 alternative techniques such as SEM and TEM10 are typically used for characterisation of monoliths prepared at microscopic (capillary) scale. SEM and TEM provide real visualisation of a single cross-section of the sample prepared either in bulk or in capillaries. Thus, direct information on the pore size (including closed pores), true pore shape, and pore connectivity can be obtained. Nevertheless, a significant number of images need to be taken at different points along the sample in order for TEM and SEM analysis results to be representative of the whole sample, which leads to a significant increase in the time and cost of the analysis.
In contrast, automated imaging techniques such as focussed ion beam-scanning electron microscopy (FIB-SEM) can provide a much faster approach for collection of typically about a hundred images by means of serial slicing and imaging. Briefly, this technique consists in the removal of thin slices (several nanometers/micrometers thick) from the target material by FIB sectioning, followed by image acquisition using SEM. By repeating this automated procedure until the desired volume has been removed from the sample, a series of parallel cross-sectional images of the material under study can be obtained. Finally, processing of the 2D-image stack via image-analysis routines allows the reconstruction of the corresponding 3D porous structure, which in turn can be used for calculation of the morphological parameters of interest. These 3D reconstructions can be further used for modelling of the material transport properties and/or designing numerical simulation tools allowing the prediction of the material performance, as well as for optimisation of the material morphology. Actually, the use of FIB-SEM for reconstruction and visualisation of 3D porous structures has been recently used for the morphological characterisation of a wide variety of samples including metamorphic rocks,11 biological tissues,12 neuronal cells,13 bones,14 ceramic mixed ionic-electronic conductors,15 and carbon materials for fuel cells, batteries and supercapacitors.16,17 SEM imaging and serial sectioning by means of an ultramicrotome has been also employed for 3D reconstruction of the macropore structure present in a polymer monolith in capillary format.18 This technique, known as serial block-face scanning electron microscopy (SBF-SEM), allowed identification of two distinct size domains within the macropore structure of the polymer monolith as well as the characterisation of the radial macroporosity profile of the capillary column. Alternatively, confocal laser scanning microscopy (CLSM) has been used for analysis of silica monoliths in capillary format. This optical “slicing” of specimens by CLSM allowed to reconstruct19 and compare20 the macropore structure of silica-based monoliths, and in turn to conduct benchmark simulations of flow21 and mass transport22 using these 3D reconstructions for derivation of quantitative morphology–transport relationships.
Although the above approaches represent a huge step towards the full characterisation of the “real” porous structure in monolithic materials, there are still several challenges associated with the use of these techniques for reconstruction of some of these materials. For example, although FIB-SEM is highly suitable for imaging electrically conducting materials (e.g. carbon monoliths), analysis of non-conducting materials (e.g. silica and polymer monoliths) is not straightforward owing to sample charging, which causes significant image drift. Due to the high magnification and precision required for sample positioning, sputtering the sample with gold or other metals after removal of each slice to avoid sample charging is not a practical approach. Thus, a common method for imaging non-conducting materials by FIB-SEM (e.g. biological samples) consists in staining the sample with an electrically conducting compound which increases the sample conductivity.23 Alternatively, a system integrating an environmental or “low vacuum” SEM (ESEM), i.e. FIB-ESEM, can be employed for serial sectioning and imaging, preventing the need for sample preparation steps directed to render the sample conductive. Despite these possible alternatives, as yet there are no reports on the use of FIB-SEM for imaging and reconstruction of silica and polymer monoliths. Similarly, only one report on the analysis of a polymer monolith using SBF-SEM was presented recently.18 It should be noted that although commercial SBF-SEM systems employ an ESEM, which allows direct analysis of insulating materials, conductive stains are still used for enhancement of the image contrast.18 In fact, SBF-SEM has the capability of slicing larger sample areas much faster than FIB-SEM, but the minimum slice thickness is generally larger than for FIB-SEM (ca. 30 nm vs. 10–15 nm). On the other hand, SBF-SEM was still found unsuitable for analysis of silica monoliths since the diamond knife of the ultramicrotome would not withstand the cutting process.24 Refractive index matching between sample and objective can also pose a significant challenge in CLSM,19 as pointed out by Tallarek's group, who found CLMS unsuitable for imaging polymer monoliths as a result of their high refractive index.18 In addition, one of the main drawbacks of CLSM compared to FIB-SEM and SBF-SEM is its lower resolution.
In the work presented herein, an in-house prepared carbon monolith and a commercial silica-based monolith were selected as samples to evaluate the potential of FIB-SEM for general application in the 3D reconstruction and morphological characterisation of porous monolithic materials. The in-house prepared carbon monolith employed in this study presented a hierarchical pore structure (macroporous/mesoporous) and excellent electrical conductivity. The commercial silica-based monolith consisted of a silica-skeleton modified with C18 groups and activated carbon (MonoTrap RCC18, GL Sciences). This commercial silica-based monolith was selected based on the assumption that the activated carbon would help to minimise the charge built-up following each slicing step. The methodology developed for imaging and 3D reconstruction of both monolithic materials is described in detail. The reconstructed volumes were further used for calculation of the average macroporosity and comparison with porosity measurements obtained by MIP. The final aim of the present work was to present an alternative method for characterisation and visualisation of porous monolithic materials that complements more conventional characterisation techniques such as MIP, BET, SEM and TEM.
2. Experimental
2.1. Chemicals
1-Butanol and ferric chloride (99%) were obtained from Riedel-De Haen (Seelze, Germany). Resorcinol (99%), formaldehyde (37 wt% in water) and concentrated hydrofluoric acid (HF, 38–40%) were obtained from Sigma-Aldrich (Gillingham, UK). Silica beads with a diameter of 5 μm (surface area, 95 m2 g−1; pore size, 16 nm) were used as templates for the preparation of the carbon monolithic rods. Carbon-modified silica-based monolithic rods (MonoTrap RCC18) were obtained from GL Sciences (Tokyo, Japan) and used as received. Deionised water (18.2 MΩ cm) was obtained from a Millipore Direct-Q™ 5 water purification system (Millipore, Bedford, MA, USA).2.2. Preparation of carbon monolith
The carbon monolith was prepared following a similar procedure to that reported previously.25–27 Briefly, 1 g of 5 μm silica particles were dispersed in 1.5 g of 1-butanol under sonication for 1 h. This was followed by the addition of 0.18 g of ferric chloride and 0.367 g of resorcinol. Then, 0.3 g of ice-cooled, 37% formaldehyde solution in water was added to the mixture and stirred for 1 h in an ice-water bath. The resultant mixture was polymerised in 6 mm ID glass tubes kept at 90 °C for 16 h in a water bath (GFL, model 1013, Laborgerateborse GmbH, Burladingen, Germany). The resulting crack-free phenolic resin/silica rods were then removed from the glass tubes and aged for 72 h in a fumehood to slowly evaporate the majority of the residual solvent. Finally, the rods were dried under vacuum at 80 °C overnight using an EHRET thermovacuum oven (Ehret Labor and Pharmatechnik GmbH, KG, Emmendingen, Germany), and further cured at 135 °C for 4 h to ensure complete polymerisation. A desktop alumina tube furnace (model GSL1300X, MTI, Richmond, VA, USA) purged with N2 was then used to pyrolise the rods. The temperature was initially ramped from room temperature to 800 °C at a rate of 2.5 °C min−1, followed by a second ramp from 800 °C to 1250 °C at a rate of 10 °C min−1. Then, the furnace was allowed to cool down to room temperature. Next, the silica particles and the iron catalyst were removed from the rods by etching with 38% HF for 5 h. Finally, the carbon monolithic rods were thoroughly washed with deionised water and dried under vacuum at 80 °C for 16 h.2.3. Image acquisition
A small portion of the carbon and silica-based monolithic rods were subjected to FIB-SEM analysis with a FEI Quanta 3D FEG DualBeam instrument (FEI Ltd, Hillsboro, USA) integrating a Ga+ liquid metal ion source (spot size <5 nm). Prior to the FIB-SEM analysis, the samples were fixed onto the stub using silver paste and subsequently coated with a thin layer of gold (thickness, ∼7 nm) using a Emitech K575X Sputter Coating Unit (Quorum Technologies, UK). FIB sectioning was carried out at an acceleration voltage of 30 kV to etch a section of approximately 60 × 50 μm to a depth of 100 nm for the carbon monolith (current, 0.3 nA), and of 55 × 40 μm to a depth of 66.7 nm for the silica-based monolith (current, 0.5 nA). A 2D image of each section was automatically acquired by the SEM at a magnification of 2000× (1500× for the silica-based monolith), an acceleration voltage of 5 kV, and a nominal current of 5.92 pA for the carbon monolith and of 1.5 pA for the silica-based monolith. This FIB-SEM sequence was automatically repeated 100 times for the carbon monolith and 150 times for the silica-based monolith in a set routine. A total of 100 images were thus collected for the carbon monolith, each one being 100 nm further into the structure compared to the preceding image. Likewise, a total of 150 images were collected for the silica-based monolith, each one being 66.7 nm further into the structure. The angle of image acquisition was 52° in relation to the direction of the FIB (Fig. 1).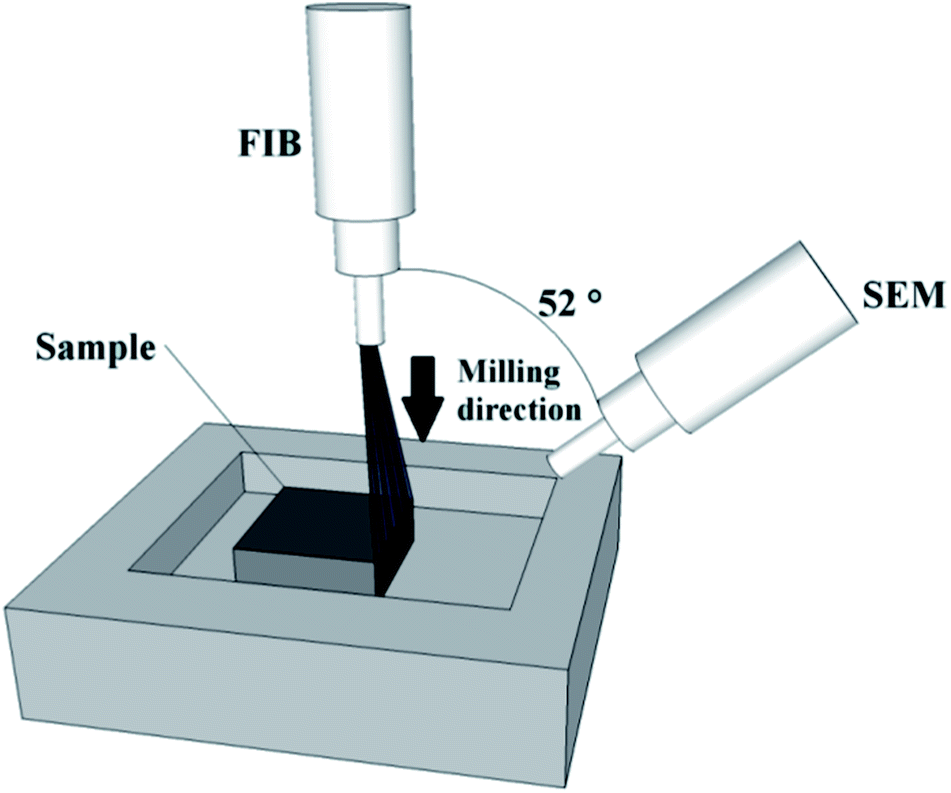 | ||
| Fig. 1 Sketch of the sample/FIB-SEM system. | ||
2.4. Image processing
The SEM images resulting from the FIB serial sectioning procedure (Fig. 2 and 3) were processed via image analysis routines to extract the pore boundaries and reconstruct the 3D structure. First, the images were resized anisotropically to correct for the image acquisition angle of 52°, after which the images were subjected to an image registration processes where landmarks were identified in the sequence of images. These landmarks were then used to calculate the necessary translation required to minimise any image alignment error. This was achieved using the formula for a rigid body translation (eqn (1)),x = {(cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ, −sinθ), (sinθ, cosθ)}·u + Δu θ, −sinθ), (sinθ, cosθ)}·u + Δu | (1) |
 | ||
| Fig. 2 Image captured by SEM after FIB sectioning of the carbon monolith. | ||
 | ||
| Fig. 3 Image captured by SEM after FIB sectioning of the carbon-modified silica-based monolith. | ||
The resulting images were then cropped to reduce the area of analysis to approximately 58.2 μm × 26.5 μm (width × height) for the carbon monolith and 44.7 μm × 33.7 μm for the silica-based monolith, with a resolution of 74 nm/pixel. From the images captured for the carbon monolith, only the first 93 were used for analysis. Thus, the depth of the final image stack was 9.3 μm, with a resolution of 100 nm per pixel in the depth direction. The depth of the final image stack employed for the reconstruction of the silica-based monolith was 10 μm, with a resolution of 66.7 nm per pixel in the depth direction. No attempt was made to increase the resolution of the captured images by increasing the SEM magnification, thus decreasing the size of the detectable pores. The scope of this work was to demonstrate the suitability of FIB-SEM analysis for reconstruction of the bulk porous structure in porous monolithic materials. Therefore, a rather large reconstructed volume was considered to be more representative of the overall monolith morphology, as compared to a reconstructed volume just a few cubic micrometers in size that would have been needed for visualisation of the mesoporous structure.
Finally, the two image stacks were binarised after manual outlining and filling of the porous structures, as shown in Fig. 4.
 | ||
| Fig. 4 From L to R: Outline of the pores, filling of pores and binarisation applied to the image stack obtained for the carbon monolith. | ||
2.5. 3D reconstruction and morphological analysis
Following binarisation, a 3D histogram of each of the image stacks was produced, enabling the calculation of the total macroporosity of the volume under analysis by eqn (2).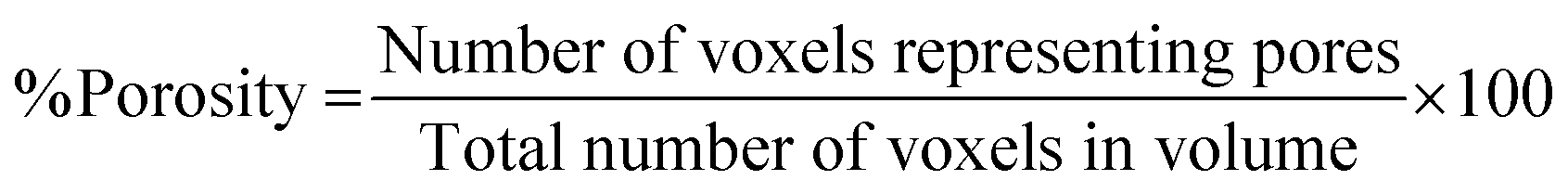 | (2) |
To asses the accuracy of the resulting macroporosity values, MIP measurements were also carried out with the carbon and silica-based monoliths. The total porosity and pore size distribution were determined with an Autopore IV 9500 Series (Micromeritics Instrument Corporation). Dried samples with weights ranging from 36 mg to 47 mg were used for these measurements.
The 3D reconstruction of both monoliths from their respective image stacks was then performed using the volume rendering technique available within the Materialise Mimics 13.0 software (Materialise NV, Belgium).
3. Results and discussion
3.1. 3D reconstruction and morphological analysis of the carbon monolith
A video showing the series of captured 2D-images obtained for the carbon monolith can be found in the ESI.† The resulting 3D reconstruction of the porous carbon monolith from the 2D-image stack consisting of 93 images is shown in Fig. 5. The volume reconstructed had a rectangular geometry with dimensions of 58.2 μm × 26.5 μm × 9.3 μm in the x, y and z directions, respectively. As shown in Fig. 5, the reconstructed carbon monolith exhibited an interconnected porous structure formed by quasi-spherical macropores with sizes mostly ranging from approximately 4 μm to 7.5 μm. Moreover, the 3D reconstruction revealed a non-homogeneous spatial distribution of the interskeleton macropores within the volume analysed. The latter could be attributed to a non-uniform packing density of the silica particles within the phenolic resin used for preparation of the carbon monolith. Likewise, the relatively broad range of macropore sizes observed in the monolith reconstruction could be ascribed to the use of silica particles with non-uniform sizes as templates, which could also largely affect the packing density of the silica particles in the silica/phenolic resin mixture. Furthermore, the presence of pores with sizes between ca. 10 μm and 15 μm is a clear indication of the formation of voids in the space between the silica particles during preparation of the monolith. This could be explained by the limited infiltration of the phenolic resin into the inter-particle space due to poor mixing of the silica particles with the phenolic resin and/or high viscosity of the phenolic resin. This non-homogeneous macropore morphology is consistent with previous results obtained by Eltmimi et al. in carbon monoliths prepared with 5 μm silica particles as templates, where the presence of inter-particle voids was already suspected based on experimental evidence.25 Therefore, the direct confirmation of the real morphology of a monolith via reconstruction and visualisation of its porous structure (including voids) represents a clear advantage of this FIB-SEM methodology, which can significantly contribute to speed up the optimisation of the monolith morphology in view of improving its analytical performance.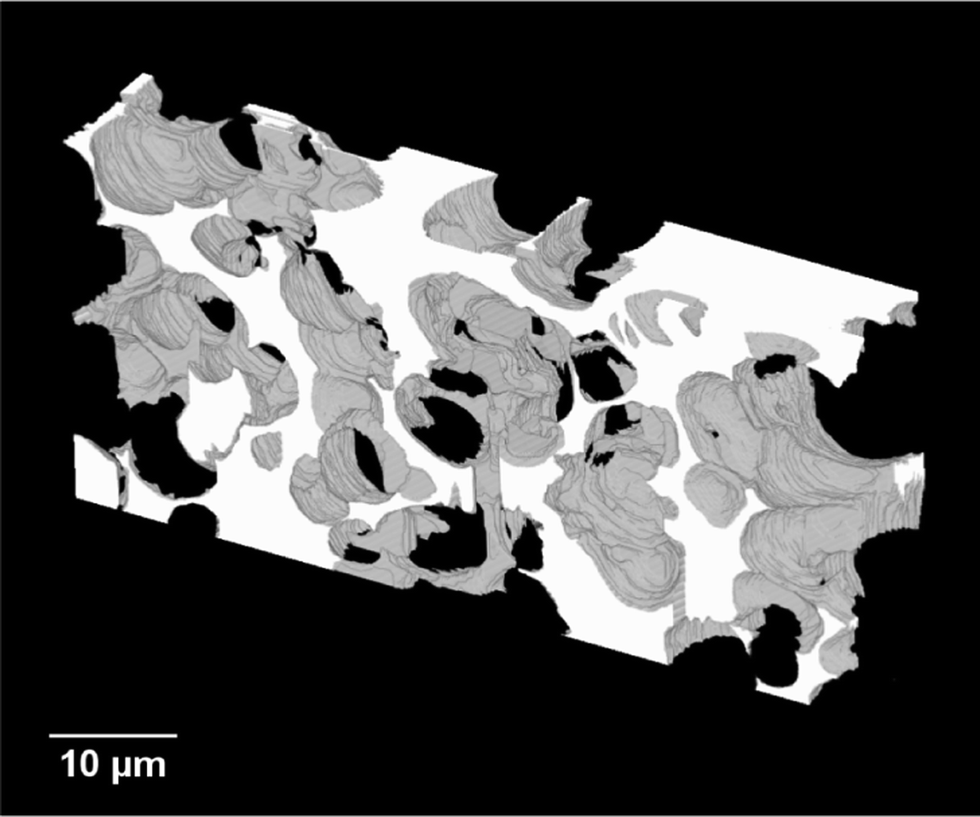 | ||
| Fig. 5 3D reconstruction of the porous carbon monolith. | ||
The total macroporosity of the volume analysed following binarisation of the corresponding image stack was found to be 72.9%. This value agrees well with the results obtained by MIP, where the total porosity was found to be 76.8% and 80.2% for two consecutive analyses. MIP measurements also allowed determination of the pore size distribution, as shown in Fig. S-1 (see the ESI†). A median pore diameter (in volume terms) of 2.6 μm and 2.9 μm was obtained for the two consecutive MIP analyses, respectively.
3.2. 3D reconstruction and morphological analysis of the silica-based monolith
The 3D reconstruction of a commercial porous silica-based monolith from the corresponding 150-image stack obtained by FIB-SEM analysis is shown in Fig. 6. The reconstructed volume also had a rectangular geometry with dimensions of 44.7 μm × 33.7 μm × 10 μm in the x, y and z directions, respectively. As can be seen in Fig. 6, the reconstructed volume exhibited a rather homogeneous spatial distribution of interconnected macropores with irregular shapes and variable sizes, as those commonly found in silica monoliths. Therefore, the modification of the silica monolith skeleton with activated carbon proved to be highly effective for successful reconstruction and visualisation of the silica-based monolith macropore structure by FIB-SEM analysis. The electrical conductivity provided by the carbon significantly minimised charge build-up, typically arising in non-conducting materials such as silica, and successfully allowed FIB-SEM analysis. Although no specific information on how this commercial carbon-modified silica-based monolith was produced, protocols for the preparation of carbon-silica composites have been already described in previous works. Shi et al. obtained a carbon-coated silica monolith with a bimodal pore structure by infiltration of a silica monolith with a styrene and divinylbenzene mixture prior to polymerisation and subsequent carbonisation of the resulting organic polymer under inert atmosphere.28 Alternatively, Alvarez et al. demonstrated the synthesis of carbon monoliths via the vapour deposition polymerisation method using furfuryl alcohol as the carbon precursor and a macro/mesoporous silica monolith as template (which in turn was prepared using polystyrene foam moulds via calcination of the resulting silica-polystyrene composite).29 Recently, the impregnation under vacuum of silica monoliths with different solutions of a phenolic resin in THF also resulted in carbon-silica composites. Those were ultimately used for the preparation of macro/microporous carbon monoliths following pyrolysis and removal of the silica template with HF.30 | ||
| Fig. 6 3D reconstruction of the porous carbon-modified silica-based monolith. | ||
The total macroporosity of the volume analysed for the carbon-modified silica-based monolith was found to be 52.9%, which agrees well with the 56.0% value for total porosity obtained by MIP. The pore size distribution of the silica-based monolith can be seen in Fig. S-2 (see the ESI†), with a median pore diameter (in volume terms) of 2.7 μm.
4. Conclusions
The 3D reconstruction of a carbon monolith and a commercial carbon-modified silica-based monolith using automated FIB serial sectioning and SEM imaging was demonstrated, showing that this methodology can be successfully employed for visualisation and characterisation of porous monolithic materials. The modification of the silica skeleton with activated carbon thus proved to be a suitable approach for FIB-SEM analysis of silica-based monoliths. The reconstructed volumes were then used for calculation of the total macroporosity demonstrating that the latter was also satisfactorily estimated from the 3D reconstructions with less than a 7.0% and a 5.5% relative error when compared to the porosity obtained by MIP for the carbon and the silica-based monoliths, respectively.Based on all the above, it is envisaged that FIB-SEM analysis followed by 3D reconstruction can be a very useful technique for direct determination of porosity and other morphological parameters in porous monoliths prepared at microscopic (capillary) scale, such as those used in capillary-LC or nano-LC, preventing errors associated with extrapolation of results obtained for bulk monoliths from MIP and nitrogen adsorption measurements. In addition, this direct visualisation method does not exclude closed pores or assume an ideal pore morphology as for MIP and nitrogen adsorption. Furthermore, the 3D reconstructions could be used for determination of additional morphological parameters such as pore sphericity, aspect ratio, hydraulic ratio, and interconnectivity, among others. It is anticipated that a more comprehensive morphological characterisation of such monolithic materials may result in a better understanding of how monolith preparation and resulting pore morphologies affect the analytical performance of stationary phases. Last but not least, 3D reconstructions have been proven to be an excellent tool for successful modelling of the “real” fluid flow and mass transport properties in porous monolithic materials.21,22 Therefore, the methodology presented here is expected to help advance research and knowledge in this area.
Acknowledgements
Financial support from Science Foundation Ireland (Grants 08/SRC/B1412 and 12/IA/1576) and the Irish Research Council for Science, Engineering & Technology is gratefully acknowledged. The authors would also like to thank Prof. Peter Myers for supplying the silica particles used as carbon-monolith templates, and Dr Ian Reid for his support with the FIB-SEM measurements.Notes and references
- B. Paull and P. N. Nesterenko, TrAC, Trends Anal. Chem., 2005, 24, 295 CrossRef CAS PubMed.
- J. Bones, K. Thomas, P. N. Nesterenko and B. Paull, Talanta, 2006, 70, 1117 CrossRef CAS PubMed.
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. X. Cui, I. Khayrullin, S. O. Dantas, I. Marti and V. G. Ralchenko, Science, 1998, 282, 897 CrossRef CAS.
- J. Wang and B. A. Freiha, Anal. Chem., 1982, 54, 861 CrossRef CAS.
- C. D. Liang, S. Dai and G. Guiochon, Anal. Chem., 2003, 75, 4904 CrossRef CAS.
- J. Lee, S. Han and T. Hyeon, J. Mater. Chem., 2004, 14, 478 RSC.
- H. Giesche, Part. Part. Syst. Charact., 2006, 23, 9 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- B. Mayr, G. Holzl, K. Eder, M. R. Buchmeiser and C. G. Huber, Anal. Chem., 2002, 74, 6080 CrossRef CAS.
- J. Courtois, M. Szumski, F. Georgsson and K. Irgum, Anal. Chem., 2007, 79, 335 CrossRef CAS PubMed.
- R. Wirth, Chem. Geol., 2009, 261, 217 CrossRef CAS PubMed.
- A. J. Bushby, K. M. Y. P'ng, R. D. Young, C. Pinali, C. Knupp and A. J. Quantock, Nat. Protocols, 2011, 6, 845 CAS.
- F. Schmidt, M. Kuhbacher, U. Gross, A. Kyriakopoulos, H. Schubert and R. Zehbe, Ultramicroscopy, 2011, 111, 259 CrossRef CAS PubMed.
- P. Schneider, M. Meier, R. Wepf and R. Muller, Bone, 2011, 49, 304 CrossRef PubMed.
- K. Matsuzaki, N. Shikazono and N. Kasagi, J. Power Sources, 2011, 196, 3073 CrossRef CAS PubMed.
- S. Thiele, R. Zengerle and C. Ziegler, Nano Res., 2011, 4, 849 CrossRef CAS.
- J. Balach, F. Miguel, F. Soldera, D. F. Acevedo, F. Mucklich and C. A. Barbero, J. Microsc., 2012, 246, 274 CrossRef CAS PubMed.
- T. Mullner, A. Zankel, C. Mayrhofer, H. Reingruber, A. Holtzel, Y. Q. Lv, F. Svec and U. Tallarek, Langmuir, 2012, 28, 16733 CrossRef PubMed.
- S. Bruns, T. Müllner, M. Kollmann, J. Schachtner, A. Höltzel and U. Tallarek, Anal. Chem., 2010, 82, 6569 CrossRef CAS PubMed.
- S. Bruns, T. Hara, B. M. Smarsly and U. Tallarek, J. Chromatogr., A, 2011, 1218, 5187 CrossRef CAS PubMed.
- D. Hlushkou, S. Bruns and U. Tallarek, J. Chromatogr., A, 2010, 1217, 3674 CrossRef CAS PubMed.
- D. Hlushkou, S. Bruns, A. Seidel-Morgenstern and U. Tallarek, J. Sep. Sci., 2011, 34, 2026 CAS.
- M. Milani, D. Drobne and F. Tatti, in Modern Research and Educational Topics in Microscopy, ed. A. Méndez-Vilas and J. Díaz, FORMATEX, Badajoz, Spain, 2007, vol. 2, pp. 787–794 Search PubMed.
- K. Hormann, T. Müllner, S. Bruns, A. Höltzel and U. Tallarek, J. Chromatogr., A, 2012, 1222, 46 CrossRef CAS PubMed.
- A. H. Eltmimi, L. Barron, A. Rafferty, J. P. Hanrahan, O. Fedyanina, E. Nesterenko, P. N. Nesterenko and B. Paull, J. Sep. Sci., 2010, 33, 1231 CAS.
- X. He, L. Zhou, E. P. Nesterenko, P. N. Nesterenko, B. Paull, J. O. Omamogho, J. D. Glennon and J. H. T. Luong, Anal. Chem., 2012, 84, 2351 CrossRef CAS PubMed.
- X. He, E. P. Nesterenko, P. N. Nesterenko, D. Brabazon, L. Zhou, J. D. Glennon, J. H. T. Luong and B. Paull, ACS Appl. Mater. Interfaces, 2013, 5, 8572 CAS.
- Z.-G. Shi, Y.-Q. Feng and S.-L. Da, Carbon, 2003, 41, 2668 CrossRef CAS.
- S. Alvarez, J. Esquena, C. Solans and A. B. Fuertes, Adv. Eng. Mater., 2004, 6, 897 CrossRef CAS.
- R. Backov, N. Brun, C. Sanchez, US Pat., US2011/0262993 A1, 2011.
Footnote |
| † Electronic supplementary information (ESI) available: Video showing the series of captured 2D-images obtained for the carbon monolith once aligned and cropped; Mercury intrusion - extrusion curves and pore size distributions for the carbon and the silica-based monoliths. See DOI: 10.1039/c3an01827j |
| This journal is © The Royal Society of Chemistry 2014 |