Fluidity and water in nanoscale domains define coacervate hydrogels†
Julia H.
Ortony‡
ab,
Soo-Hyung
Choi§
b,
Jason M.
Spruell
b,
Jasmine N.
Hunt
ab,
Nathaniel A.
Lynd
b,
Daniel V.
Krogstad
bc,
Volker S.
Urban
e,
Craig J.
Hawker
abc,
Edward J.
Kramer
bcd and
Songi
Han
*abd
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: songi@chem.ucsb.edu
bMaterials Research Laboratory, University of California, Santa Barbara, California 93106, USA
cDepartment of Materials, University of California, Santa Barbara, California 93106, USA
dDepartment of Chemical Engineering, University of California, Santa Barbara, California 93106, USA
eCenter for Structural Molecular Biology and Chemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 10th October 2013
Abstract
Coacervate-based hydrogels, formed in aqueous solution by simple mixing of two oppositely charged ABA block copolyelectrolytes represent a new and versatile approach to the design of bio-inspired gelators. While coacervate-based hydrogels provide high tunability of a range of desirable properties, little is understood about the molecular-level makeup of the nanometer-scale domains. Small angle neutron scattering was employed to quantify the effective polymer density and water content of each domain. Further, electron paramagnetic resonance and Overhauser dynamic nuclear polarization of block-specific spin labels elucidate domain-specific, local, polymer and water dynamics. This unique combination of techniques reveals that the charged A blocks segregate into spherical domains with a radius of 8 nm, and are dispersed in a continuous matrix of water soluble, PEO B blocks. The edges of the spherical A block domains are found to be soft and diffuse, and the B block matrix exhibits higher water and polymer dynamics than the A block domains. The selective measurement of the local water and polymer dynamics shows a viscous and dense, but fluidic environment in the spherical A block domains, thus permitting the designation as a complex coacervate phase. Further, the physical properties of the analogous homopolymers mixed at equal composition to that of the triblock copolyelectrolytes leads to the conclusion that “the whole is greater than the sum of its parts”: nanometer scale complex coacervates only form when the two charged A blocks are covalently linked by a PEO midblock that serves as an intrinsic osmolyte.
Introduction
The molecular design of new hydrogelators has recently gained substantial attention due to their function in a wide range of biomedical and industrial technologies.1–3 For these applications, the presence of nanoscale domains or heterogeneities can provide enhanced function over traditional homogeneous bulk materials.4–7 For example, multi-domain hydrogels are currently being developed for cell scaffolding, tissue engineering, and as drug delivery systems.8–12 In these systems, the requirements of biocompatibility and facile processing in aqueous media are important. Another design objective involves the capability of the hydrogel to be switched upon receiving specific signals for their formation or disintegration. For these and other biologically inspired objectives it is critical for the material to be hydrated throughout the molecular network so that functional biomolecules can be hosted or environmental triggers (such as pH) reach the target site.13 With these goals in mind, we recently developed a stimuli-responsive hydrogel platform whose bio-inspired design is based on marine organisms and the self-assembly of oppositely charged ABA triblock copolymers in simply water.14 Here, hydrogelation is proposed to occur through domain partitioning of the charge-complementary copolyelectrolyte A blocks and the neutral B blocks, to result in hydrogels with nanometer scale features and enhanced mechanical properties.This new design motif, in particular the association of the A blocks to form a nanoscale phase, was inspired by the biological process of complex coacervation, by which oppositely charged polyelectrolytes, secreted by marine organisms, associate into liquid droplets suspended in water with high internal polyelectrolyte concentrations. These complexes are believed to be involved in the formation of adhesive plaques under water by sessile species, including the mussel and sandcastle worm.15,16 The molecular characteristics of freely suspended complex coacervates in bulk solutions have previously been elucidated by both fluorescence anisotropy17 and electron paramagnetic resonance (EPR) spectroscopy,18 revealing remarkably high polymer segment dynamics and highly fluidic properties. Furthermore, the recently established magnetic resonance relaxometry technique, Overhauser dynamic nuclear polarization (ODNP), clearly demonstrates that free floating complex coacervates are hydrated, while presenting considerable translational mobility of around 1/10 of the diffusivity of bulk water.39 Such diffusion represents a high degree of fluidity, comparable to the water loosely associated with (or bound to) protein surfaces exposed to bulk solvent, or in the molten globule interior of folded proteins.18 These molecular insights provide clues to the expected polymer and water dynamics in the A block domains within what are proposed to be nanoscale, coacervate-based hydrogels. Moreover, local diffusion dynamics of hydration water have been measured by ODNP of a wider variety of biomolecular and colloidal surfaces and interfaces that provide us with a scale to categorize the local hydration dynamics within the hydrogel domains (Fig. 2).19–22
Initial structural characterization of coacervate-based hydrogels shows the presence of spherical domains by small angle X-ray scattering (SAXS) with domain spacing reported on the order of 10 nm.14 Both, domain spacing and domain dimensions may be tuned by varying the block copolymer composition, molecular weight, and concentration23 with the ionic strength of the charged A blocks influencing the mechanical properties of the hydrogel.14 This structural model, A blocks forming dense coacervate domains and the B blocks forming a less dense continuous matrix, suggests that the domain-specific dynamics of both the polymer and the water play a critical role in defining the properties of these materials. However, directly measuring the dynamics of the solvent and polymer with nanometer length scale specificity (smaller than the 10 nm diameter of the spherical domains in these systems) presents an experimental challenge that requires the development of alternative strategies.
Here, a new combination of techniques of continuous wave (cw) EPR lineshape analysis, ODNP analysis and small angle neutron scattering (SANS) is introduced as a generally applicable approach for polymer domain-specific and dynamics-based characterization on the nanometer scale. The access to direct characterization is particularly important, as the properties of these contemporary hydrogels cannot be intuitively derived from that of traditional coacervates that tend to occupy volumes of roughly 0.5 μm3, which is in direct contrast to the A block domains in these nanoscale hydrogels that occupy roughly 5 nm.3 This dramatic difference in volume suggests the hydrogel-“trapped” A block domains, even if confirmed to be complex coacervates, may display crucially different physical and functional properties compared to suspensions of free floating coacervate droplets.
The specific experimental strategy was three-fold. Cw EPR lineshape analysis of block-specific nitroxide-based spin labels was employed to measure the motional restriction by polymer packing16via quantification of the spin labels' rotational diffusion rates from spectral simulations.24 Next, ODNP relaxometry of the same, block-specifically spin labeled polyelectrolytes was used to determine the local water diffusion coefficients, within molecular length scales of the spin labels attached to one of the polyelectrolyte blocks (here the A blocks), encompassing several layers of water (within 5–15 Å) around the spin labels. Finally, SANS measurements and analysis were employed to determine the domain-specific density of polymer and water.
Experimental section
Synthetic characterization methods
Gel permeation chromatography (GPC) was performed in chloroform (with 0.25% triethylamine) on a Waters 2695 Separation Module equipped with a Waters 2414 Refractive Index Detector and a Waters 2996 Photodiode Array Detector. The molecular weights of the polymers were calculated relative to linear polystyrene and poly(ethylene oxide) standards. 1H and 13C solution-state NMR were recorded on a Varian VNMRS 600 (600 MHz for 1H and 150 MHz for 13C) spectrometer. Chemical shifts are reported relative to residual solvent peaks (δ 7.26 for CDCl3 in 1H NMR and δ 77.2 for CDCl3 in 13C NMR).Materials
Triblock copolyelectrolytes were synthesized by oxyanionic ring-opening polymerization of allyl glycidyl ether from poly(ethylene oxide) macroinitiators to provide poly[(allyl glycidyl ether)-b-(ethylene oxide)-b-(allyl glycidyl ether)] (P(AGE-b-EO-b-AGE)) triblock copolymers. These triblock copolymers were then functionalized with ionic groups through thiol–ene chemistry, as previously described.14 Poly(ethylene oxide) (Sigma-Aldrich) was used as a macroinitiator after vacuum drying. Monomethyl poly(ethylene oxide) was used as received (Sigma-Aldrich). Allyl glycidyl ether (TCI America) was degassed by freeze–pump–thaw, and then purified with butyl magnesium chloride. The neutral precursor, P(AGE-b-EO-b-AGE), was prepared by initiation of PEO-diol with potassium naphthalenide (0.4 M in tetrahydrofuran) at 40 °C under dry argon, followed by addition of allyl glycidyl ether (AGE) and polymerization for 20 h. P(AGE-b-EO-b-AGE) was precipitated in hexane and characterized by size exclusion chromatography (SEC) and 1H NMR. The combination of SEC and NMR characterization showed the molecular weights of the blocks of the polymer precursor, P(AGE-b-EO-b-AGE), to be 5.9 kg mol−1 for each AGE block, and 35 kg mol−1 for the EO block. P(AGE-b-EO-b-AGE) was obtained with PDIs of 1.1.Ionic functionalization of P(AGE-b-EO-b-AGE) was carried out by thiol–ene “click” chemistry with functional groups (8 equiv. per alkene) dissolved in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH–water. The radical photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (0.05 equiv. per alkene), was added to the reaction mixture, which was then sparged for 30 min with argon. The degassed solution was irradiated at 365 nm with stirring for 3 h. The reaction mixture was then dialyzed in 6–8 kDa molecular weight cut-off (MWCO) cellulose membranes against ultra-high-purity water. The product was obtained by lyophilization as a white powder.
:1 MeOH–water. The radical photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (0.05 equiv. per alkene), was added to the reaction mixture, which was then sparged for 30 min with argon. The degassed solution was irradiated at 365 nm with stirring for 3 h. The reaction mixture was then dialyzed in 6–8 kDa molecular weight cut-off (MWCO) cellulose membranes against ultra-high-purity water. The product was obtained by lyophilization as a white powder.
Spin labelling
Cationic polyelectrolytes were spin labeled by coupling of the guanidinium functional groups to 4-carboxy-2,2,6,6-tetramethylpiperidinyloxy (4-carboxy-TEMPO) in the presence of excess 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) in water. The spin labeling reactions were highly efficient (>90%), as determined by EPR lineshape analysis of functionalized vs. free spin labels. Functionalization by 4-carboxy-TEMPO was controlled stoichiometrically and the TEMPO-based spin labels were randomly incorporated at 1 mol% by repeat unit.PEOSL preparation
To a solution of monomethyl PEO (180 mg, 550 g mol−1, 0.36 mmol), 4-carboxy-2,2,6,6-tetramethylpiperidinyloxy (4-carboxy-TEMPO, 110 mg, 0.55 mmol), and 4-dimethylaminopyridine (4 mg, 0.04 mmol) dissolved in dry dichloromethane (3.6 mL) was added to dicyclohexylcarbodiimide (113 mg, 0.55 mmol) under N2 atmosphere. After stirring for 24 h at room temperature the mixture was filtered, the solvent was removed in vacuo and the residue purified through flash chromatography (SiO2; eluent 1:1 ethyl acetate–hexane initially, then acetone to elute the product) to afford PEOSL, an orange oil (194 mg, 97% yield). PEOSL: 1H NMR (600 MHz, CDCl3, taken with an excess of pentafluorophenyl hydrazine (NMR silent) to reduce the TEMPO radical): δ 4.23 (t, 2H), 3.70–3.53 (m, 50H), 3.38 (s, 3H), 2.77–2.68 (m, 1H), 1.89–1.68 (m, 4H), 1.30–1.06 (m, 12H); GPC (0.25% NEt3 in CHCl3versus poly(ethylene oxide) standards): Mn = 1770 g mol−1, PDI = 1.06.
Sample preparation and turbidity
Polymer solutions were made at 20 wt% in HPLC-grade H2O and were allowed to dissolve overnight. Hydrogels were obtained upon mixing two oppositely charged triblock copolyelectrolyte solutions in a 1:1 volume ratio at room temperature. Gelation was observed within 15 seconds of mixing. Homopolymer complex coacervates were obtained upon mixing two oppositely charged homopolymers in solution at 20 wt%. It was observed that consistent mixing methods was important to minimize sample-to-sample variations, and that the hydrogels mature over several hours, requiring the comparison of samples with similar incubation times to ensure consistency. Thus, all samples studied here were equilibrated for 1 h before measurements were carried out. The homopolymer complexes were analyzed by light microscopy, as well as turbidity measurements (carried out with a Shimadzu UV-1800 UV/vis spectrometer at 600 nm).
Small-angle neutron scattering (SANS)
SANS was conducted with the CG-3 Bio-SANS instrument at the High Flux Isotope Reactor in the Oak Ridge National Laboratory with a neutron wavelength of λ = 6.09 Å.25 Sample to detector distances of 0.3 m and 14.5 m were used to cover the range of a wave vector, 0.008 Å−1 < q < 0.60 Å−1, defined as q = 4πλ−1sin(θ/2), where θ is the scattering angle. The resulting two-dimensional scattering patterns were corrected for detector sensitivity, sample transmission and empty cell scattering, and then azimuthally averaged and reduced to absolute intensity based on the aluminum standard. The coherent scattering intensity was obtained by subtraction of the solvent scattering signature.
Hydrogels prepared from 20 wt% aqueous polymer solutions were placed between two quartz plates with a path length of 0.5 mm, and exposed to the neutron beam at r.t. for a total exposure time of 30 min per sample. The neutron scattering length density (SLD) of the isotopic solvent mixture was modified by mixing water (SLDH2O = −5.6 × 109 cm−2) and D2O (SLDD2O = 6.4 × 1010 cm−2) so that the SLDbackground = fH2OSLDH2O + fD2OSLDD2O, where fH2O (fD2O) is the fraction of H2O (D2O) in the solvent mixture. Based on the mass density of 1.12 g mL−1 for PEG, SLDPEG was calculated as 6.4 × 109 cm−2. Since it is hypothesized that the core domains of the hydrogel are formed by two oppositely charged A blocks, the SLDcore for the denser core domain was experimentally measured to be 1.18 × 1010 cm−2. The data analysis that yielded these numbers will be further explained in the following section.
Electron paramagnetic resonance (EPR)
Cw EPR spectra were acquired with a Bruker EMX X-band spectrometer and a cylindrical (TE011) cavity. Samples were irradiated at 9.8 GHz with the magnetic field set at 3400 G to excite the center resonance of the nitroxide spin label, and sweep width set at 200 G. The field modulation amplitude was kept below 0.2 times the center peak linewidth to acquire authentic EPR lineshapes and amplitudes. N2 gas was streamed through the cavity at 14 L min−1 for temperature control and all spectra were acquired at 298 K.EPR lineshape analysis
Cw EPR spectra were fit using the microscopic order macroscopic disorder (MOMD) model implemented by Freed et al. in their non-linear least squares stochastic Liouville (NLSL) program.24 With the spherical component of the rotational diffusion tensor (rbar) obtained from each fit, the rotational diffusion rates, kr, were calculated as kr = 10rbar. Fits were performed with initial fitting parameters both above and below the final values to ensure convergence to global minima. Final NLSL fits all exhibited ≥99.5% correlation with the experimental EPR spectral lineshape.Overhauser dynamic nuclear polarization enhanced NMR (ODNP)
The local water diffusion coefficients within 5–15 Å range from nitroxide radical spin labels were measured by ODNP using the same sample and the same instrument as for X-band EPR experiments. The magnetic field and frequency for irradiation was set to the center resonance of the nitroxide EPR spectrum. Samples contained in a capillary of 0.03′′ I.D. and 0.06′′ O.D. were placed in a home-built U-shaped NMR coil (Cu wire, 28 AWG) tuned to 14.8 MHz. The NMR signal was acquired with a broadband channel of a Bruker Avance NMR spectrometer, as described in detail elsewhere.26,27 Nuclear T1 relaxation of the 1H of water, in the absence and presence of the nitroxide spin label functionalized polymers, were measured using a standard inversion recovery pulse sequence, to determine the leakage factor expressing the contribution to 1H relaxation not driven by dipolar coupling to the electron spins. The 1H NMR signal enhancement of water was recorded as a function of increasing microwave power that was varied using a home-built X-band microwave amplifier with a power output between 0.1 mW and 1.5 W. The acquired 1H NMR spectra were integrated over their absorption peak, and the absolute values plotted versus the input microwave power. From the extrapolated 1H NMR signal enhancement value at infinitely high microwave power and the leakage factor, the key ODNP parameter termed the coupling factor was determined, presenting the electron–1H cross relaxation efficiency. From the ODNP coupling factor, using the appropriate molecular dynamics model and expression for the spectral density function, the correlation time, τ, for translational diffusion dynamics and the diffusion coefficient (D) for the local water in the dipolar coupling vicinity of the spin label was computed.19,26,27Results and discussion
The synthetic versatility of employing the ABA motif as structural building blocks for the construction of well-defined hydrogel networks is further enhanced by the ability to have control over the number of ionic end groups coupled with the opportunity to introduce secondary functionality (spin labels) into these materials. A consequence of the anionic and cationic ABA triblock copolymers being derived from the same starting PAGE-b-PEO-b-PAGE triblock is that the ionic end blocks have the same degree of polymerization. This allows a family of materials to be easily prepared with the ABA triblock copolyelectrolytes being formulated as 20 wt% aqueous solutions. In this study, the charged anionic and cationic functional groups on the A blocks were chosen such that their pKa values differ sufficiently enough to find pH conditions under which hydrogelation occurs upon mixing. Here, guanidinium and sulfonate pendant groups exhibit a large ΔpKa of 15.1, where the mixture of these triblock copolyelectrolytes has previously been shown to result in hydrogelation.14 A different ABA triblock copolyelectrolyte pair with lower ΔpKa of 5.8 was found to form viscous solution complexes, not hydrogels, in which the copolyelectrolytes weakly associate, as discussed in the ESI.† The cationic copolyelectrolyte (ABA+) was spin labeled at 1% incorporation relative to its A block repeat units with 4-carboxy-TEMPO to yield ABASL+. Upon mixing aqueous solutions of ABASL+ and ABA−, hydrogelation occurs, while the spin label's location within the A block permits domain specific probing by magnetic resonance techniques. It should be noted that no changes in mechanical or physical properties could be observed on comparison of the spin-labeled derivatives with the parent, unlabeled materials indicating little if any perturbation due to spin label introduction at 1% incorporation (Fig. 3).The observed sensitivity of the bulk polymer dynamics and rheology to the acid/base strength of the A blocks suggests that intermolecular interactions of the oppositely charged A blocks play a key role in guiding the materials assembly process in solution. Therefore, the heterogeneous morphology originating from the segregation of A vs. B blocks as depicted in Fig. 1, is suggested to be representative of ABA+/ABA− hydrogels, but not of the weak copolyelectrolyte solution complexes, thus presenting clearly testable systems with contrasted polymer and water dynamics properties. However, while we can conclude that no structure is observed when delta pKa is low, we cannot differentiate whether this is due to different types of structure forming, such as truly amorphous materials, or whether similar structures are forming that are dynamically dissociating on timescales unobtainable by SAXS experiments.
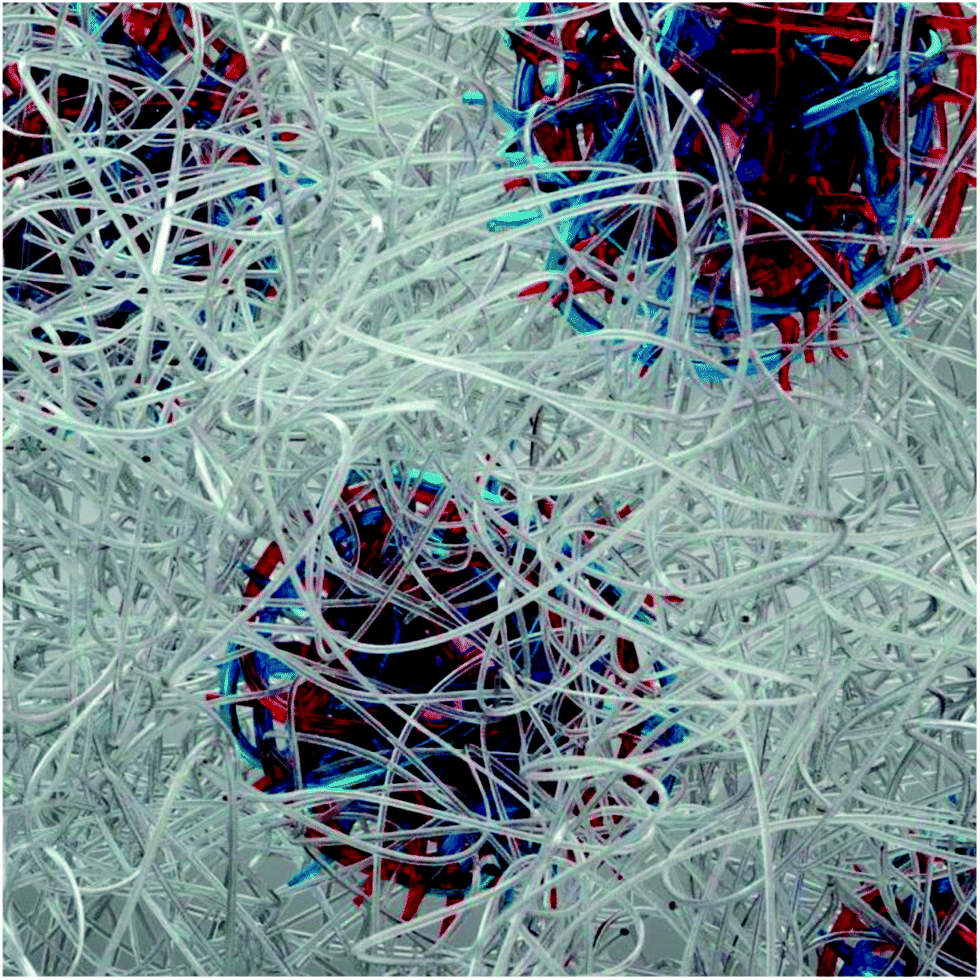 | ||
| Fig. 1 Hypothesized assembly motif of ABA hydrogels where spheres are composed of oppositely charged A blocks indicated by red and blue lines and are embedded in a matrix of bridging PEO B blocks indicated by silver lines. | ||
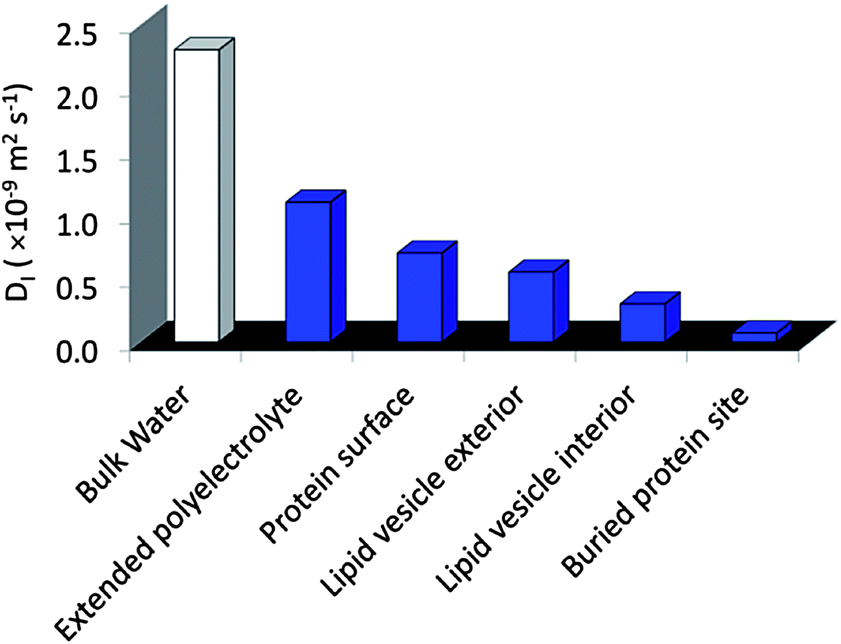 | ||
| Fig. 2 Typical water diffusion coefficients, D, measured by Overhauser dynamic nuclear polarization enhanced NMR spectroscopy (ODNP). | ||
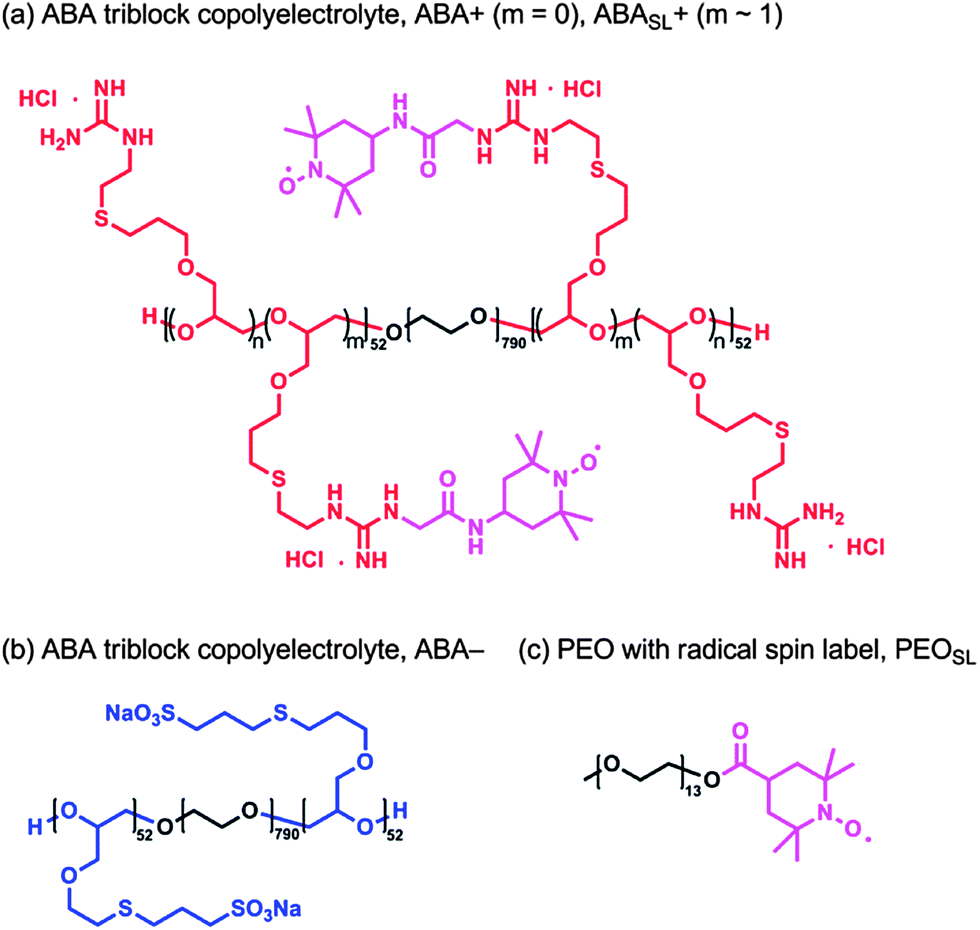 | ||
| Fig. 3 (a) Chemical structure of the cationic ABA triblock copolyelectrolyte, where m = 0 is denoted by ABA+ and the spin labeled analogue, where m ≈ 1, is denoted by ABASL+; (b) chemical structure of the anionic ABA triblock copolyelectrolyte, ABA−; (c) chemical structure of PEO functionalized with terminal TEMPO radical, PEOSL. The polyelectrolytes depicted exhibit an ABA motif, where the A blocks are comprised of 52 repeat units and the B blocks are comprised of 791 repeat units. | ||
Contrast-variation SANS experiments were carried out to quantify the hydrogel structure and morphology, including the domain-specific polymer density and water content, as described in more detail in the ESI† section. Fig. 4a displays the SANS profiles obtained from the ABA+/ABA− hydrogels in varying mixtures of H2O and D2O. The structure peak is evident at nearly the same position for all solutions, indicating that spherical endblock domains are present with the same inter-domain spatial correlation under all solvent conditions. Since the intensity of the structure peak is related as I ∼ (SLDcore − SLDbackground)2, SLDcore was estimated as 1.18 × 1010 cm−2 where the normalized intensity around the structure peak (0.018 Å−1 < q < 0.031 Å−1) disappears as shown in Fig. 4b. Here, we note that scattering contributions from the highly solvated PEO matrix, which should be insignificant compared to the contribution of scattering at the charged and denser core domains made of the A blocks, have been discounted.
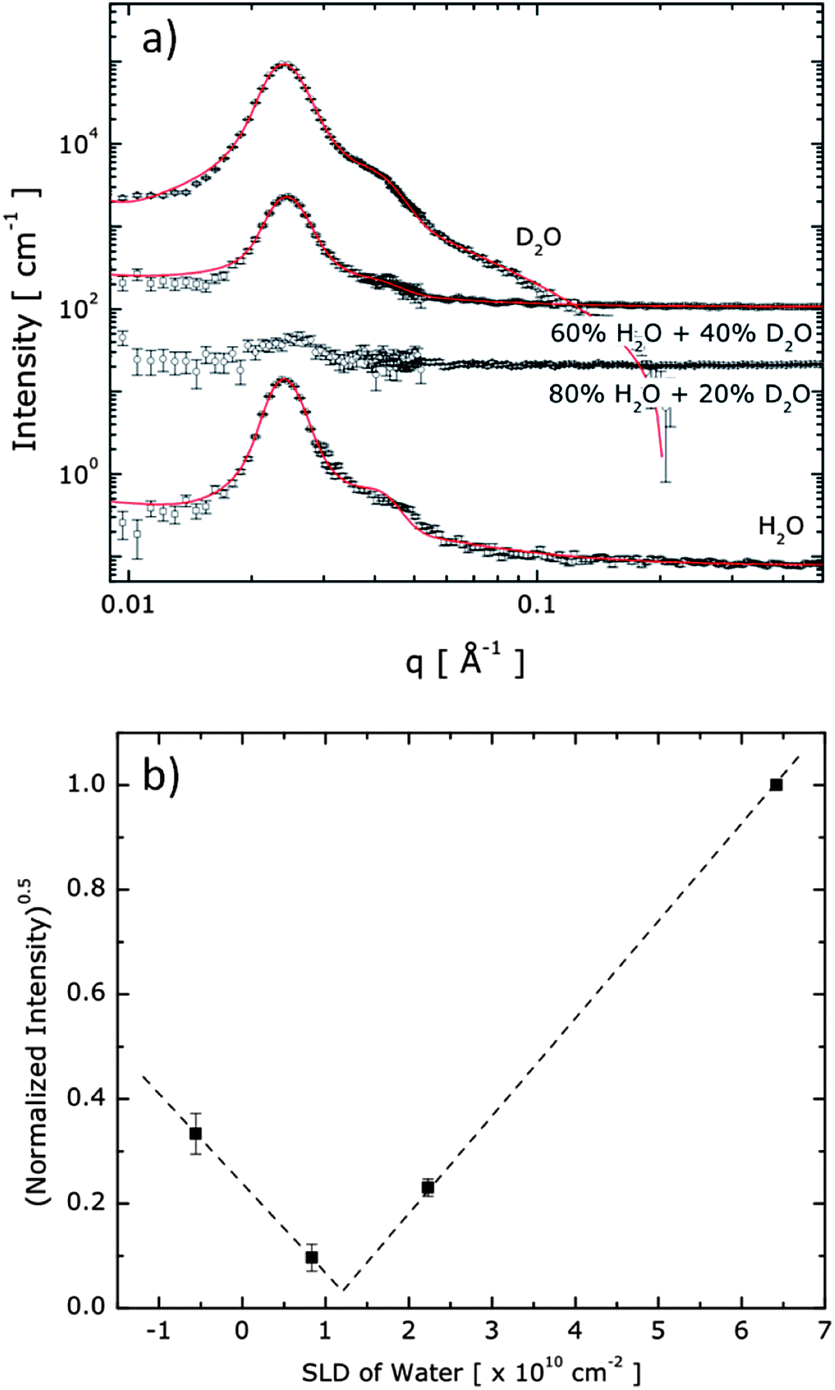 | ||
| Fig. 4 (a) SANS data obtained from 20 wt% hydrogels composed of ABA+ and ABA− in various mixing ratios of H2O and D2O. For clarity, data are shifted vertically (×102, ×5 × 102, and ×103, for 20% D2O, 40% D2O, and D2O solvent, respectively). The symbols are the SANS data and the solid lines are the model fits. Data are vertically offset for clarity. (b) Normalized intensities around the structure peak (0.018 Å−1 < q < 0.031 Å−1) were plotted upon SLD of background to extract SLDcore experimentally. | ||
The hydrogel system is modelled as spherical cores with polydisperse core radii and Gaussian chains attached to the core surface.28 The curves in Fig. 4a represent the best fits to the model (the mathematical form is described in the ESI†). We found an averaged number of 57 for the polymer chains in a core (Nagg), a core radius (Rcore) of 8 ± 1 nm, and an average distance between A block domains (2 × Rhs, where Rhs is the hard sphere radius) of 30 nm to provide the best fit to the data, as shown in Fig. 4a. To obtain good fits to the SANS scattering profiles, it was necessary that the model accommodates a soft, diffuse boundary between the core and the continuous phase. With this model, the interfacial thickness of the core domains was found to be 3 nm, which is roughly 38% of the core radius, confirming that the A block domains have soft boundaries of significant proportion.
The water content in the core domain was estimated by a detailed structural characterization method, which is an approach that has been widely and successfully applied,29–32 and is an alternative to the contrast matching experiment that has also been shown to precisely quantify water content.33 When the mass density of a hypothetical pure A block chain is assumed to be identical to that of a pure PEO midblock chain (i.e., 1.12 g mL−1), and all A block chains are assumed to be segregated into the spherical domains, then the water content in the core can be estimated by comparing the fraction of the coacervate domain in the solution (∼(Rcore/Rhs)3) and the fraction of A block in the solution. With this estimate the water content in the spherical domains and the hydrophilic matrix are found to be 47% and 86%, respectively, confirming that water is not homogeneously distributed through the hydrogel, in which case both domains would contain 80% water—a level of hydration as typically found for conventional hydrogels.34 The assumed model also allows us to estimate the polymer density in the coacervate and matrix domains to be significantly different, with 0.59 g mL−1 and 0.14 g mL−1, respectively. These observations suggest an interesting nanostructure with heterogeneity found at the nanometer scale, in which the spherical domains are significantly swollen by water, while these dense, yet fluidic, domains are covalently connected with the continuous PEO matrix that displays yet higher water content, at a level expected for cross-linked PEO hydrogels.
In addition to the definite presence of internal water, the designation of the spherical A domains as a coacervate hinges on the observation of clearly measurable polymer chain and solvent, here water, dynamics, characteristic of an unequivocal liquid, not a solid phase.35–37 This is in direct contrast to the restricted chain dynamics as typically found for the more common, ionic, precipitates. The fluidity within these coacervate-inspired hydrogels was quantified by EPR and ODNP using domain-selective nitroxide radical spin-labels, as will be discussed in the following.
EPR lineshape analysis was used to measure the rotational diffusion rates of the spin labeled polymer segments (kr), while ODNP was used to measure the local water translational diffusion coefficient (D) around the spin label. The cationic polymer solution of ABASL+, without addition of the anionic counterparts (i.e.ABA−), was initially probed. By fitting the EPR lineshape, as shown in Fig. 5a, molecular motion at the spin labeled A block was extracted and found to exhibit a rotational diffusion rate of kr = 0.52 GHz (Table 1). This rate is typical for spin labels covalently bound to unconfined polyelectrolyte chains that are freely dispersed in solution.27 Upon formation of a hydrogel by mixing solutions of ABASL+ and ABA−, the rotational diffusion rate of the cationic A blocks significantly decreases, while a two-component EPR spectral feature becomes apparent (Fig. 5b). Here, 27% of the spin labels exist in a somewhat more hindered environment compared to the freely dissolved polymer with kr = 0.34 GHz, while 73% of the spin labels exhibit a much more hindered motion with kr = 0.03 GHz. These highly restricted dynamics likely result from the dense polymer packing driven by electrostatic attraction (and close proximity) of the oppositely charged A block segments. To provide a point of reference, in the non hydrogel-forming weak triblock copolyelectrolyte solution complexes, the polymer dynamics slowed from 0.81 GHz to only 0.45 GHz, while the EPR spectrum maintained its single component character (see ESI, Fig. S2†). Another point of reference is that the slow-component of the rotational diffusion rate observed in the A block domain of the ABA+/ABA− hydrogels is still two orders of magnitude greater than that found in dry polymers, glasses, and frozen polymer solutions, indicating that the A block domains experience motional freedom, unlike in a solid-like phase.
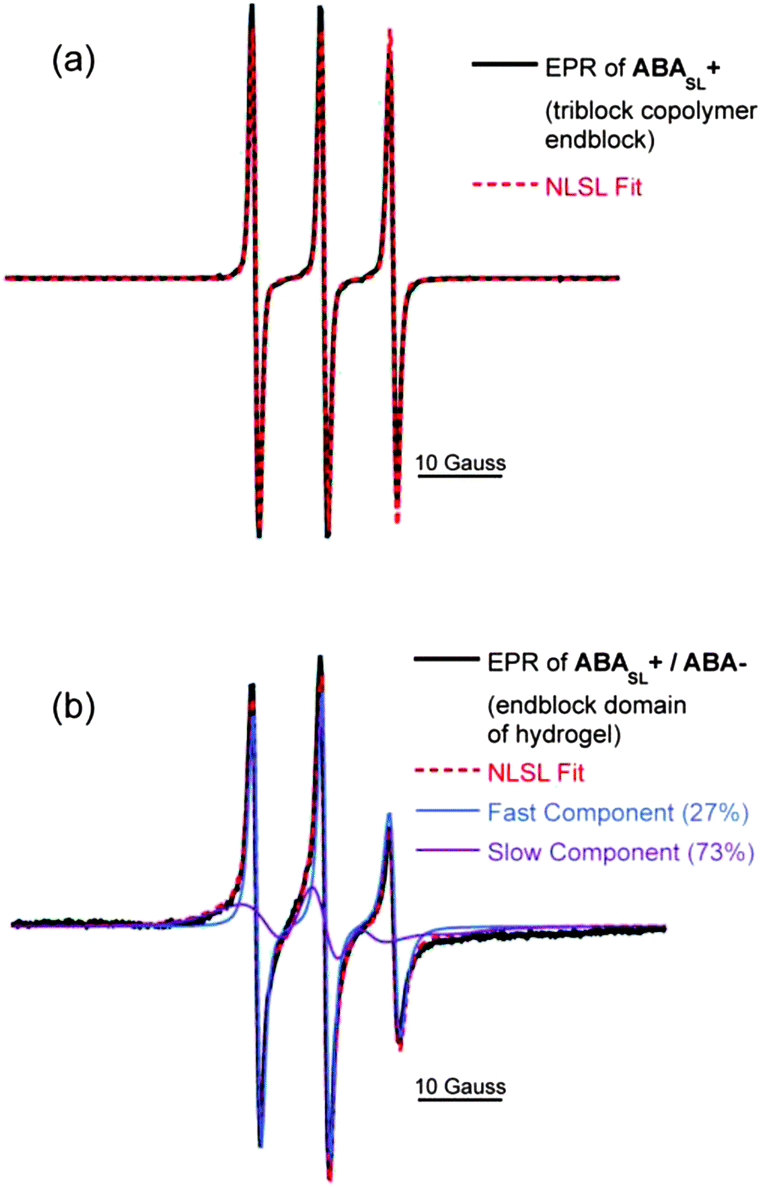 | ||
| Fig. 5 EPR spectra (black lines) of (a) triblock copolyelectrolyte solution (ABASL+), and (b) the hydrogel composed of oppositely charged triblock copolyelectrolytes (ABASL+/ABA−). Dotted red lines indicate NLSL fits. Blue and purple dotted lines indicate each component of a two component fit. | ||
| ABASL+ (solution) | ABASL+/ABA− (hydrogel) | |
|---|---|---|
| Rotational diffusion rate, kr (GHz) | 0.53 | 0.03 (73%) |
| 0.34 (27%) | ||
| Water diffusion coefficient, D (×10−9 m2 s−1) | 0.71 | 0.24 |
Mirroring the trend observed with the rotational diffusion rate, Table 1 also presents the local water diffusion coefficients as determined by ODNP. Around the spin labels of ABASL+ in solution, the polymer surface water diffusion coefficient is found to be high with D = 0.71 × 10−9 m2 s−1. Values of this order of magnitude are typical for spin labels covalently attached to polyelectrolytes that are fully exposed to bulk water. When ABASL+ is gelled by mixing with ABA−, the water diffusion coefficient is reduced by a factor of ∼3, indicating that water becomes more viscous in the coacervate domains of the hydrogel but is still clearly mobile. This finding is particularly surprising and insightful, given that the volume of the core domains is largely (47%) composed of water, eliminating the possibility that the retardation of water dynamics is merely due to spatial confinement caused by collapsed polymer chains. Given that the A block domains of the hydrogels contain a substantial amount of water (47%), fluid-like polymer dynamics, and mobile water, the A block domains satisfy the requirements to be designated as coacervate domains. Here, the significantly reduced water dynamics found in the ABA+/ABA− hydrogels compared to ABA+ alone in solution or even compared to the larger free homopolymer coacervates may represent a key physical property of the dense, yet fluid, spherical domains, exerting surprisingly high mechanical stability to these hydrogels without involving covalent cross-linking.
The midblock rich domains of the triblock complexes were also selectively probed by the incorporation of spin labeled PEO (PEOSL) at low concentrations (1 wt%). By quantitative EPR lineshape analysis of aqueous solutions of ABA+ with addition of PEOSL, a high rotational diffusion rate of 0.80 GHz is found for PEOSL, as presented in Table 2. Upon gelation by mixing ABA+ with ABA−, the rotational diffusion rate of PEOSL remains unchanged, indicating that PEOSL resides in the more dilute B block matrix where the local polymer rotational dynamics are unchanged from that of the local B block in ABA+ solution. Thus, the PEO polymer dynamics are decoupled from that of the spherical coacervate domains, confirming that distinctly differential biphasic behavior is observed at the nanometer length scale. This morphology defines the internal structure of the gel and the associated physical properties.
| ABA+/PEOSL (solution) | ABA+/ABA−/PEOSL (hydrogel) | |
|---|---|---|
| Rotational diffusion rate kr (GHz) | 0.80 | 0.80 |
| Water diffusion coefficient D (×10−9 m2 s−1) | 0.95 | 0.70 |
In contrast to the polymer chain dynamics, the local water diffusion coefficient values around PEOSL upon formation of the hydrogels (ABA+ to ABA+/ABA−) reduce measurably by 26%. This observed slowing of water in the PEO-rich domain signifies the expected behavior of B blocks approaching each other more closely, concurrent to the sequestering of the charged A blocks. We can conclude that the continuous PEO matrix experiences a more subtle collapse that does not alter the rotational diffusion of the spin label itself attached to the polymer chain, but clearly affects the local water dynamics further away from the spin label (at up to 15 Å, or 3–4 water layers away from the spin label).38
Having elucidated the hydration and polymer dynamics of or around the ABA chains in solution and coacervate hydrogel domains, an important question remains as to what is the role of the triblock geometry, distinct from the overall chemical composition. Homopolymers analogous to the A blocks alone, without the central PEO B block, were synthesized, as shown in Fig. 6 and the corresponding anionic and cationic homopolymers, A+ and A−, mixed to form freely suspended coacervate droplets, as verified by light microscopy and turbidity measurements (Fig. 6c). As expected, maximum coacervation is observed upon charge matching at a 1:1 mole ratio of A+ to A−.
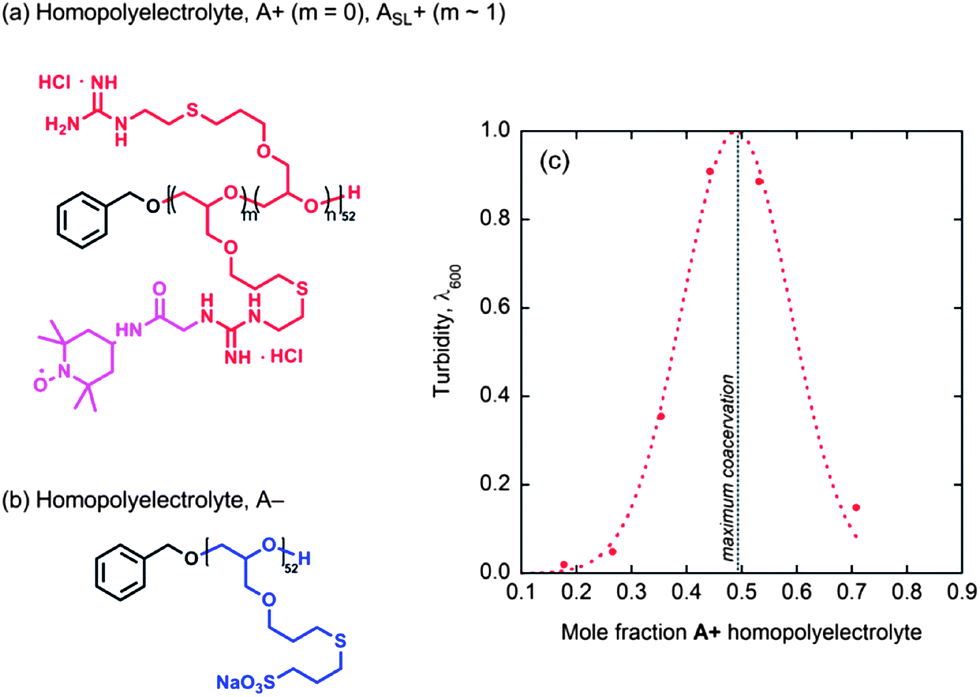 | ||
| Fig. 6 (a) Chemical structure of the cationic homopolyelectrolyte, where m = 0 is denoted by A+ and the spin labeled analogue, where m ≈ 1, is denoted by ASL+; (b) chemical structure of the anionic homopolyelectrolyte, A−; (c) turbidity of homopolymer coacervate suspensions made from homopolymers A+/A−, shown as a function of mole fraction A+. | ||
The polymer segment rotational diffusion rates (kr) and water diffusion coefficients (D) of and around these homopolymers and their complexes are presented in Table 3. The homopolymer ASL+ shows exceptionally fast polymer segment dynamics (and thus low packing density) with kr = 1.39 GHz (nearly three times faster than kr = 0.53 GHz of the ABASL+ triblock solution, Table 1). Interestingly, the water diffusion coefficient around the spin label of the homopolymer, with D = 0.88 × 10−9 m2 s−1, is similarly high as compared to that around the ABASL+ triblock where D = 0.71 × 10−9 m2 s−1. When homopolymers ASL+ and A− are mixed under maximum coacervation conditions, with or without the addition of PEO homopolymer at the same proportion as present in the triblock polyelectrolytes, the local polyelectrolyte density increases, and therefore the polymer rotational diffusion rate is expected to decrease. However, Table 3 shows the opposite trend. Upon coacervate formation, the spin label mobility of ASL+/A− as measured by EPR increases by more than two-fold, to kr = 3.58 GHz in the absence of PEO. Similarly, a high value of kr = 3.37 GHz is observed within the ASL+/A− coacervates, in the presence of PEO. The local water diffusion coefficient also increases, corroborating the unexpected behavior for the dramatic increase in local polymer dynamics in ASL+/A− coacervates, as compared to ASL+ in solution.
| ASL+ (solution) | ASL+/A− (coac.) | ASL+/A−/PEO (coac. + PEO) | |
|---|---|---|---|
| Rotational diffusion rate kr (GHz) | 1.39 | 3.58 | 3.37 |
| Water diffusion coefficient D (×10−9 m2 s−1) | 0.88 | 1.34* | 0.85 |
Upon closer inspection, we find a clear time-dependent behavior for the water diffusion coefficient within the homopolymer coacervates (ASL+/A−) (no added PEO homopolymer), where the value reported in Table 3 is the quasi-equilibrium value that establishes ∼2 h after mixing. The water diffusion coefficient within the homopolymer coacervates (ASL+/A−) dramatically increases within the first hour of mixing (Fig. 7a), while the plateau value of 1.34 × 10−9 m2 s−1, established after 2 h of mixing, corresponds to very high dynamics that is only two-fold retarded compared to the high bulk water diffusivity. Such high water dynamics have only been observed by ODNP on very “smooth” or hydrophobic polymer or protein surfaces.27 The systematic measurement of the time-dependent behavior provides confidence that the unexpected observation of increased hydration dynamics upon complexation is not an experimental artifact. The plateau value of 1.34 × 10−9 m2 s−1 represents a 50% increase in local water diffusivity within coacervate complexes compared to that of the surface of dissolved homopolymers in solution. It should be noted that when poly(ethylene oxide) is added to the ASL+/A− homopolymer coacervates, such that the overall chemical composition is the same as that of the triblock copolymer hydrogel ABASL+/ABA−, the polymer segment rotational diffusion rate decreases only slightly from 3.58 GHz to 3.37 GHz while the water diffusion coefficient decreases dramatically from 1.34 × 10−9 m2 s−1 (plateau value) to 0.85 × 10−9 m2 s−1, which corresponds to a typical value observed on the surfaces of homopolymer solutions. Thus, the extremely high hydration dynamics in the coacervate regions, without the addition of PEO, is suggested to be due to high osmotic pressure differential. This results in an increased volume of water in the coacervates and therefore increased water dynamics, which may approach that of the bulk water diffusion coefficient of D = 2.4 × 10−9 m2 s−1. The addition of free PEO balances the osmotic pressure differential and thus reduces the volume of water within the coacervate domains, since PEO will not permeate into the coacervate phase but resides in the dilute solution phase. The coacervate formation appears to be a much faster process than its equilibration of osmotic pressure.
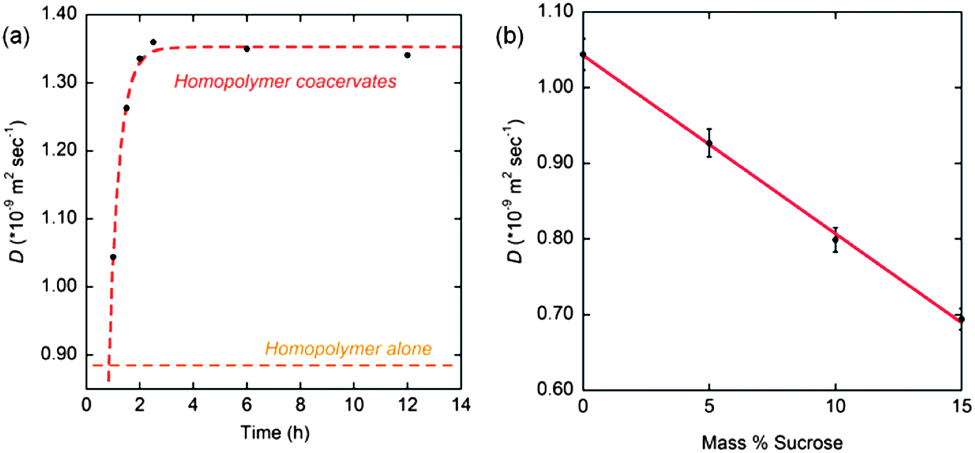 | ||
| Fig. 7 (a) Time course of water diffusion coefficients of ASL+ homopolymer alone and ASL+/A− complex coacervates. Water diffusion coefficient in the complex coacervates increases over 3 h, at which point it equilibrates at D ≈ 1.34 × 10−9 m2 s−1; (b) effect on water diffusion coefficient upon addition of an osmolyte (sucrose) to homopolyelectrolyte complex coacervates suspension (ASL+/A−). | ||
To further test the effect of osmotic pressure and gain clarity on this unexpected behavior, coacervates derived from homopolymers were titrated with concentrated sucrose, a well-known osmolyte, and the surface water diffusion coefficients around the homopolymer chains were measured by ODNP. Fig. 7b shows the water diffusion coefficients within homopolymer coacervates as a function of percent sucrose added. The initial water diffusion coefficient was measured within an hour of preparation of the coacervates, and from this sample, aliquots were removed and small volumes of sucrose added in increasing concentrations to each aliquot. The water diffusion coefficients showed an inverse linear relationship to mass percent sucrose added, implying that sucrose again reduces the osmotic pressure differential by depressing the concentration gradient across the coacervate interface to the dilute solution phase. The depression of the water diffusion within these bulk coacervates, to a value of 0.69 × 10−9 m2 s−1, is achieved when the osmotic pressure is balanced by the addition of the small molecule osmolyte sucrose, and roughly agrees with the 0.85 × 10−9 m2 s−1 value achieved with the addition of an entirely different macromolecular osmolyte, PEO. While the water diffusion coefficients in free floating complex coacervates are depressed by addition of an external osmolyte, this depression is still less than that observed in the A blocks of ABA triblocks upon gelation. This effect can be explained by the different effective PEO concentrations in the volume around the nm-scale coacervate domains in the triblock hydrogels versus in the dilute PEO solution surrounding the μm-scale complex coacervates. The nm-scale coacervate domains in triblock hydrogels are surrounded by an environment displaying a much higher effective concentration of the PEO blocks due to the molecular architecture of the triblock chains binding them closer to the coacervate interface. In contrast, the distribution of freely diffusing osmolytes surrounding the large μm-scale coacervates is more dilute and uniform throughout the solution. Thus, the propensity for water in the nm-scale coacervates to diffuse towards the higher concentration PEO domain is greater than in the case of free-floating coacervates surrounded by osmolytes free in solution.
Conclusions
Herein we have shown that coacervate-derived hydrogels, formed by mixing solutions of cationic and anionic triblock copolyelectrolytes, yield hydrogels with well-defined nanoscale domains. Experimental proof for fluidity and high polymer density within the spherical domains are consistent with coacervate formation.Molecular level characterization, combining SANS, EPR lineshape and ODNP analyses with domain-specific resolution, allowed insight into the morphology and dynamics of these novel hydrogels. Phase separation of the ionic A blocks leads to dense and spherical coacervate domains of 8 nm in radius that are further surrounded by a diffuse (∼3 nm), soft boundary. The uncharged, central B blocks compose the much more dynamic and dilute, continuous PEO matrix. The spherical A domains exhibit fluidic polymer and water dynamics, distinctly different from solid-like precipitates dispersed in solutions. Contrast variation SANS analysis showed that the coacervate domains show significant polymer density (0.59 g mL−1), yet retain high water concentration (47% water by volume). Furthermore, EPR spectral simulations and ODNP-enhanced NMR analysis of hydration water, employing strategically positioned spin labels, show that upon formation of mechanically robust hydrogels, their A block segment and hydration dynamics both decrease, while still remaining unequivocally fluidic. In contrast, the B block region maintains high water diffusion and displays nearly unchanged polymer segment mobility, regardless of the state of the polyelectrolytes (solution, solution complex, or gel).
While homopolymer coacervates and triblock copolymer core domains are presumed to have the same chemical composition, the internal dynamics measured in these systems was found to be significantly different. This can be understood by examining the dimension of each and also the native environment in which each exists. Homopolymer coacervates are 1 μm in diameter and are suspended in water, whereas triblock hydrogel core domains are 8 nm in diameter and are suspended in a PEO matrix, in which the A block movement, translation and partitioning is restricted by its anchoring to the PEO B blocks. It is apparent from the reduced dynamics in the hydrogel coacervate domains that PEO behaves as an intrinsic osmolyte by imparting a high effective polymer concentration surrounding the coacervate interface, and therefore leading to the transport of mobile, bulk-like, water from the core domains outwards, causing a partial dehydration. Interestingly, the water diffusion coefficient within the homopolymer coacervates, after balancing the osmotic pressure, is still higher than that observed in the hydrogel core domains. While the molecular origin of this effect may be complex, the residual water in the denser, 8 nm sized, coacervate domains has less opportunity to avoid the close interaction with and distortion by the polyelectrolyte chains compared to within μm-sized suspended homopolymer coacervates, possibly leading to further arrest of water dynamics. This is interesting, given that the polyelectrolyte interaction is thought to be comparable in the homopolymer vs. the triblock coacervate domains, when measured via the spin label dynamics reported by EPR. We hypothesize that the ABA chemical structure could be varied further to tune the osmolytic effect of the B block domain or the charge density of the A block domain, presenting a new tunable parameter for the rational design and understanding of coacervate hydrogels.
Having established that the ABA hydrogels are composed of fluidic, hydrated coacervate core domains and continuous PEO domains, we can hypothesize that a variety of polar and non-polar molecular cargos can be stored in the coacervate core domains, and can readily receive environmental cues (changes in pH, temperature and concentration gradients, etc.). The stable water structure in the coacervate domain will likely contribute to the overall mechanical stability of the hydrogel material, as well as allow for the stable storage and carriage of molecular cargo, while ensuring full hydration, which should be particularly critical for protein-based cargo. Finally, we present a novel characterization toolbox, by combining cw EPR lineshape analysis of polymer segment dynamics, ODNP analysis of water dynamics and contrast-variation SANS analysis as a generally applicable approach for domain-specific, dynamics-based characterization of nanoscale materials.
Abbreviations
| NMR | Nuclear magnetic resonance |
| ODNP | Overhauser dynamic nuclear polarization |
| EPR | Electron paramagnetic resonance |
| SANS | Small angle neutron scattering |
Acknowledgements
This work was supported by the MRSEC Program of the National Science Foundation (NSF) under Award DMR-1121053 (JHO, SHC, JMS, JNH, NAL, CJH, EJK and SH). We also acknowledge fellowship support from the NSF (JHO, JNH), Packard Foundation (SH) and the California Nanosystems Institute, Elings Fellowship to JMS. Facilities support from the NSF-funded Materials Research Facilities Network (http://www.mrfn.org). We thank Jerry Hu for his help with ODNP instrumentation, and the Oak Ridge National Laboratory is gratefully acknowledged.References
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- F. A. Leibfarth, M. Kang, M. Ham, J. Kim, L. M. Campos, N. Gupta, B. Moon and C. J. Hawker, Nat. Chem., 2010, 2, 207–212 CrossRef CAS PubMed.
- R. Shrestha, M. Elsabahy, H. Luehmann, S. Samarajeewa, S. Florez-Malaver, N. S. Lee, M. J. Welch, Y. Liu and K. L. Wooley, J. Am. Chem. Soc., 2012, 134, 17362–17365 CrossRef CAS PubMed.
- T. O. McDonald, P. Martin, J. P. Patterson, D. Smith, M. Giardiello, M. Marcello, V. See, R. K. O'Reilly, A. Owen and S. Rannard, Adv. Funct. Mater., 2012, 22, 2469–2478 CrossRef CAS.
- M. C. Chiappelli and R. C. Hayward, Adv. Mater., 2012, 24, 6100–6104 CrossRef CAS PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- J. B. Matson, R. Helen Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed.
- A. A. Aimetti, A. J. Machen and K. S. Anseth, Biomaterials, 2009, 30, 6048–6054 CrossRef CAS PubMed.
- P. C. Nicolson and J. Vogt, Biomaterials, 2001, 22, 3273–3283 CrossRef CAS.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de La Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- B. P. Lee, P. Messersmith, J. Israelachvili and J. Waite, Annu. Rev. Mater. Res., 2011, 41, 99 CrossRef CAS PubMed.
- D. S. Hwang, H. Zeng, A. Srivastava, D. V. Krogstad, M. Tirrell, J. N. Israelachvili and J. H. Waite, Soft Matter, 2010, 6, 3232–3236 RSC.
- A. Srivastava, J. H. Waite, G. D. Stucky and A. Mikhailovsky, Macromolecules, 2009, 42, 2168–2176 CrossRef CAS PubMed.
- R. Kausik, A. Srivastava, P. A. Korevaar, G. Stucky, J. H. Waite and S. Han, Macromolecules, 2009, 42, 7404–7412 CrossRef CAS PubMed.
- E. R. McCarney, B. D. Armstrong, R. Kausik and S. Han, Langmuir, 2008, 24, 10062–10072 CrossRef CAS PubMed.
- A. Pavlova, E. R. McCarney, D. W. Peterson, F. W. Dahlquist, J. Lew and S. Han, Phys. Chem. Chem. Phys., 2009, 11, 6833 RSC.
- S. Hussain, J. M. Franck and S. Han, Angew. Chem., Int. Ed., 2013, 52, 1953–1958 CrossRef CAS PubMed.
- B. D. Armstrong, P. Soto, J. E. Shea and S. Han, J. Magn. Reson., 2009, 200, 137 CrossRef CAS PubMed.
- D. V. Krogstad, N. A. Lynd, S.-H. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2013, 46, 1512–1518 CrossRef CAS.
- D. E. Budil, S. Lee, S. Saxena and J. H. Freed, J. Magn. Reson., Ser. A, 1996, 120, 155 CrossRef CAS.
- G. Lynn, W. Heller, V. Urban, G. Wignall, K. Weiss and D. A. A. Myles, Phys. B, 2006, 385, 880–882 CrossRef PubMed.
- B. D. Armstrong and S. Han, J. Chem. Phys., 2007, 127, 104508 CrossRef PubMed.
- J. H. Ortony, C. Y. Cheng, J. M. Franck, R. Kausik, A. Pavlova, J. Hunt and S. Han, New J. Phys., 2011, 13, 015006 CrossRef.
- S.-H. Choi, F. S. Bates and T. P. Lodge, J. Phys. Chem. B, 2009, 113, 13840–13848 CrossRef CAS PubMed.
- J. Bang, K. Viswanathan, T. P. Lodge, M. J. Park and K. Char, J. Chem. Phys., 2004, 121, 11489 CrossRef CAS PubMed.
- T. P. Lodge, J. Bang, M. J. Park and K. Char, Phys. Rev. Lett., 2004, 92, 145501 CrossRef.
- S.-H. Choi, F. S. Bates and T. P. Lodge, J. Phys. Chem. B, 2009, 113, 13840–13848 CrossRef CAS PubMed.
- R. Lund, L. Willner, P. Lindner and D. Richter, Macromolecules, 2009, 42, 2686–2695 CrossRef CAS.
- I. Goldmints, G.-e. Yu, C. Booth, K. A. Smith and T. A. Hatton, Langmuir, 1999, 15, 1651–1656 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef PubMed.
- J. T. G. Overbeek and M. Voorn, J. Cell. Comp. Physiol., 1957, 49, 7–26 CrossRef CAS.
- F. Weinbreck, R. De Vries, P. Schrooyen and C. De Kruif, Biomacromolecules, 2003, 4, 293–303 CrossRef CAS PubMed.
- C. G. de Kruif, F. Weinbreck and R. de Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340–349 CrossRef CAS PubMed.
- C.-Y. Cheng, J.-Y. Wang, R. Kausik, K. Y. C. Lee and S. Han, Biomacromolecules, 2012, 13, 2624–2633 CrossRef CAS PubMed.
- J. H. Ortony and D. S. Hwant, Biomacromolecules, 2013, 14, 1395–1402 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sc52368c |
| ‡ Present address: Institute for BioNanotechnology in Medicine, Northwestern University, Chicago, IL 60611, USA. |
| § Present address: Department of Chemical Engineering, Hongik University, Seoul, 121-791, Korea. |
| This journal is © The Royal Society of Chemistry 2014 |