Effects of surface charges of graphene oxide on neuronal outgrowth and branching†
Qin
Tu‡
a,
Long
Pang‡
a,
Yun
Chen
a,
Yanrong
Zhang
a,
Rui
Zhang
b,
Bingzhang
Lu
c and
Jinyi
Wang
*a
aColleges of Science and Veterinary Medicine, Northwest A&F University, Yangling, Shaanxi 712100, China. E-mail: jywang@nwsuaf.edu.cn; Fax: +86-298-708-2520; Tel: +86-298-708-2520
bDepartment of Chemistry & Biochemistry, Florida International University, Miami, Florida 33199, USA
cDepartment of Chemical Physics, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 8th October 2013
Abstract
Graphene oxides with different surface charges were fabricated from carboxylated graphene oxide by chemical modification with amino- (–NH2), poly-m-aminobenzene sulfonic acid- (–NH2/–SO3H), or methoxyl- (–OCH3) terminated functional groups. The chemically functionalized graphene oxides and the carboxylated graphene oxide were characterized by infrared spectroscopy, X-ray photoelectron spectroscopy, UV-Vis spectrometry, ζ potential measurements, field emission scanning electron microscopy, and contact angle analyses. Subsequently, the resulting graphene oxides were used as substrates for culturing primary rat hippocampal neurons to investigate neurite outgrowth and branching. The morphological features of neurons that directly reflect their potential capability in synaptic transmission were characterized. The results demonstrate that the chemical properties of graphene oxide can be systematically modified by attaching different functional groups that confer known characteristics to the substrate. By manipulating the charge carried by the functionalized graphene oxides, the outgrowth and branching of neuronal processes can be controlled. Compared with neutral, zwitterionic, or negatively charged graphene oxides, positively charged graphene oxide was found to be more beneficial for neurite outgrowth and branching. The ability to chemically modify graphene oxide to control neurite outgrowth could be implemented clinically, especially in cases wherein long-term presence of outgrowth modulation is necessary.
Introduction
Graphene, a 2D monolayer of sp2-bonded carbon atoms, has elicited much interest due to its excellent electrical, optical, mechanical, and thermal properties.1 To date, much effort has been exerted to explore some of the novel properties of graphene-based materials, such as their superior electrical and photoelectrical attributes, as well as the availability of chemical functionalization for use in biological applications. Research has been focused on biological applications of graphene-based materials, such as drug/gene delivery,2,3 photothermal cancer therapy,4–6 imaging,7 and biosensing8–11 due to their characteristic properties with facile functionalization. For example, Chen et al.12 synthesized a hydrophilic and carboxylated graphene oxide (GO–COOH)–Au nanoparticle hybrid in situ. Subsequently, glucose oxidase (GOD) was bound to the surface of the hybrid through a condensation reaction between the amino groups on the lysine residues of GOD and the carboxylic acid groups on the Au nanoparticles. The hybrid provided a suitable microenvironment for GOD to retain its biological activity, after which direct electron transfer between the GOD and the electrode was observed. The blood sugar concentration in human serum samples was tested by using this novel glucose biosensor, and the results were consistent with measurements performed in a hospital. The study provided new information on the interactions between GO and biological molecules, which could lead to the fabrication of chemically functionalized GOs for biomedical applications.Biomedical applications of graphene-based materials have grown at a rapid pace and exhibited immense potential for future applications. Unlike other nanomaterials, graphene-based nanomaterials have advantages over the presently used cell growth substrates (for example carbon nanotubes) because of the high mobility of charge carriers,13 intrinsic low electrical noise,14 and reduced cytotoxicity.15,16 The nervous system would be an ideal breakthrough model for biomedical applications of graphene-based materials.17 Neuronal stimulation and monitoring are necessary for a variety of clinical diagnostics and treatments.18–20 Thus, the unique electrical properties of graphene-based materials offer great advantages for therapeutic or other purposes. The electronic properties of graphene can be tailored to match the charge transport required for electrical cellular interfacing.21 In addition, the chemically stable properties of graphene facilitate integration with neural tissues. However, studies on the interactions between graphene-based materials and neurons are still in their infancy. Li et al.17 reported the feasibility of using unmodified graphene as a substrate for neuronal growth. The number and average neurite length on the unmodified graphene were significantly enhanced 2 days to 7 days after cell seeding compared with the conventional tissue culture polystyrene. This work suggested the biocompatibility of graphene as a substrate for neurons. Zhou et al.22 prepared graphene–heparin/poly-L-lysine polyelectrolytes as neural scaffolds, and cell culture experiments have shown that the resulting graphene–polyelectrolyte scaffold supports neuron adhesion and neurite outgrowth, with no appreciable cell death. Therefore, electroactive scaffold modification may assist in neuronal regeneration for creating functional and biocompatible polymer scaffolds for electrical entertainment or biosensing applications. The above-mentioned studies show that graphene-based materials can potentially be used for neural prostheses. Given that graphene-based materials are not biodegradable, they could be used as implants where long-term extracellular molecular cues for neurite outgrowth are necessary, such as in regeneration after nerve injury.
Previous studies have reported that the ends of carbon nanotubes can be chemically modified in a defined manner (changing the surface charge of carbon nanotubes) for neurite outgrowth and branching.23 Considering that graphene-based materials and carbon nanotube have similar properties, we hypothesized that the surface charge of graphene-based materials (particularly GO) can also modulate neurite outgrowth and branching. To test this hypothesis, the carboxylated GO (GO–COOH) was chemically modified with amino- (–NH2), poly-m-aminobenzene sulfonic acid- (–NH2/–SO3H), or methoxyl- (–OCH3) terminated functionalities to change its surface charge. The surface modification of GO can be used to successfully control the characteristics of neurite outgrowth.
Experimental
Materials and reagents
Carboxylated graphene oxide (GO–COOH) was purchased from Nanoon (Hebei, China). Polyoxyethylene bis(amine) (Mn = 4000) was purchased from Aladdin (Shanghai, China). Poly(ethylene glycol) monomethyl ether (Mn = 5000) was purchased from Sigma-Aldrich (St. Louis, MO). Poly(m-aminobenzene sulfonic acid) (PABS, Mn = 4083) utilized in this study was synthesized as previously described.24N-Hydroxysuccinimide (NHS) and 1-ethyl-3-(3-(dimethylamino)propyl) carbodi-imide (EDC) were obtained from GL Biochem, Ltd. (Shanghai, China). Hoechst dye H33258, propidium iodide (PI), acridine orange (AO), and cytosine β-D-arabinofuranoside (Ara-C) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The cell culture medium, fetal calf serum, and newborn calf serum were purchased from Gibco Invitrogen Corporation (CA, USA). The specific primary antibodies against tau (mouse monoclonal IgG) and MAP-2 (mouse monoclonal IgG), goat anti-mouse (IgG-fluorescein isothiocyanate), and goat anti-rabbit antibodies (IgG-rhodamine B isothiocyanate) were purchased from Boster Biotechnology (Wuhan, China). All solvents and other chemicals were purchased from local commercial suppliers and were of analytical reagent grade, unless otherwise stated. All solutions were prepared using ultra-purified water (18.4 MΩ cm) supplied by a Milli-Q water system (Millipore, Billerica, MA).Preparation of chemically functionalized GO
The GO–COOH solution (1 mg mL−1) was ultrasonicated for approximately 8 h before the modification process was performed.25 The resulting solution was treated with 2.2 mmol L−1 EDC and 1.5 mmol L−1 NHS in phosphate-buffered saline (PBS; pH 7.4). After the carboxyl groups were activated, the solution was treated with 130.6 μmol L−1 polyoxyethylene bis(amine) (NH2-PEG-NH2), poly(ethylene glycol) monomethyl ether (CH3O-PEG-OH), or PABS for 1 h and stirred at room temperature for 48 h. The raw product was purified by centrifugation (four times at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 4 min each) and washed thrice with ultra-purified water. Finally, chemically functionalized GOs (GO–NH2, GO–OCH3, and GO–PABS) were obtained.
000 rpm for 4 min each) and washed thrice with ultra-purified water. Finally, chemically functionalized GOs (GO–NH2, GO–OCH3, and GO–PABS) were obtained.
Preparation of chemically functionalized GO substrates
GO films were prepared by deposition of the chemically functionalized GOs onto polyethyleneimine (PEI)-coated glass coverslips for neuronal growth. The chemically functionalized GO samples were separately dispersed by sonication in 95% ethanol (1 mg mL−1) for 30 min and then sprayed onto preheated (65 °C to 75 °C) glass coverslips (round, 10 mm in diameter) that were pre-coated with a thin layer of PEI.26–28 After ethanol evaporation, the chemically functionalized GO formed films on the surface of PEI-coated glass coverslips. GO-coated glass coverslips were then inlaid into glass culture dishes (round, 35 mm in diameter) and sterilized under UV light, followed by application of the cell culture medium to the dishes/coverslips prior to use for neuronal growth. The pre-coating of glass coverslips with PEI was necessary because the GO films formed on glass coverslips without PEI pre-coating exhibited short retention (minutes to hours) when exposed to aqueous culture medium, resulting in their peeling off and floating on the culture medium.23Analysis of chemically functionalized GOs and GO substrates by FT-IR spectroscopy, XPS, UV-Vis spectroscopy, ζ potential, FE-SEM, and contact angle measurement
Fourier-transform infrared spectroscopy (FT-IR) was recorded on a Nicolet 5700 spectrometer at wavelengths ranging from 500 cm−1 to 4000 cm−1 and a resolution of 3 cm−1 in 32 scans. The FT-IR spectra were measured using KBr pellets. X-ray photoelectron spectroscopy (XPS) analyses were performed on an Axis Ultra X-ray photoelectron spectrometer with an Al X-ray source operating at 150 W (15 kV, 13 mA). The vacuum in the main chamber was kept above 3 × 10−6 Pa during XPS data acquisitions. UV-Vis absorption spectra of chemically functionalized GO (GO–COOH, GO–NH2, GO–OCH3, and GO–PABS) ethanol solutions (0.5 mg mL−1) were obtained using a NICOLET300 Scan UV-Vis spectrophotometer at wavelengths ranging from 200 cm−1 to 700 cm−1. The surface charges of the chemically functionalized GOs in ethanol solutions (0.05 mg mL−1) were analyzed by ζ potential measurements using a Zetasizer Nano ZS (Malvern Instruments, Malvern, U.K.). The morphology of the prepared chemically functionalized GOs was observed under an S-4800 field emission scanning electron microscope (FE-SEM) at an acceleration voltage of 5.0 kV. The GO–COOH, GO–NH2, GO–OCH3, and GO–PABS ethanol solutions (0.5 mg mL−1, 1 drop) were dripped onto clean mica sheets, which were dried overnight at room temperature. The samples were gold plated for 30 s prior to imaging. The contact angles of the prepared chemically functionalized GOs were measured using Dropmeter 100 equipment (Marist Vision, Ningbo, China) using the sessile drop method.Primary rat hippocampal neuron culture
The primary hippocampal-dissociated neurons were prepared from postnatal Sprague-Dawley (SD) rat pups (aged 1 day to 3 days) following the methods reported previously.29,30 All animal treatment procedures were conducted according to the Institutional and Ethical Guidelines of The Committee of Science and Technology of the People's Republic of China (1988). Briefly, the hippocampi of the rats were dissected in calcium-free and magnesium-free Hank's balanced salt solution (CMF-HBSS). After rinsing with CMF, the hippocampi were resuspended in trypsin solution (0.125% trypsin in CMF-HBSS) for 15 min at 37 °C. Trypsinization was stopped with Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum. The cells were mechanically dissociated through trituration in CMF-HBSS using a fire-polished Pasteur pipette, and the resulting supernatant was centrifuged at 1000 rpm for 5 min. The hippocampi were then resuspended, counted, diluted with DMEM, and supplemented with 10% fetal calf serum (Invitrogen), 0.5 mmol L−1L-glutamine (Invitrogen), 0.03 mmol L−1 glucose, 100 U mL−1 penicillin, and 0.1 mg mL−1 streptomycin (Sigma-Aldrich). To investigate the primary hippocampal neuronal growth behavior on the chemically functionalized GO substrates, the hippocampal neurons were cultured on different chemically functionalized GO substrates at an initial cell density of 1 × 105 cells per mL. After one day, the culture medium was replaced with complete growth medium (DMEM containing 0.5 mmol L−1L-glutamine and 1% B-27). Non-neuronal cell division was halted by following the methods reported previously.31,32 Briefly, feed cells on day 3, adding Ara-C (to kill dividing cells) by replacing complete growth medium containing Ara-C (a final Ara-C concentration of 5 μmol L−1). Feed the cells again on day 5 by replacing with complete growth medium (without Ara-C). From then on, feed cells every half day by replacing half of the medium.32 On day 7, we observed the primary hippocampal neuronal growth behavior on the chemically functionalized GO substrates. Each test was repeated at least three times. Control experiments were run simultaneously during each experiment.Cell viability analysis
After the samples were cultured on chemically functionalized GO substrates for one week, cell viability was assessed according to the AO/PI double staining protocol.33 After rinsing the cultures thrice with PBS, AO (final concentration of 5 mg mL−1) and PI solutions (final concentration of 1.0 mmol L−1) were successively introduced into the neuronal culture dishes. After the cultures were incubated for 10 min at room temperature and washed thrice with PBS, cell viability was analyzed by counting live (green) and dead (red) cells.SEM observation of neuron morphology
The cells were cultured on the chemically functionalized GO substrates for 7 days, fixed with 4% paraformaldehyde in PBS for 1 h, and then rinsed thrice with PBS. Dehydration was performed by rinsing the samples with graded ethanol–water mixtures (50%, 70%, 80%, 90%, and 100%; each step was performed for 10 min at 4 °C). Ethanol was slowly exchanged successively by amyl acetate and liquid CO2. The samples were dried using the critical point method, and then sputter coated with a thin gold layer.34Immunochemistry staining of the cultured neurons
After the cultured neurons were washed thrice with PBS (pH 7.4) and fixed with 4% paraformaldehyde for 30 min at room temperature, 0.2% Triton X-100 solution in PBS was introduced to permeabilize the nerve cells cultured on chemically functionalized GO substrates for 30 min. The permeabilized nerve cells were then incubated with 0.2% Triton X-100 and 10% newborn calf serum (NBS) in PBS for 30 min at 37 °C to block nonspecific binding during immunochemistry staining. Sequentially, the nerve cells were incubated at 4 °C overnight with specific primary antibodies, namely, monoclonal mouse anti-MAP2 antibody (for neuron-specific cytoskeletal protein MAP2 determination)35 and monoclonal rabbit anti-tau antibody (for neuron-specific cytoskeletal protein tau determination),36 in PBS containing 0.2% Triton X-100 and 10% NBS. Dilution of the primary antibodies utilized for immunochemistry staining was performed as follows: before utilization, the original monoclonal mouse anti-MAP2 antibodies (Boster, Wuhan) and monoclonal rabbit anti-tau antibodies (Boster, Wuhan) were diluted using PBS with a volume ratio of 1:50 (primary antibody:PBS). After rinsing thrice with PBS, fluorescence-labeled secondary goat anti-rabbit antibodies (IgG-FITC; Boster, Wuhan) and goat anti-mouse (IgG-RBITC; Boster, Wuhan), that were diluted with PBS at a volume ratio of 1:20, were added into the nerve cells cultured in Petri dishes and incubated at 37 °C for 1 h. The immunochemistry-stained nerve cells were then washed thrice with PBS. Cell nuclei staining was performed by following the procedures described above using Hoechst dye H33258 (0.05 mg mL−1; Sigma-Aldrich).
Image acquisition and analysis
Bright-field and fluorescence images were obtained using an inverted microscope (Olympus, CKX41) equipped with a CCD camera (QIMAGING, Micropublisher 5.0 RTV) and a mercury lamp (Olympus, U-RFLT50). Software Image-Pro Plus® 6.0 (Media Cybernetics) and SPSS 12.0 (SPSS Inc.) were used in image and statistical data analyses, respectively. The results and error bars in the graphs were expressed as the mean ± S.D. Tests of data significance were performed using one-way ANOVA.Results and discussion
Synthesis and characterization of the functionalized GOs
To investigate the effect of the surface charge of GO on neurite outgrowth and branching, chemically functionalized GOs (GO–NH2, GO–OCH3, and GO–PABS; Scheme 1) were synthesized by activating the carboxyl (–COOH)-rich groups of the GO–COOH with 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide/N-hydroxysuccinimide (EDC/NHS) chemistry.25 All the prepared GO materials were characterized by FT-IR, XPS, UV-Vis spectroscopy, ζ potential measurements, field FE-SEM, and contact angle analyses.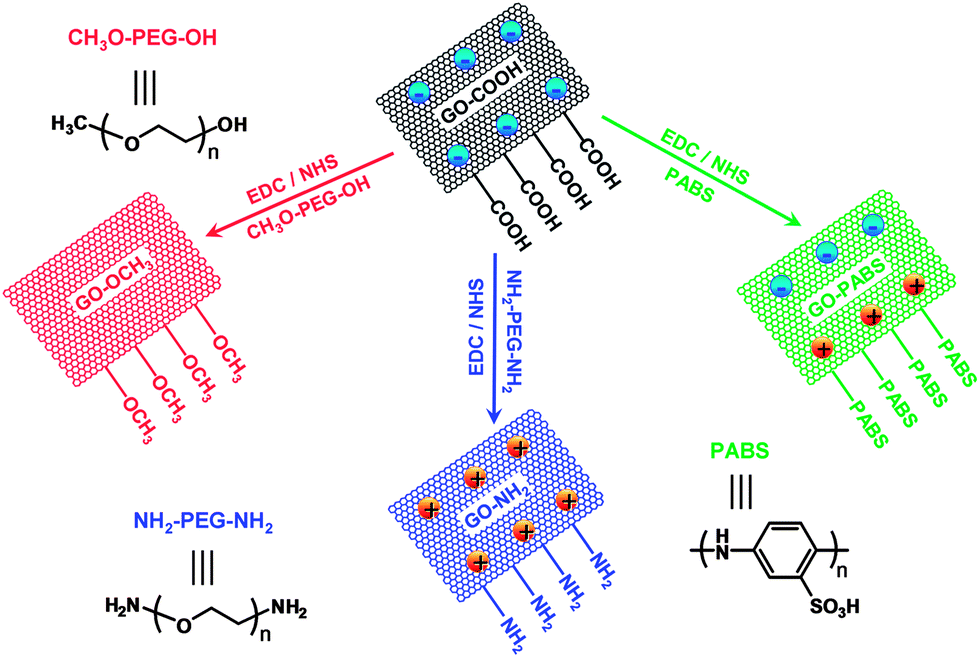 | ||
| Scheme 1 Chemical functionalization of graphene oxides. At the 7.4 physiological pH used to grow neurons, GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 exhibit different surface charges ranging from negative to neutral/zwitterionic to positive. | ||
Fig. 1 shows the FT-IR spectra of GO–COOH, GO–NH2, GO–OCH3, and GO–PABS. The GO–COOH spectrum (Fig. 1, black line) shows the presence of O–H (νO–H at 3412 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (νCO at 1731 cm−1 in carbonyl groups), and CC (νCC at 1622 cm−1). The typical adsorption peaks of GO–NH2 (Fig. 1, blue line) and GO–OCH3 (Fig. 1, red line) became visible when GO–COOH was modified with NH2-PEG-NH2 or CH3O-PEG-OH. The GO–NH2 and GO–OCH3 spectra exhibited the characteristic PEG absorption features at 2872 and 2854 cm−1 due to C–H symmetric and asymmetric stretching of methyl and methylene groups, respectively.37 The peaks (Fig. 1, blue and red lines) at around 1085 cm−1 were identified as C–O–C stretching vibrations in PEG units.37 Compared with the spectra of the polymers NH2-PEG-NH2 and CH3O-PEG-OH (Fig. S1 and S2, ESI†), the characteristic NH–CO (1640 cm−1) and O–CO (1721 cm−1) stretching vibrations also appeared in the polymer functionalized GOs (GO–NH2 and GO–OCH3, respectively). The GO–PABS spectrum (Fig. 1, green line) showed a strong quinoid band at 1580 cm−1 and weak benzenoid absorption at 1500 cm−1. The symmetric stretching vibration of SO2 appeared between 1170 and 1028 cm−1, and the peaks at 701 and 693 cm−1 were assigned to the characteristic S–O and C–S stretching modes, respectively.24 The strong quinoid band at 1580 cm−1 may overlap with the characteristic CC stretching (1622 cm−1) of GO. Compared with the polymer PABS spectrum (Fig. S3, ESI†), the peak at around 1640 cm−1 was attributed to the amide carbonyl vibration of the target GO–PABS. All the results above indicate that the chemically functionalized GOs (GO–NH2, GO–OCH3, and GO–PABS) were successfully synthesized.
O (νCO at 1731 cm−1 in carbonyl groups), and CC (νCC at 1622 cm−1). The typical adsorption peaks of GO–NH2 (Fig. 1, blue line) and GO–OCH3 (Fig. 1, red line) became visible when GO–COOH was modified with NH2-PEG-NH2 or CH3O-PEG-OH. The GO–NH2 and GO–OCH3 spectra exhibited the characteristic PEG absorption features at 2872 and 2854 cm−1 due to C–H symmetric and asymmetric stretching of methyl and methylene groups, respectively.37 The peaks (Fig. 1, blue and red lines) at around 1085 cm−1 were identified as C–O–C stretching vibrations in PEG units.37 Compared with the spectra of the polymers NH2-PEG-NH2 and CH3O-PEG-OH (Fig. S1 and S2, ESI†), the characteristic NH–CO (1640 cm−1) and O–CO (1721 cm−1) stretching vibrations also appeared in the polymer functionalized GOs (GO–NH2 and GO–OCH3, respectively). The GO–PABS spectrum (Fig. 1, green line) showed a strong quinoid band at 1580 cm−1 and weak benzenoid absorption at 1500 cm−1. The symmetric stretching vibration of SO2 appeared between 1170 and 1028 cm−1, and the peaks at 701 and 693 cm−1 were assigned to the characteristic S–O and C–S stretching modes, respectively.24 The strong quinoid band at 1580 cm−1 may overlap with the characteristic CC stretching (1622 cm−1) of GO. Compared with the polymer PABS spectrum (Fig. S3, ESI†), the peak at around 1640 cm−1 was attributed to the amide carbonyl vibration of the target GO–PABS. All the results above indicate that the chemically functionalized GOs (GO–NH2, GO–OCH3, and GO–PABS) were successfully synthesized.
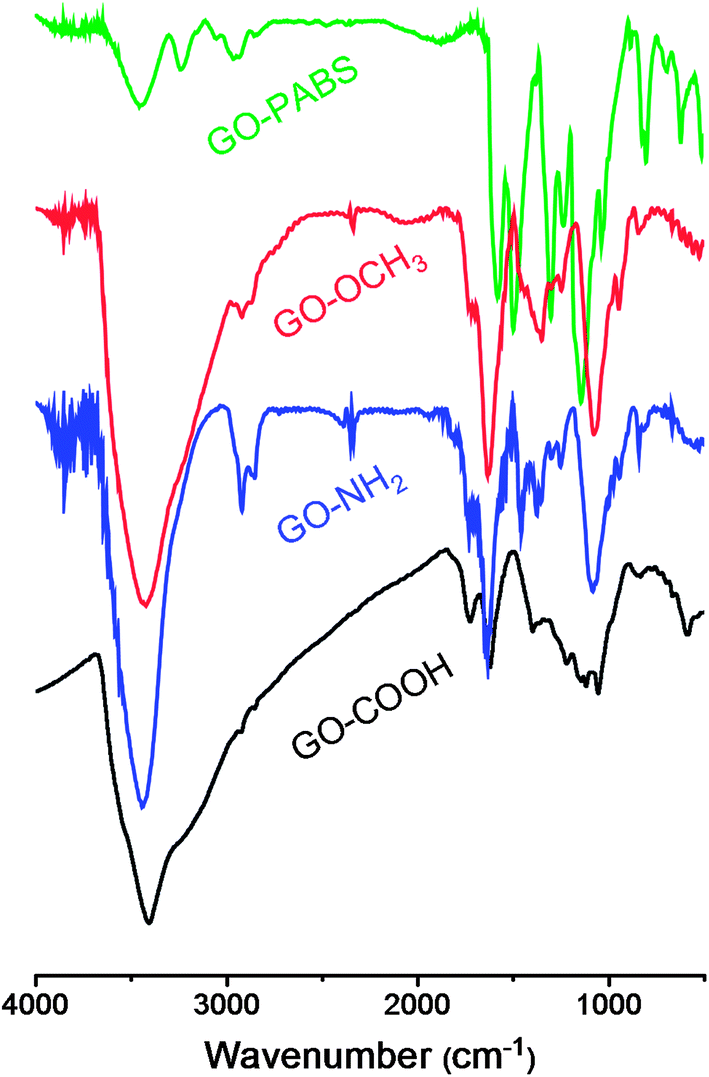 | ||
| Fig. 1 FT-IR spectra of GO–COOH (black line), GO–NH2 (blue line), GO–OCH3 (red line), and GO–PABS (green line). | ||
The formed GO–NH2, GO–OCH3, and GO–PABS were further confirmed by XPS measurements (Fig. 2 and 3), which are used as effective tools to characterize the presence of different elements, such as carbon, nitrogen and oxygen. Only carbon (C1s at 284.5 eV) and oxygen (O1s at 532.0 eV) appeared in the wide-scan spectrum of GO–COOH (Fig. 2, black line) and GO–OCH3 (Fig. 2, red line). After GO–NH2 was formed, nitrogen (N1s at 400.0 eV) appeared in the wide-scan spectrum. A certain amount of sulfur (S2s at 227.9 eV) was evidently present in the GO–PABS, in addition to C, O, and N (Fig. 2, green line). This phenomenon was attributed to the sulfonic acid groups in the PABS units. Further evidence of these composites was obtained through high-resolution XPS spectra (Fig. 3). The high-resolution C1s XPS spectra of GO–COOH (Fig. 3a) exhibited peaks at 284.6 eV (C–C), 286.7 eV (C–O), 287.8 eV (CO), and 288.7 eV (O–CO). After CH3O-PEG-OH was conjugated, the C1s XPS spectrum of GO–OCH3 (Fig. 3b) showed a significant increase in signals at 284.6 eV, indicating the increase in C–C functionalities. The results suggest that GO–COOH was functionalized well by CH3O-PEG-OH. Fig. 3c shows the C1s XPS spectrum of GO–NH2. The additional absorbance peaks appearing at 285.6 and 287.8 eV correspond to carbon in the C–N bond and OC–N bond, respectively. The high-resolution C1s XPS spectra of GO–PABS (Fig. 3d) have peaks at 284.6 eV (C–C), 286.7 eV (C–O/C–S), 285.6 eV (C–N), and 287.8 eV (OC–N). The appearance of the C–S and C–N species, as well as OC–N, confirms the formation of PABS-functionalized GO. A comparison of the above XPS results of functionalized GOs with those (Fig. S4, ESI†) of the polymers (NH2-PEG-NH2, CH3O-PEG-OH, and PABS) also suggests that the GO–NH2, GO–OCH3, and GO–PABS were successfully prepared.
 | ||
| Fig. 2 XPS wide-scan spectra of GO–PABS (a), GO–NH2 (b), GO–OCH3 (c), and GO–COOH (d). | ||
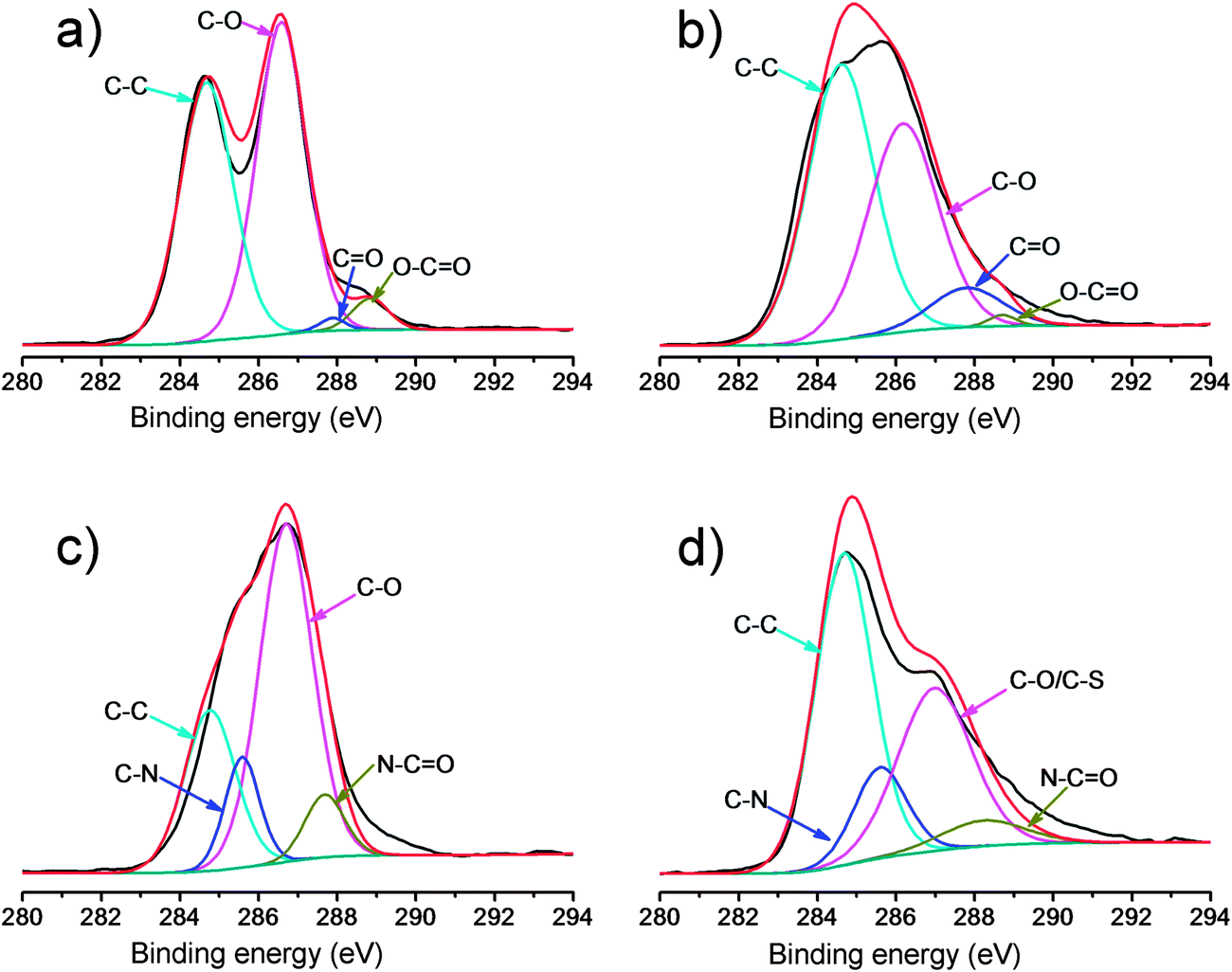 | ||
| Fig. 3 High-resolution XPS C1s spectra of GO–COOH (a), GO–OCH3 (b), GO–NH2 (c), and GO–PABS (d). | ||
The UV-Vis spectra of GO–COOH, GO–NH2, GO–OCH3, and GO–PABS are shown in Fig. 4. The absorption peak of GO–COOH originating from the π-plasmon of carbon38,39 was observed at 212 nm. Considering that NH2-PEG-NH2 and CH3O-PEG-OH cannot be detected by UV-Vis absorption, the absorption peaks of NH2-PEG-NH2 and CH3O-PEG-OH functionalized GOs (GO–OCH3 and GO–NH2) remained essentially unchanged. However, the absorption of GO–PABS redshifted from 212 nm to 226 nm due to the introduction of PABS, as well as the interaction between the GO sheets and the benzene ring of PABS (Fig. 4a). In addition, at 0.5 mg mL−1 concentration, the chemically functionalized GO solutions were very stable (Fig. 4b), even after several weeks of storage without the formation of precipitates, which is very favorable for further application of the functionalized GOs.40
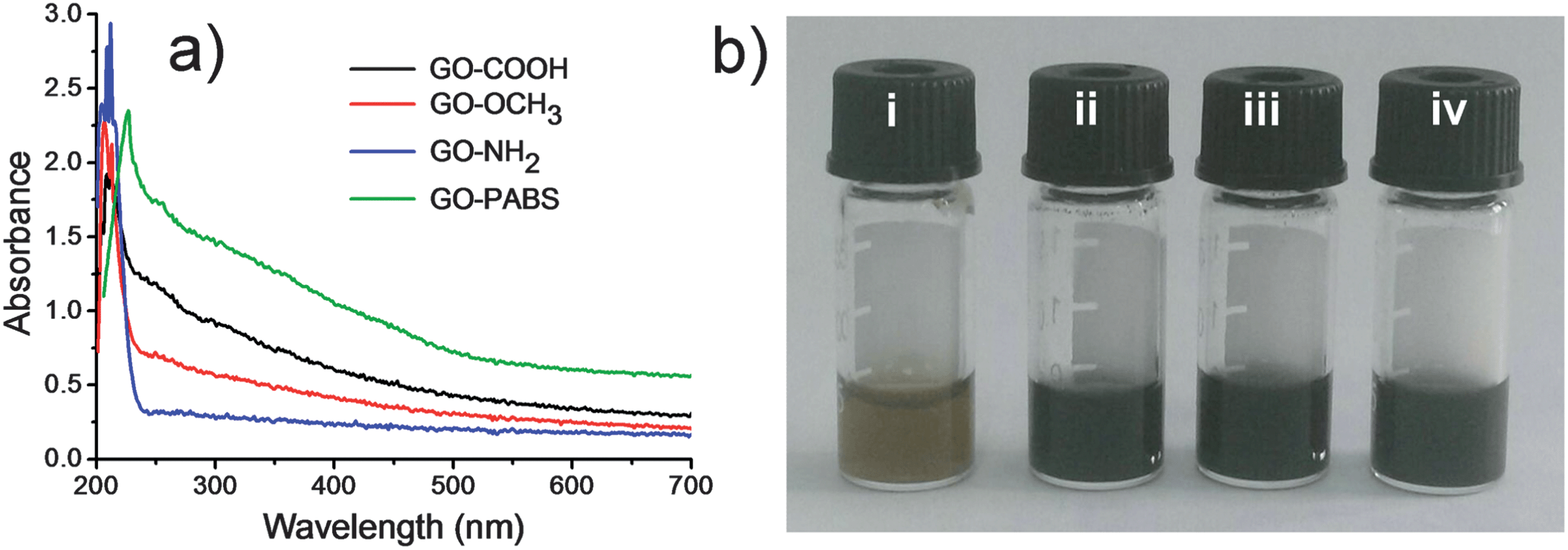 | ||
| Fig. 4 (a) UV-Vis spectra of GO–COOH (black line), GO–OCH3 (red line), GO–NH2 (blue line), and GO–PABS (green line); (b) images of GO–COOH (i), GO–OCH3 (ii), GO–NH2 (iii), and GO–PABS (iv) ethanol solutions. | ||
The surface charges of GO–COOH, GO–NH2, GO–OCH3 and GO–PABS were analyzed by ζ potential measurements following the method reported previously.41 The results showed that GO–COOH had a negatively charged surface with a ζ potential of −49.1 mV. After reaction with NH2-PEG-NH2, the prepared GO–NH2 possessed a positively charged surface with a ζ potential of 40.4 mV due to the introduction of –NH2 groups. However, the prepared GO–PABS and GO–OCH3 with polymers PABS and CH3O-PEG-OH almost showed neutral surfaces due to the introduction of neutral –OCH3 and zwitterionic –NH2/–SO3H groups. The ζ potential of GO–PABS and GO–OCH3 was −0.71 and −0.34 mV, respectively. The small ζ potential of the GO–PABS and GO–OCH3 may be caused by the residual carboxyl groups of the GO–COOH surface due to the incomplete modification.
The morphologies of the substrates of GO–COOH, GO–NH2, GO–OCH3, and GO–PABS were studied by field emission (FE)-SEM (Fig. 5a). The 0.5 mg mL−1 dispersions of GO–COOH, GO–NH2, GO–OCH3, or GO–PABS (in ethanol) were placed on mica sheets to form a thin layer. The mica sheets were then dried under ambient conditions for 24 h and directly examined using an FE-SEM. The distortions caused by the oxygen groups and the extremely small thicknesses of the resulting functionalized GO sheets led to a wrinkled topology (Fig. 5a). Compared with GO–COOH [Fig. 5a(i)], the stacking of every layer of GO–OCH3 [Fig. 5a(ii)], GO–PABS [Fig. 5a(iii)], and GO–NH2 [Fig. 5a(iv)], was considerably more desultory. The crumpled and wrinkled edges of chemically functionalized GOs are believed to be possibly rolled into irregular structures and may be folded at times, which may be attributed to the strong interactions between the chemically functionalized chains and the carboxyl groups of the GO–COOH sheet themselves.42
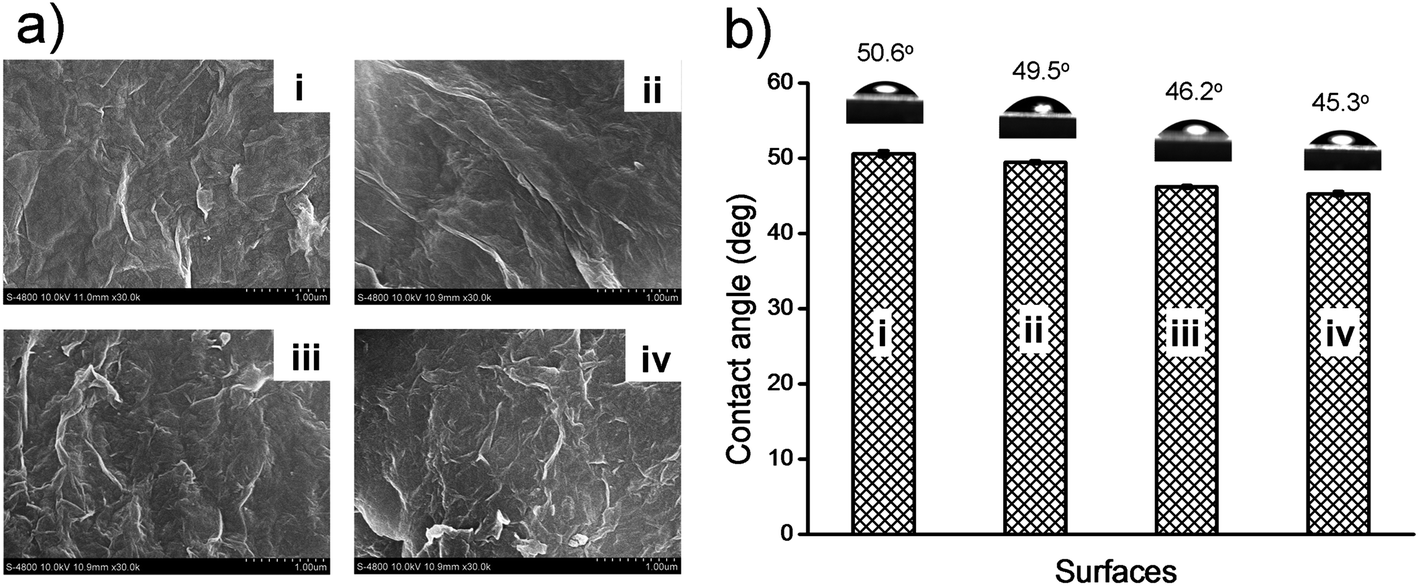 | ||
| Fig. 5 (a) FE-SEM (30000× magnification) images of GO–COOH (i), GO–OCH3 (ii), GO–PABS (iii), and GO–NH2 (iv); (b) wettability of GO–COOH (i), GO–OCH3 (ii), GO–PABS (iii), and GO–NH2 (iv) films; inset: images of water drops on the surfaces of the GO films. | ||
The hydrophilicities of the substrates of GO–COOH, GO–NH2, GO–OCH3, and GO–PABS were evaluated according to the sessile drop method.43Fig. 5b shows that the contact angles of the GO–COOH [Fig. 5b(i)], GO–OCH3 [Fig. 5b(ii)], GO–PABS [Fig. 5b(iii)], and GO–NH2 [Fig. 5b(iv)] substrates were 50.6°, 49.5°, 46.2°, and 45.3°, respectively. These results suggest that the functionalized GOs possess good hydrophilicity. The slight differences in the contact angles also provide supporting evidence that GO–NH2, GO–OCH3, and GO–PABS were successfully prepared.
In summary, different GO modifications were employed at 7.4 physiological pH for use in growing neurons. Due to their association constants, the resulting functionalized GO products exhibited different surface charges ranging from negatively charged GO–COOH, neutral GO–OCH3, and zwitterionic GO–PABS to positively charged GO–NH2.
Culture of primary rat hippocampal neurons on the chemically functionalized GOs
Based on the characterization and analysis of the functionalized GOs, the effect of the surface charges on the chemically functionalized GOs on neurite outgrowth and branching were investigated. The primary hippocampal neurons from postnatal 1 day to 3 day SD rat pups with an initial cell density of 1 × 105 cells per mL were cultured on the functionalized GO films. The cell viability assays using the acridine orange and propidium iodide (AO/PI) double-staining protocol33 showed that the hippocampal neurons remained alive (stained green) for 7 days in culture on glass coverslips coated with the chemically functionalized GOs (Fig. 6, top). Quantification analysis (Fig. 6, bottom) of the total number of adherent neurons indicated that the viability of neurons can reach over 96%.17 The results indicate that the resulting GOs exhibit excellent biocompatibility for use in the culture of primary hippocampal neurons.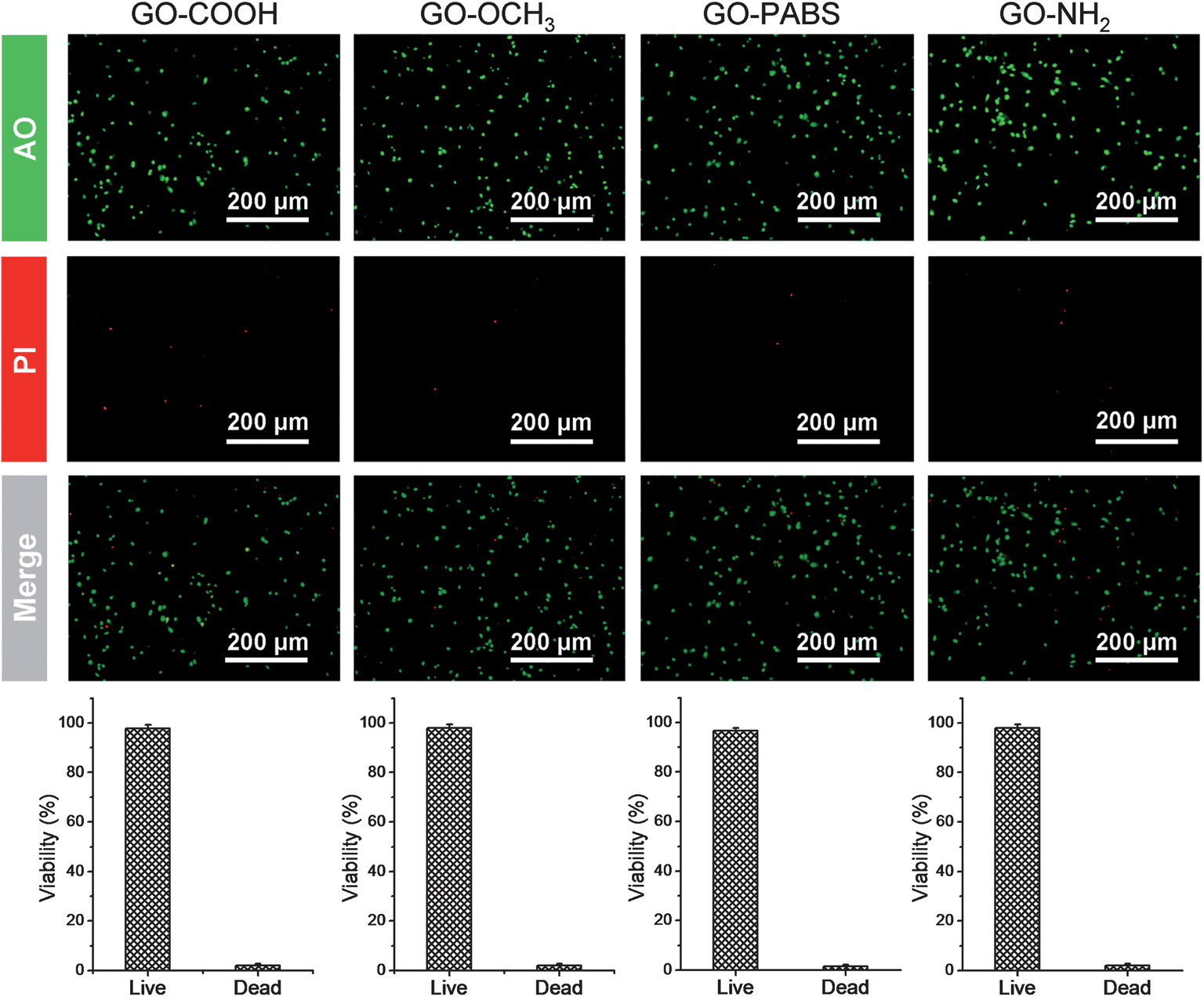 | ||
| Fig. 6 Cell viability analysis. Top: one set of the images of AO/PI double-staining hippocampal neurons after culturing for 7 days on GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 (from left to right); bottom: quantitative analysis of the viability of hippocampal neurons after culturing for 7 days on GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 (from left to right). | ||
After the initial evaluation of the biocompatibility of functionalized GOs, tests were carried out to determine whether the surface charge of the GOs can modulate neurite outgrowth and branching. Neurons were grown on the functionalized GO films. Neuron attachment was assessed by light microscopy. Fig. 7 (first row) shows one set of the representative images of neurons cultured on different films after incubating the samples for 7 days. The results indicate that hippocampal neurons grew well on any of the substrates tested. All the rat hippocampal neurons displayed a regular cobblestone morphology, apparent neurites, and formation of extensive neurite networks. To evaluate the potential modulatory effect of the charges, several different parameters related to neurite outgrowth were quantified (Fig. 8). Although no obvious differences were found between the number of neurites per neuron when these cells were grown on different GOs (Fig. 8a), the maximum neurite length and total outgrowth per neuron were longer when the neurons where plated onto positively charged GO–NH2 (conjugate acid at physiological pH), instead of zwitterionic GO–PABS, neutral GO–OCH3, or negatively charged GO–COOH (conjugate base) (Fig. 8b and c). The cell body area per neuron was higher in neurons grown on GO–NH2 than those on GO–PABS, GO–OCH3, or GO–COOH (Fig. 8d). Branching of neurites indicates the graded dependency of GO charge in the order GO–NH2 > GO–PABS/GO–OCH3 > GO–COOH (Fig. 8e and f), suggesting that chemically modified GOs can be used to control neurite branching, which may be attributed to the charge on the different types of chemical functionalities attached to GO. In the culture media (pH = 7.4), the carboxylic groups on GO–COOH became deprotonated, resulting in negatively charged GO–COOH. By contrast, the amino groups on GO–NH2 became positively charged due to the higher pKa of the amino groups. GO–PABS (containing both amino and sulfonic groups, nearly zwitterionic) and GO–OCH3 were almost electroneutral at pH 7.4. The SEM image of typical neurons (Fig. 7, second row) after 7 days in culture on different functionalized GOs shows that neurons cultured on chemically functionalized GOs exhibited normal adhesion and neurite formation. Previous studies have demonstrated that substrate qualities are involved in growth cone motility and neurite branching.44,45 In the present study, neurons grown on GO–OCH3, GO–PABS, and GO–NH2 were shown to have higher number of growth cones than neurons grown on GO–COOH (Fig. 7, second row), indicating that negatively charged surfaces are ineffective in promoting the initiation of the growth cones. Moreover, a comprehensive comparison of the hippocampal neuron growth modality on different GO films showed the same conclusion with the above analysis.
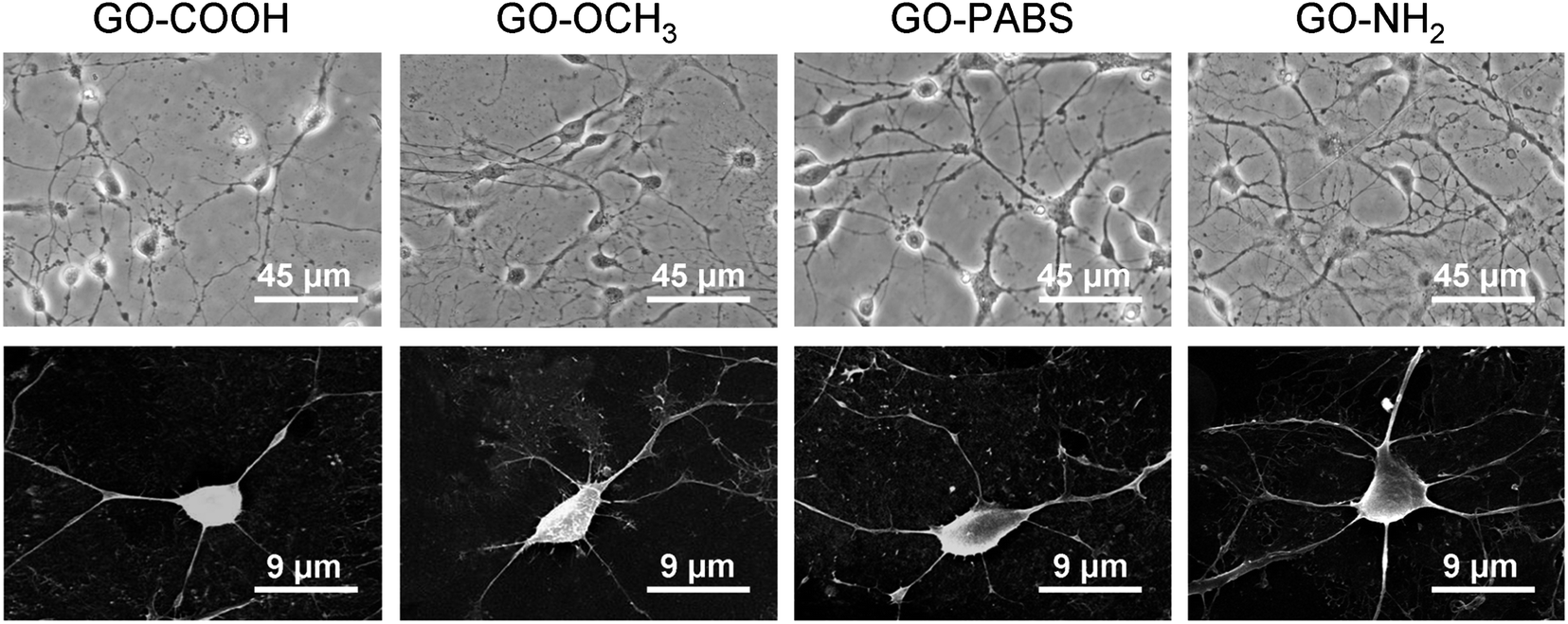 | ||
| Fig. 7 Observation of the neuron morphology. First row: optical images of hippocampal neurons after 7 days of culture on GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 (from left to right); second row: SEM images of single hippocampal neurons after 7 days of culture on GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 (from left to right). | ||
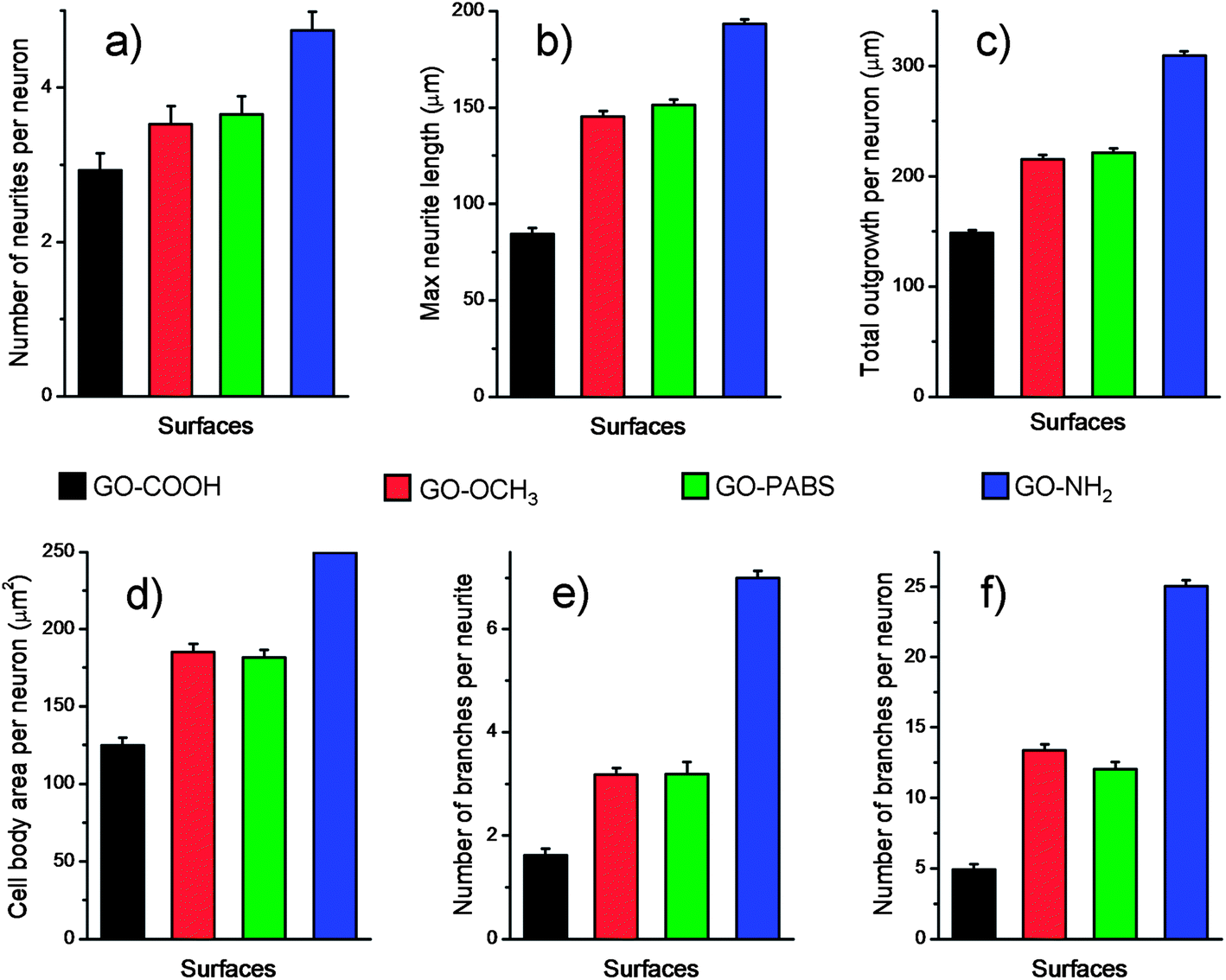 | ||
| Fig. 8 Parameters of hippocampal neuron growth after 7 days of culture on GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 (from left to right). (a) Number of neurites per neuron, (b) maximum neurite length, (c) total outgrowth per neuron, (d) cell body area per neuron, (e) number of branches per neurite, and (f) number of branches per neuron. For the growth characteristics, more than 150 cells were analyzed for each parameter under each experimental condition. | ||
Immunochemistry staining assay of the cultured neurons
Immunochemistry staining analysis of the cultured hippocampal neurons was performed by following the method46 reported previously using the neuron-specific cytoskeletal protein mouse anti-MAP2 antibodies, and rabbit anti-tau antibodies, as well as their corresponding fluoro-labeled secondary antibodies, namely, goat anti-rabbit IgG-FITC and goat anti-mouse IgG-RBITC. The cell nuclei were stained using the conventional Hoechst dye H33258 fluorochrome. The results (Fig. 9) show that most of the cells were hippocampal neurons. In addition, to better observe the morphology of hippocampal neurons, a set of typical fluorescent images (Fig. 10) of single hippocampal neurons were provided. Fig. 10 shows that the micrographs of hippocampal neurons exhibited the marked cell body and longer neurites that spread on the GO–OCH3, GO–PABS, or GO–NH2 films compared with neurons grown on the GO–COOH film. A comparison of the positively charged GO–NH2 with zwitterionic GO–PABS and neutral GO–OCH3 indicates that the positively charged GO–NH2 was more beneficial for neurite outgrowth and branching. The findings agree with the results obtained from optical phase-contrast image analysis. In addition, the immunochemistry analysis provided supporting evidence that the surface charge of the GOs can modulate neurite outgrowth and branching.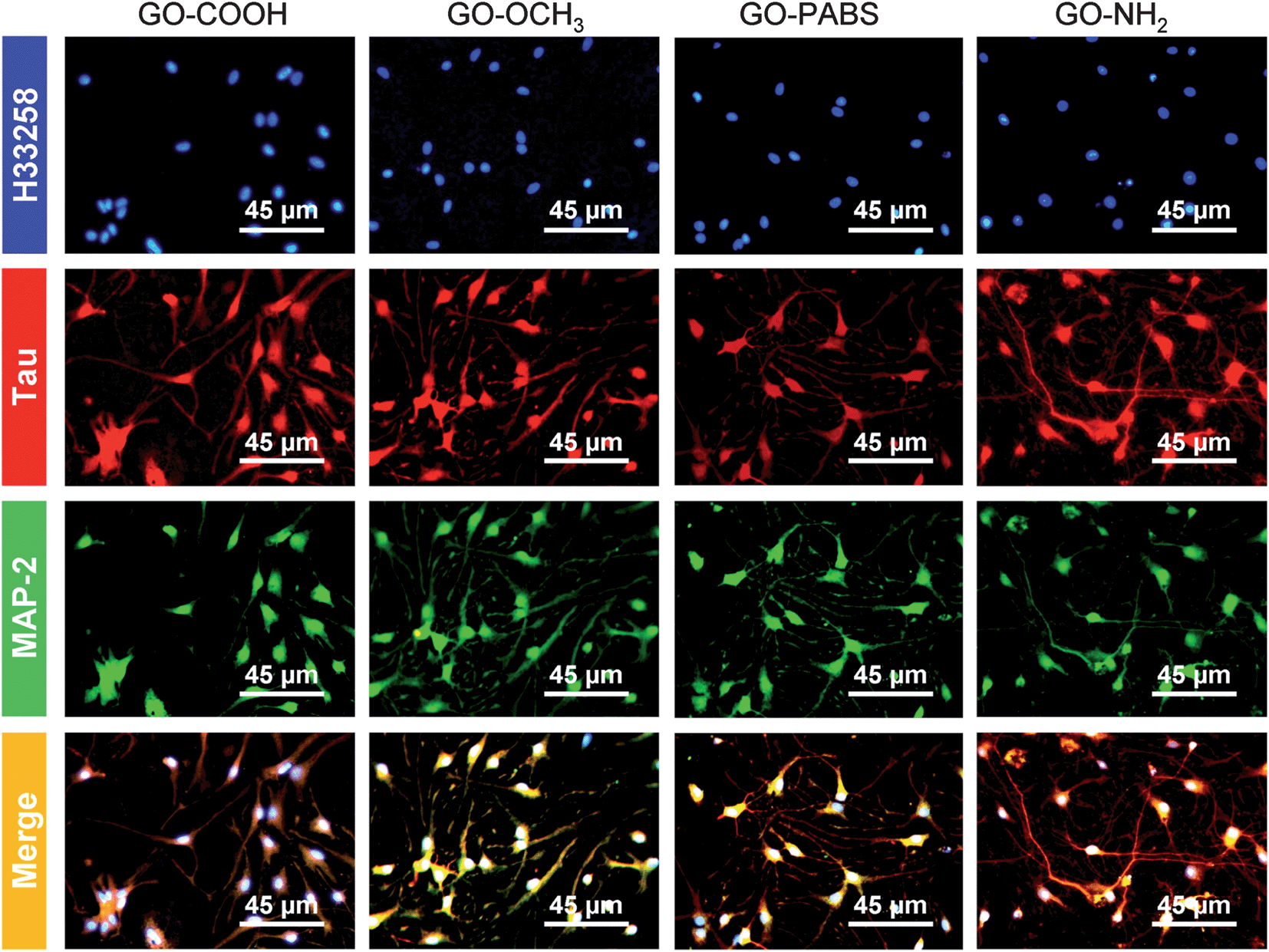 | ||
| Fig. 9 Immunochemistry staining images of hippocampal neurons after 7 days of culture on GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 (from left to right). | ||
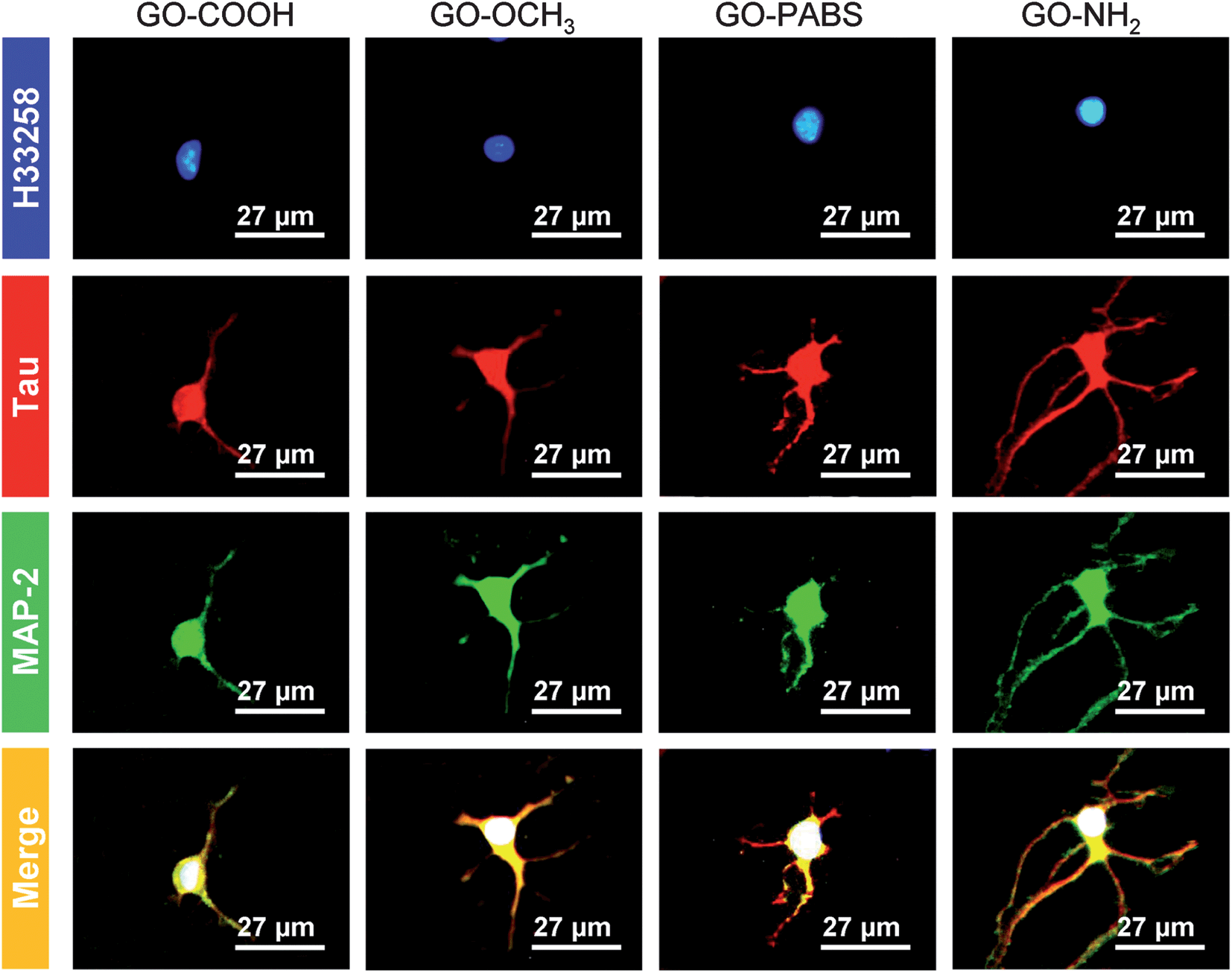 | ||
| Fig. 10 One set of typical immunochemistry staining images of single hippocampal neurons after 7 days of culture on GO–COOH, GO–OCH3, GO–PABS, and GO–NH2 (from left to right). | ||
Conclusions
GO was modified by changing its surface charge using amino- (–NH2), poly-m-aminobenzene sulfonic acid- (–NH2/–SO3H), or methoxyl- (–OCH3) terminated functionalities. Using the resulting functionalized GOs as substrates for the culture of primary rat hippocampal neurons, the effects of the surface charges of GOs on neurite outgrowth and branching were investigated. The results show that the surface charge of GOs can be used to control neurite outgrowth and branching. Compared with neutral, zwitterionic, or negatively charged GO, positively charged GO was more beneficial for neurite outgrowth and branching. We believe that the ability to chemically modify GO to control neurite outgrowth could be implemented in the clinical setting, especially in cases wherein long-term outgrowth modulation is necessary.Acknowledgements
This study was supported by the National Natural Science Foundation of China (21175107, 20975082, and 21375106), the Ministry of Education of the People's Republic of China (NCET-08-602 0464), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, the State Education Ministry, and the Northwest A&F University.Notes and references
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- L. Zhang, Z. Lu, Q. Zhao, J. Huang, H. Shen and Z. Zhang, Small, 2011, 7, 460–464 CrossRef CAS PubMed.
- O. Akhavan, E. Ghaderi and H. Emamy, J. Mater. Chem., 2012, 22, 20626–20633 RSC.
- K. Yang, J. Wan, S. Zhang, B. Tian, Y. Zhang and Z. Liu, Biomaterials, 2012, 33, 2206–2214 CrossRef CAS PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. Sanchez Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- K. P. Liu, J. J. Zhang, F. F. Cheng, T. T. Zheng, C. M. Wang and J. J. Zhu, J. Mater. Chem., 2011, 21, 12034–12040 RSC.
- O. Akhavan, E. Ghaderi and R. Rahighi, ACS Nano, 2012, 6, 2904–2916 CrossRef CAS PubMed.
- Y. Liu, D. Yu, C. Zeng, Z. Miao and L. Dai, Langmuir, 2010, 26, 6158–6160 CrossRef CAS PubMed.
- J. S. Lee, H. A. Joung, M. G. Kim and C. B. Park, ACS Nano, 2012, 6, 2978–2983 CrossRef CAS PubMed.
- S. Y. Lim, J. Ahn, J. S. Lee, M. G. Kim and C. B. Park, Small, 2012, 8, 1994–1999 CrossRef CAS PubMed.
- Y. Chen, Y. Li, D. Sun, D. Tian, J. Zhang and J. J. Zhu, J. Mater. Chem., 2011, 21, 7604–7611 RSC.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS PubMed.
- B. Zhang and T. Cui, Appl. Phys. Lett., 2011, 98, 073116 CrossRef.
- S. Y. Park, J. Park, S. H. Sim, M. G. Sung, K. S. Kim, B. H. Hong and S. Hong, Adv. Mater., 2011, 23, H263–H267 CrossRef CAS PubMed.
- Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS PubMed.
- N. Li, X. Zhang, Q. Song, R. Su, Q. Zhang, T. Kong, L. Liu, G. Jin, M. Tang and G. Cheng, Biomaterials, 2011, 32, 9374–9382 CrossRef CAS PubMed.
- B. S. Wilson, D. T. Lawson, J. M. Muller, R. S. Tyler and J. Kiefer, Annu. Rev. Biomed. Eng., 2003, 5, 207–249 CrossRef CAS PubMed.
- J. D. Weiland, W. T. Liu and M. S. Humayun, Annu. Rev. Biomed. Eng., 2005, 7, 361–401 CrossRef CAS PubMed.
- X. Navarro, T. B. Krueger, N. Lago, S. Micera, T. Stieglitz and P. A. J. Dario, Peripher. Nerv. Syst., 2005, 10, 229–258 CrossRef PubMed.
- N. A. Kotov, J. O. Winter, I. P. Clements, E. Jan, B. P. Timko, S. Campidelli, S. Pathak, A. Mazzatenta, C. M. Lieber, M. Prato, R. V. Bellamkonda, G. A. Silva, N. W. S. Kam, F. Patolsky and L. Ballerini, Adv. Mater., 2009, 21, 3970–4004 CrossRef CAS.
- K. Zhou, G. A. Thouas, C. C. Bernard, D. R. Nisbet, D. I. Finkelstein, D. Li and J. S. Forsythe, ACS Appl. Mater. Interfaces, 2012, 4, 4524–4531 CAS.
- H. Hu, Y. Ni, V. Montana, R. C. Haddon and V. Parpura, Nano Lett., 2004, 3, 507–511 CrossRef PubMed.
- B. C. Roy, M. D. Gupta, L. Bhowmik and J. K. Ray, Synth. Met., 1999, 100, 233–236 CrossRef CAS.
- B. Gulbakan, E. Yasun, M. Ibrahim Shukoor, Z. Zhu, M. You, X. Tan, H. Sanchez, D. H. Powell, H. Dai and W. Tan, J. Am. Chem. Soc., 2010, 132, 17408–17410 CrossRef CAS PubMed.
- U. T. Ruegg and F. Hefti, Neurosci. Lett., 1984, 49, 319–324 CrossRef CAS.
- P. C. Letourneau, Dev. Biol., 1975, 44, 92–102 CrossRef CAS.
- P. C. Letourneau, Dev. Biol., 1975, 44, 77–91 CrossRef CAS.
- K. J. Ivins, E. T. Bui and C. W. Cotman, Neurobiol. Dis., 1998, 5, 365–378 CrossRef CAS PubMed.
- A. M. Taylor, S. W. Rhee, C. H. Tu, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Langmuir, 2003, 19, 1551–1556 CrossRef CAS PubMed.
- O. Nicole, C. F. Ali, L. Docagne, E. T. Plawinski, D. V. MacKenzie and A. J. Buisson, J. Neurosci., 2001, 21, 3024–3033 CAS.
- L. C. Doering, Protocols for Neural Cell Culture, McMaster University Press, Canada, 2010, pp. 137–159 Search PubMed.
- G. P. H. Dietz, B. Dietz and M. Bähr, Brain Res., 2007, 1164, 136–141 CrossRef CAS PubMed.
- O. du Roure, A. Saez, A. Buguin, R. H. Austin, P. Chavrier, P. Silberzan and B. Ladoux, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2390–2395 CrossRef CAS PubMed.
- M. Buddle, E. Eberhardt, L. H. Ciminello, T. Levin, R. Wing, K. DiPasqualea and K. M. Raley-Susman, Brain Res., 2003, 978, 38–50 CrossRef CAS.
- C. Mailliot, T. Bussière, M. Caillet-Boudin, A. Delacourte and L. Buée, Neurosci. Lett., 1998, 255, 13–16 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- B. W. Reed and M. Sarikaya, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 195404 CrossRef.
- S. Attal, R. Thiruvengadathan and O. Regev, Anal. Chem., 2006, 78, 8098–8104 CrossRef CAS PubMed.
- C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska and L. Niu, Langmuir, 2009, 25, 12030–12033 CrossRef CAS PubMed.
- X. Zhang, L. Meng and Q. Lu, ACS Nano, 2009, 3, 3200–3206 CrossRef CAS PubMed.
- S. Zhang, P. Xiong, X. Yang and X. Wang, Nanoscale, 2011, 3, 2169–2174 RSC.
- L. Y. Yang, L. Li, Q. Tu, L. Ren, Y. R. Zhang, X. Q. Wang, Z. Zhang, W. Liu, L. Xin and J. Wang, Anal. Chem., 2010, 82, 6430–6439 CrossRef CAS PubMed.
- M. P. Mattson, R. C. Haddon and A. M. Rao, J. Mol. Neurosci., 2000, 14, 175–182 CrossRef CAS.
- J. H. Lustgarten, M. Proctor, R. I. Haroun, A. M. Avellino, A. A. Pindzola and M. Kliot, J. Biomech. Eng., 1991, 113, 184–188 CrossRef CAS.
- A. M. Taylor, M. Blurton-Jones, S. W. Rhee, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Nat. Methods, 2005, 2, 599–605 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: FT-IR and XPS spectra of the polymers NH2-PEG-NH2, CH3O-PEG-OH, and PABS. See DOI: 10.1039/c3an01796f |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |