Ultra-fast framework stabilization of Ge-rich zeolites by low-temperature plasma treatment†
Mohamad
El-Roz
a,
Louwanda
Lakiss
a,
Aurelie
Vicente
a,
Krassimir N.
Bozhilov
b,
Frederic
Thibault-Starzyk
a and
Valentin
Valtchev
*a
aLaboratoire Catalyse et Spectrochimie, ENSICAEN – Université de Caen – CNRS, 6 bd du Maréchal Juin, 14050 Caen, France. E-mail: valentin.valtchev@ensicaen.fr
bCentral Facility for Advanced Microscopy and Microanalysis, University of California, Riverside, CA 92521, USA
First published on 16th October 2013
Abstract
A novel one-step post-synthesis method for structural stabilization of germanium (Ge) silicate molecular sieves is presented. The modification includes low-temperature SiCl4 and TiCl4 plasma treatment of as-synthesized Ge-silicates. During plasma reaction template removal, partial extraction of Ge from the framework and incorporation of Si and Ti in micropore space takes place. The modified material is further subjected to high-temperature calcination, which completes the incorporation of replacing cations in the zeolite framework. The feasibility of this approach for stabilization and modification of Ge-silicate molecular sieves is exemplified by employing Ge-rich BEC-type zeolite as model material. In situ IR spectroscopy is used to monitor the interactions of SiCl4 and TiCl4 precursors with zeolite material and to follow the structural changes provoked by Ge substitution. Ultimate products and their intermediates were subjected to comprehensive characterization employing complementary methods (XRD, SEM/TEM, TG/DSC, NMR, N2 sorption and chemical analysis). The set of experimental data unambiguously proves successful stabilization of the zeolite framework. Stabilized Ti-containing BEC-type materials were used for methanol photooxidation in order to verify catalytic activity of zeolites obtained by this innovative approach.
Introduction
Over the last fifty years crystalline microporous catalysts have played continuously increasing role in addressing global issues such as increasing energy demand, depletion of fossil energy resources and improving environmental standards for industrial pollution. The steady interest in these materials is due to their unique properties and hence the sustainable solutions of global issues they can bring. More specifically zeolite-type materials, which are crystalline tectosilicates built from TO4 tetrahedra (T = tetrahedral atom, e.g., Si and Al) have found a wide spectrum of applications in industrial chemical technology and in applied and fundamental research with their diverse catalytic, absorption, ion exchange, and molecular sieve properties. The importance and widespread use of zeolites are due to the versatility of their properties as well as to the fact that in general they are environmentally-friendly substances.New standards and challenges require a constant improvement of known and discovery of new microporous zeolite type materials. For example today the need for processing heavy and extra heavy crude oils demands the utilization of large and extra-large pore zeolites. The interest in the synthesis of zeolite materials with extra-large pores is increasing constantly by demands not only from traditional fields such as catalysis, ionic exchange and separation1,2 but also from emerging applications in medical, optical and electronic fields.3,4 The main avenues for discovery and synthesis of new zeolite structures are typically development of new gel compositions, and utilization of novel organic structure directing agents. In line with these aims, partial or full replacement of conventional T (Si, Al) atoms with Zn and Ge have been explored.5–7 For instance Ge4+, which replaces Si4+ easily, is indispensable for the synthesis of extra-large pore molecular sieves due to its structure directing effect toward small 4- and 3-membered ring units. The latter lead to the formation of structures with low framework density.8–10 Thus, the use of Ge has resulted in a wide variety of novel or iso-type structures,11–20 as well as Ge-containing extra-large pore zeolites, which fill the gap between conventional zeolite materials with pore openings below 1 nm and mesoporous materials with pores larger than 2 nm.13,14,20 Although Ge-rich zeolite materials possess extremely useful features, their practical use is still a distant reality because of the limited stability of framework Ge. Unfortunately, this element hydrolyses readily in water-vapor atmosphere and is extracted from the tetrahedral positions in the zeolite structure. Thus, once the organic structure directing agent is removed and Ge is exposed to air humidity the most open frameworks typically lose crystallinity.21 The fact that Ge is sensitive to air humidity is particularly difficult to deal with, since immediately after template combustion the zeolite should remain in water-free atmosphere. This obstacle renders post-synthesis methods that could be employed to replace germanium for a more stable framework cation, almost impossible to apply. Further practical limitation in utilizing Ge-rich zeolite materials is the high price of germanium which limits its extensive use.
Besides the drawbacks, low hydrothermal stability of Ge in zeolite frameworks offers advantages in structural transformation of zeolites. For instance, Roth et al. demonstrated the possibility to extract Ge-rich double four rings (D4R) from UTL-type structure and thus to obtain lamellar two-dimensional (2D) zeolites.22a Again UTL Ge-silicate was used to generate new tri-dimensional structures by inverse σ transformation.22b A new 3D zeolite structure was generated by removal of the Ge-rich layer in UTL structure and contraction of the remaining part of the structure. The same top-down approach was used to generate two new zeolite materials from UTL-type Ge-silicate.22c The two new materials exhibit 12- and 10 × 8-member ring systems, while the parent zeolite contains 14-member ring channels.
In general, the chemical composition of zeolite material during synthesis depends on the ratio of the components in the initial gel.23 The variation of the initial gel composition is possible in certain, usually very narrow limits, which are determined by the crystallization field of the zeolite. In order to prepare zeolitic materials with compositions overstepping these limits, post-synthesis modification approaches have been developed. The post-synthesis treatments can be applied to all important parts of a microporous material, that is, the framework composition, the species situated in the channel system and the zeolite crystal features. Post-synthesis modification of zeolite frameworks is particularly difficult to control because of the limited access to framework cations and the possible amorphisation of the material. Nevertheless, post-synthesis substitution of framework cation is used abundantly. For instance, dealumination,24 alumination25 and even consecutive dealumination and re-alumination of the same material are employed to modify the zeolite properties.26 These modifications are usually performed in two stages, first removal of the organic structure directing agent and then substituting framework species. Unfortunately, such an approach cannot be applied to materials that cannot withstand the first step of the process.
In the present work, we report a novel low-temperature plasma approach which combines in one step the processes of template elimination and Ge substitution. The approach is demonstrated on Si and Ti modification of BEC-type zeolite. BEC-type zeolite denoted ITQ-17 is the polymorph C of zeolite Beta.10 It displays three-dimensional (3D) twelve-membered ring (12MR) channels that are linear and intersect perpendicularly. Its large pore apertures (6.3 × 7.5 Å [001] and 6.0 × 6.9 Å [100]) favor the diffusion of relatively large guest species. Thus it represents the ideal candidate to develop, investigate and demonstrate the feasibility of our approach. Besides, Moliner et al. have successfully prepared Ti-BEC zeolite materials through a direct synthesis approach assisted by molecular modeling.27 Moreover, in a previous study micron- and nano-sized BEC-type crystals prepared with tetraethylammonium hydroxide have been used for hydrothermal post-synthesis substitution of Ge for Al.28 The analysis of the resulting products and their intermediates revealed that the partial substitution of Ge by Al was successfully achieved. However, only the peripheral part of the micron-sized crystals was stabilized, whereas the nano-size crystals showed total stabilization of the framework. Obviously the short diffusion path is a key factor for successful template removal and replacement of Ge for heteroatoms in the zeolite framework. Hence, nano-size zeolite crystals have been used in the present study.
Herein we report the stabilization of BEC-type germanium silicate via low-temperature plasma treatment using TiCl4 and SiCl4 precursors. Template extraction and framework substitution of Ge for Si and Ti in zeolite structure is discussed as well as the characteristics of the final products and their intermediates.
Experimental section
Synthesis of BEC-type material
First, a specific amount of colloidal silica (Ludox, HS-40, Aldrich) was dissolved in hexamethonium hydroxide (Hex(OH)2, Fluka). Then, hexamethonium bromide (HexBr2, Fluka) and germanium dioxide (GeO2, Aldrich) were added and the solution was stirred until complete dissolution of the solid phase. Ammonium fluoride (NH4F 98%, Aldrich) was then introduced into the mixture. The gel was stirred at room temperature for 2 h and freeze dried. An appropriate amount of water was added to obtain a gel with the following composition 0.85SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.33GeO2:0.15Hex(OH)2:0.1HexBr2:0.5NH4F:2.0H2O. The gel was then transferred to an autoclave and the synthesis was performed at 180 °C for 6 h.
:0.33GeO2:0.15Hex(OH)2:0.1HexBr2:0.5NH4F:2.0H2O. The gel was then transferred to an autoclave and the synthesis was performed at 180 °C for 6 h.
TiCl4-plasma, SiCl4-plasma and O2-plasma treatment
The plasma treatment of 200 mg of as-synthesized BEC-type zeolite has been performed in a Pyrex reactor (glowing discharge plasma with a 50 Hz sinusoidal power supply (2 kV). After introducing the powder in the reactor, the system was evacuated until the pressure stabilized at ∼10−2 mbar. Then TiCl4 or SiCl4 was introduced under continuous vacuum in order to reach an equilibrium pressure close to 0.5 mbar and to purge the plasma reactor. Further the cold plasma was turned on to decompose the TiCl4 precursor in the absence of any other gas. The duration of the reaction was 10 s after which the valve of TiCl4 or SiCl4 tube was closed. The process was repeated three times. It should be noted that in our conditions the plasma shuts off when the partial pressure of TiCl4 or SiCl4 exceeds 0.5 mbar. TiCl4-plasma treatment was followed by O2-plasma treatment for 10 min at low pressure (350 Pa) without any temperature alteration in order to completely remove the residual template. This process (TiCl4/SiCl4-plasma followed by O2-plasma treatments) is called method I. During the experiments, the temperature increase using the cold plasma process was below 393 K. The plasma treated materials were denoted “Ti-BEC”, “Si-BEC” and “Ti,Si-BEC” for the samples treated by TiCl4-plasma, SiCl4-plasma and consecutive TiCl4- and SiCl4-plasma, respectively. The as-synthesized BEC material was treated by O2-plasma in order to be used as a reference and compare with Si- and Ti-modified materials. The calcination process consisted in heating the samples from room temperature to 550 °C with a heating rate of 1.75 °C min−1, kept at 550 °C for 5 h and then cooling down to room temperature in 5 h.Consecutive O2-plasma followed by TiCl4/SiCl4-plasma treatments have also been performed (method II) under the conditions used in method I. This process allows an efficient removal of the template prior to TiCl4/SiCl4 plasma treatment.
Characterization of the obtained materials
The crystallinity and framework stability of the zeolite samples are investigated using a X-ray diffractometer (PANanalytical, X'Pert Pro MPD). The morphological features of zeolite crystals were investigated with a scanning (SEM, Tescan MIRA FEG) and a transmission (TEM, FEI-Philips CM300, LaB6 cathode) electron microscope. The latter was operated in a low-dose mode at 300 kV accelerating voltage. The TEM instrument was equipped with an energy dispersive X-ray spectrometer (EDS). Thermogravimetric (TGA) analyses were performed with a Setaram TG-ATD Labsys thermal analyzer at a heating rate of 5 °C min−1 in air. Prior to the thermogravimetric analyses, the samples were exposed to 77% humidity for a day. Nitrogen adsorption/desorption measurements were performed with an ASAP 2020 MP instrument after degassing of the sample at 300 °C overnight. The specific surface area was calculated with the BET equation while the microporous and mesoporous volumes were distinguished by the t-plot method.UV-Vis DR measurements were performed on a Thermo-electron evolution 500 UV-Vis spectrometer equipped with RSA-CU40 Diffuse Reflectance cell. From UV-Vis DR spectra information over the band gap energy can be obtained.
In order to monitor the changes in BEC-type zeolite during plasma treatment, in situ FTIR spectroscopy was performed on self-supporting wafers (9–10 mg cm−2) placed in a FTIR-plasma reactor (Fig. S1†). First, the atmospheric gas present in the ionization chamber and the water weakly adsorbed on zeolite surface were evacuated under vacuum. After the evacuation step, the first dose of TiCl4/SiCl4 was introduced under vacuum by opening the valve for ∼10 s, to reach a pressure of ∼50 Pa at equilibrium. This step allowed purging the plasma reactor from the residual gases that could remain after evacuation. The IR spectra were collected in transmission mode on a Bruker Vertex 80v equipped with a cryogenic MCT detector (4 cm−1 resolution, 8 scans, and acquisition time ∼1.2 s).
Solid-state MAS NMR spectra were recorded on a Bruker Avance-400 (9.4T) spectrometer using 4 mm OD zirconia rotors and a spinning frequency of 12 kHz. Single pulse excitation (30° flip angle) and 30 s recycling delay was used for 29Si MAS NMR experiments. Tetramethylsilane (TMS) was used as chemical shift reference for 29Si nuclei.
Photocatalytic operando test
In order to test the photocatalytic activity of Ti-BEC materials, a photocatalytic oxidation of methanol was performed and was followed by IR spectroscopy. Ti-BEC catalyst was pressed into self-supported wafers (Ø = 16 mm, m ∼ 10 mg cm−2). IR spectra were collected with a Nicolet 5700 FT-IR spectrometer (64 scans/spectrum) equipped with a MCT detector. The operando system was connected to a flow set-up. Gases were introduced into the lines (heated at 60 °C) by mass flow controllers. The system allows the two gas mixtures, so called “activation” and “reaction” flows, to be prepared and sent independently to the reactor cell. The “sandwich” type reactor-cell used in this study was an upgraded variant of the operando cell that has proven its reliability over many years of operando studies. It was made of a stainless-steel cylinder that carries a toroidal sample holder in its centre, where the catalyst self-supporting wafer was placed. Tightness was obtained by Kalrez© O-rings, and the dead volume (typically defined as the residual space between each sample face and the windows) was reduced to about 0.4 ml by filling the empty space with KBr windows placed on each side of the sample holder. The surface analysis was made possible without superposition of the gas phase signal. Gases were introduced to the sample by a 1/8′′ OD pipe and collected on the opposite side of the sample holder. For this specific photocatalytic oxidation study, UV irradiation was carried out with a polychromatic light of a Xe–Hg lamp (LC8 spot light Hamamatsu, L10852, 200 W). It was performed by using a UV-light guide (A10014-50-0110) mounted at the entrance of modified IR operando cell as presented in Fig. S2† in order to establish a “homogeneous” irradiation. UV irradiation intensity (I0) was measured using a light power meter (366 nm) (Hamamatsu).The employed configuration allowed a low partial pressure of methanol to be achieved using a saturator at controlled temperature. The gas mixture composition was then fixed at 0.5 vol% methanol and 20 vol% O2 in Ar and the total flow was adjusted to 25 cm3 min−1. The analysis of the outlet gases was performed by means of a Pfeiffer Omnistar mass spectrometer. Likewise, FT-IR spectra of the gas phase were collected using a gas microcell.
Results and discussion
X-Ray diffraction study
In order to investigate the framework stability and verify the unit-cell parameters of the plasma treated materials (Si-BEC, Ti-BEC and Ti,Si-BEC) X-ray diffraction analysis was used. The XRD patterns of non-treated BEC-type material before and after calcination were also studied. As can be seen in Fig. 1, the XRD patterns of plasma treated samples are identical and correspond to the BEC-type framework topology. Only Bragg reflections of crystalline BEC phase are present in the patterns and no indication of other titanium- or silicon-containing phases were found. The resulting diffraction patterns were indexed and refined successfully using Werner's indexing method (TREOR 90),29 despite moderate broadening of the XRD peaks due to the nanosized nature of the crystals. The recorded patterns were all indexed according to a tetragonal unit-cell (P42/mmc). The refined and the previously determined BEC-type zeolite unit-cell parameters, reported in Table 1 demonstrate that the plasma treated samples do not differ structurally from the as-synthesized material. Although a slight 2θ shift was observed between the plasma substituted BEC-type materials and the as-synthesized Ge-rich ITQ-17 zeolite, there is no indication for any substantial changes and transformation of the framework during plasma treatment. | ||
| Fig. 1 XRD patterns of the initial BEC-type material (a) and the treated samples: Ti,Si-BEC (b), Si-BEC (c), Ti-BEC (d), water-exposed TiSi-BEC-calcined (e) and water-exposed calcined BEC-type material (f). | ||
| Sample | Previously reported | As-synthesized | Ti-BEC | Si-BEC | Ti,Si-BEC | Si-BEC calcined | Ti-BEC calcined | Ti,Si-BEC calcined |
|---|---|---|---|---|---|---|---|---|
| a (= b)/Å | 12.7700 | 12.758(5) | 12.744(7) | 12.71(1) | 12.74(7) | 12.445(9) | 12.573(9) | 12.48(1) |
| c/Å | 12.9770 | 13.01(1) | 13.04(2) | 13.06(2) | 13.05(1) | 13.08 (2) | 12.94(2) | 13.00(3) |
| V/Å3 | 2116.2 | 2117 | 2118 | 2110 | 2118 | 2026 | 2046 | 2024 |
The as-synthesized and plasma-treated samples were subjected to high-temperature calcination (550 °C for 7 h). The XRD study demonstrates clearly the increased stability of the plasma treated sample. This stabilization might be due to incorporation of substituting cations (Si and Ti) in germanium positions in the zeolite structure. However, physicochemical characterization of plasma-treated sample and in particular XRD study did not provide any clues for such a mechanism of isomorphous substitution.
The fact that plasma treated samples retained their crystallinity after heating while the non-treated BEC structure collapsed (Fig. 1) provides a clear indication that Si and Ti introduced by plasma treatment in the zeolite material stabilize the structure, although no direct evidence by XRD study about the structural position of the incorporated atoms could be extracted due to the presence of appreciable but unknown amount of amorphous material and the extremely fine grain size of the zeolite crystals. Still the XRD data provide substantial indirect evidence about the structural changes taking place. All three samples showed a 2θ shift corresponding to a reduction of the unit-cell parameters after calcination. Such reduction is expected due to the collapse of the organic molecules and loss of water molecules in the channels. The differential response to the calcination process of the structures substituted by different cations is the telltale sign for interpretation of the structural changes due to substitution. While all the recorded patterns were consistent with space group P42/mmc, the unit-cell volume was reduced by about 5% for Ti,Si-BEC and Si-BEC, and the decrease was less pronounced for Ti-BEC (Table 1).
A possible interpretation of the observed stabilization of the structure after plasma treatment could be that Si and Ti are incorporated in the zeolite channels in close vicinity of Ge and thus stabilize the framework. Such a scenario could be a possible explanation of the observed stabilization of the structure but cannot account for the observed systematic changes in the structural parameters of the treated materials after calcination. A more plausible explanation of the plasma treatment is that Si and Ti ions are randomly substituting Ge in the zeolite framework. These substitutions do not lead to any observable changes in the unit-cell parameters because of the random distribution of Si and Ti among the tetrahedral sites and the presence of structure directing molecules in the channels. During calcination a possible ordering of Si and Ti among the T sites could take place that would affect the unit-cell parameters in a systematic fashion. The observed substantial structural changes expressed in unit-cell volume reduction of the calcinated materials are consistent with such changes. The unit-cell volume reduction is not isotropic, rather it is expressed mainly in clear shortening of the symmetrically equivalent a and b axes and very insignificant reduction along the c axis. The pure siliceous BEC-type material from our experiments exhibits a smaller unit-cell volume with respect to previously reported study.10 The reduction of the unit-cell volume should be expected since the average Si–O bond length is 8.1% shorter with respect to the Ge–O bond length in tetrahedral coordination in BEC-type materials as reported by Conradsson et al.12 and Cantin et al.30 The reduction of the tetrahedral bond length in the pure Si form of BEC compared to the Ge-rich form is expressed in overall reduction of the unit-cell parameters by 3.3%.12,29 The changes in the unit-cell parameters can be understood better by considering the occupancy of different crystallographic sites in the BEC-type structure. More specifically a clue to the possible structural changes taking place during the treatment is suggested by analysis of the ITQ-17 structure as refined by Corma et al.10 The latter ITQ-17 crystals contain 46% Ge in the T1 site, 28% in the T2 site and 23% in the T3 site. The presence of 46% Ge in the T1 site causes the T1–O bond to increase to 1.64 Å or by 1.9% compared to the nominal value of 1.61 Å for Si–O tetrahedral bonding.12 This increase in the T1–O bonding translates into an increase of the symmetrically equivalent a and b axes by 1.6% in the ITQ-17 structure.10
The different degree of reduction of the unit-cell parameters along a and b axes observed after plasma treatment and calcination of the BEC structure in this study supports strongly our interpretation that Si and Ti are replacing Ge in the zeolite framework. One can understand the structural response to Ge substitution by considering closely the topology of the BEC zeolite. The structure can be described as layers linked via D4R units along the a-, b- and c-axes. However, the layers along a- and b-axes differ substantially from that along the c direction where single four-ring units, occupied by T3 sites, are also involved (Fig. 2). Thus a preferential substitution of smaller/larger cations in the T1 sites will lead to reduction/augmentation of the unit-cell parameters mainly along the a and b axes. Since the Si–O bonds are shorter in tetrahedral coordination, then the substitution of Ge for Si at the T1 sites is expected to reduce the unit-cell parameters along the a and b axes. Such interpretation is consistent with the observed changes in the structural parameters of the plasma treated and calcinated samples substituted by Si and Si/Ti.
 | ||
| Fig. 2 Projections of the layers along b axis (a) and c axis (b). For clarity oxygen atoms have been omitted. T1 sites are represented in red, T2 in white and T3 in green. | ||
The substitution by Ti should lead to minimum reduction of the cell dimensions or a slight increase. The nominal average Ti–O bond length in tetrahedral coordination is reported to be in the range of 1.763 Å.31 The similarity in bond lengths between Ge–O and Ti–O in tetrahedral coordination (1.74 vs. 1.76 Å, respectively) should lead to insignificant changes in the unit-cell dimensions when Ge4+ is substituted by Ti4+. After calcination it is expected that preferential occupation of T1 sites by Ti should result in unit-cell dimensions along a and b axes larger than the form substituted by Si, which is consistent with the observations in the present experiments. Further evidence of the preferential ordering of the substituting ions at the T1 sites is the fact that the c axes are essentially identical for all three substituted samples.
Another important issue related with the isomorphous substitution of Ge is the hydrothermal stability. Plasma treated and calcined samples were exposed to an atmosphere of 77% humidity at room temperature for 60 days. The samples were then subjected to XRD measurements. No difference between the XRD patterns of the freshly calcined and water-exposed materials was observed (Fig. 1e) revealing the high framework stability of plasma modified materials. The results of the XRD study interpreted in the light of the changes in physicochemical properties of the treated materials suggest strongly that the improved (hydro)thermal stability of BEC-type Ge-silicate is a consequence of isomorphous substitution of Ge in the framework. Further analyses combining complementary methods were performed in order to corroborate the XRD data interpretation and to shed more light on the properties of the studied materials.
TEM-EDS
The TEM study of the initial untreated BEC sample showed individual crystallites with a particle size of about 20–30 nm that form complex aggregates (Fig. 3). It is worth noting that the individual crystals are loosely attached and the aggregates exhibit a significant number of textural pores. No changes in particle size or morphology of crystallites and aggregates were observed after plasma treatment (Fig. 3b and c). Careful inspection of treated samples did not provide any evidences for formation of individual TiO2 or SiO2 particles. Obviously the plasma treatment method leads to very uniform distribution of Si and Ti within the zeolite crystals. The homogeneous nature of the plasma treated samples was confirmed by SEM (Fig. S3†). | ||
| Fig. 3 TEM micrographs of initial BEC-type material (a), Si-BEC (b) and Ti-BEC (c). | ||
EDS analysis of the initial and plasma treated samples was performed. All treated materials showed higher Si/Ge ratio in respect to the initial one (Table 2). Thus the Si/Ge ratio from 2.9 changed to 3.6, 4.2 and 4.1 for Ti-BEC, Si-BEC and Ti,Si-BEC, respectively (Table 2). The decrease in Ge content is due most probably to the elimination of Ge in the form of GeCln species (with n ≤ 4) during plasma treatment. The very short reaction time, however, did not allow a more substantial extraction of Ge from the zeolite. For instance, the substitution of Al for Si in FAU-type zeolite requires 3–5 h treatment in flow of SiCl4 at temperatures ranging between 400 and 550 °C.32 The short reaction time in the present study has limited the extent of replacement of Ge and its extraction from BEC-type crystals. Consequently, it is very difficult to determine precisely the amount of Ge extracted from the zeolite framework and substituted by Si and/or Ti. The presence of extra framework species, especially in the case of silicon substitution, makes quantitative evaluation of framework cation substitutions impossible. On the basis of chemical analyses we could evaluate to some extent only the incorporation of Ti. The measured Si/Ge ratio of 2.9 in the as-synthesized BEC-type material translates into approximately 11 Ge atoms per formula unit, based on a T32O64 formula. After plasma treatment the average total number of Ge atoms, remaining in the zeolite was reduced to approximately 6.8 atoms per formula unit in the case of the Ti,Si-BEC sample. The EDS analysis of Si-BEC sample did not show the presence of fluorine in the zeolite. Although the level of detection of EDS analysis in respect to fluorine is relatively low this result still shows that the major part of the fluoride ions, integrated in the structure during the crystallization process, was eliminated during plasma treatment.
| Sample | Si:Ge |
S BET/m2 g−1 | V micro/cm3 g−1 |
|---|---|---|---|
| a Standard deviation (σ). b N2 adsorption data for the materials obtained by method II. | |||
| BEC-initial | 2.9(0.3)a | 64 | 0.004 |
| Ti-BEC-plasma | 3.5(0.5) | 373 (357b) | 0.145 (0.131b) |
| Si-BEC-plasma | 4.2(0.6) | 417 (370b) | 0.150 (0.135b) |
| Ti,Si-BEC-plasma | 4.1(0.5) | 411 | 0.150 |
| Ti-BEC-calcined | — | 263 | 0.122 |
| Si-BEC-calcined | — | 279 | 0.11 |
N2-sorption
The porosity and surface area characteristics of the parent and plasma treated samples was characterized by N2 adsorption/desorption measurements. The initial non-calcined material exhibits a relatively large external surface area (89 m2 g−1), which is indicative of the small crystal size (Fig. 4). The term external surface area is employed to distinguish the surface of crystallites from total surface area, which includes also the pores of zeolite crystals. In order to determine the external surface area, the material was degassed at relatively low temperature (130 °C) in order to preserve the organic structure directing agent that occupies the micropore volume. High temperature calcination (500–600 °C) is the conventional approach used to liberate micropores from the organic structure directing agent and thus to study the micropore volume and surface area. In our case the calcination at 550 °C of the initial material resulted in a decrease of the specific surface area (64 m2 g−1), which indicated the low thermal stability of the material. Besides the decrease in specific surface area that most probably was due to the condensation of initial nanoparticles, no presence of microporous type material was found. The isotherm is close to type III, typical of non-porous materials (Fig. 4). In contrast, all plasma treated samples showed type I isotherms, typical of microporous materials. The fast uptake at low relative pressure was combined with a large hysteresis loop at high relative pressure. The later is indicative of textural porosity of nanosized particles. Plasma treated materials showed notable increase of total surface area (SBET) reaching values four times higher than the starting sample (Table 2). The increase of the specific surface area was accompanied by an increase of the pore volume of the material. The latter proves that the plasma treatment led to a degradation of the hexamethonium structure directing agent present in the zeolite channels. The micropore volume obtained is below that expected for a highly crystalline BEC-type zeolite. The most plausible explanations for the lower micropore volume are: (i) partial amorphization; (ii) the presence in the micropore space of residual organic template due to incomplete removal by plasma treatment; and (iii) the presence of products of the reaction between zeolite and SiCl4/TiCl4 precursors.32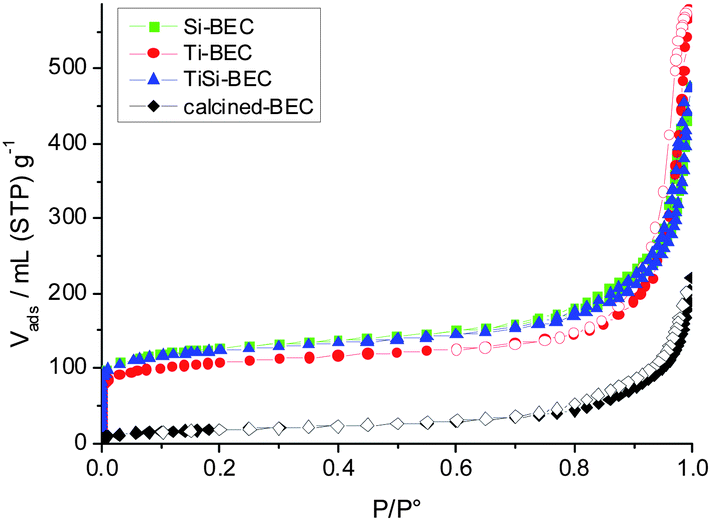 | ||
| Fig. 4 N2 adsorption/desorption isotherms of TiCl4-, SiCl4-, and consecutive Ti–Si-plasma stabilized BEC-type materials subjected to O2-plasma treatment. The adsorption/desorption isotherm of calcined at 550 °C BEC-type material is also provided (black rhombuses). | ||
Thus the plasma treatment affects the extent and efficiency of template removal and framework stabilization, which will be discussed in the next section. The calcination of Si-BEC and Ti-BEC led to a decrease of 20 to 30% of the specific surface area and micropore volume. This decrease is most probably related to localized partial collapse of the structure, in places where Ge was not substituted to the extent required to stabilize the zeolite framework. Similar results were obtained for the materials treated by method II, where first O2-plasma was employed to destroy the template and then TiCl4/SiCl4-plasma treatment was performed. The later approach did not improve the crystallinity of the final products.
Thermogravimetrical (TG) results
The amount of template remaining in the structure after SiCl4 and TiCl4 plasma treatment was estimated by TG analysis (Fig. 5). The initial material contained 14.9 wt% template and 3.2 wt% water. After plasma treatment the amount of template was reduced to 4.9 and 5.2 wt% in the case of SiCl4 and TiCl4 treatment, respectively. The degradation and extraction of debris from the template was coupled with an increase in water content, which reached 8.4 wt% in both samples. Thus the data from TG analysis are in good agreement with N2 sorption measurements.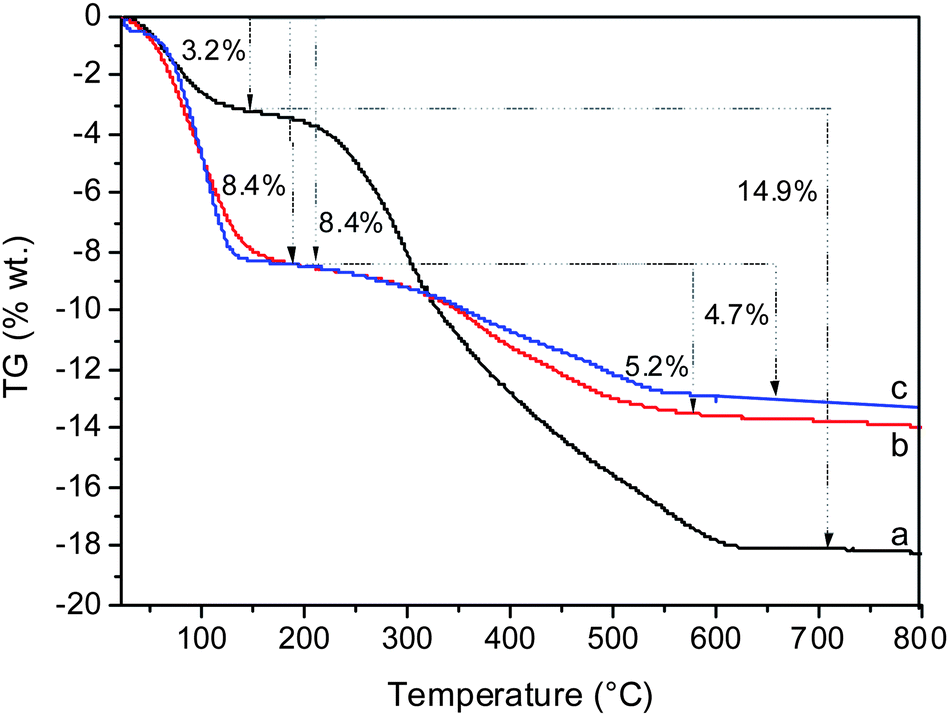 | ||
| Fig. 5 TG analyses of the as-synthesized (a), Ti-BEC plasma treated (b) and Si-BEC plasma treated (c) materials. | ||
NMR results
29Si MAS NMR spectra of the initial and treated BEC samples are shown in Fig. S4.† A broad band between −100 and −120 ppm was observed for all samples revealing the complex chemical environment of the Si atoms. In addition, a very small peak at about −95 ppm was present. BEC-type material has three well defined crystallographic positions with chemical shifts of −116, −112 and −110 ppm.10 As known, the replacement of Si for a heteroatom results in a chemical shift to higher field.33,34A random substitution of Si in the T1–3 positions by other atoms results in three additional peaks shifted to higher field. Thus, the framework substitution usually complicates the NMR spectra and makes the decomposition of the signal more difficult. The initial material employed in this study comprised a substantial amount of Ge located preferentially in the T1 position. T2 and T3 positions incorporate noticeable amounts of Ge, which makes the spectrum more complex, comprising broad overlapping peaks centered at about −115, −109 and −103 ppm (Fig. S4†). Partial extraction and replacement of Ge for Si and Ti change substantially the NMR spectra of the material and lead to the appearance of additional peaks which overlap with the resonances of the initial BEC-type material (Fig. S4†). We have used the calcined BEC-type sample that was kept under inert atmosphere as a reference for the investigation of the substituted forms of zeolite (Fig. S4a†).
In order to make clearer the effect of Ge substitution in the zeolite framework, decomposition of 29Si MAS NMR spectra was performed. Fig. 6 shows the detailed decomposition spectra of the initial Ge-rich BEC-type material and Si-substituted counterpart. As can be seen the intensity ratios between the peaks corresponding to the three symmetrically non-equivalent T sites in the structure change substantially. The spectrum of Si-substituted BEC zeolite showed a visible increase in intensity of the peaks in the range −110 and −116 ppm, which corresponds to Si(4Si) sites. In contrast, the intensity of the three peaks in the region −102 and −110 ppm corresponding to Si(1Ge, 3Si) sites decreased. The intensity of the peak corresponding to Si(2Ge, 2Si) environment did not change substantially, which might be a consequence of the presence of defect sites such as Si(1OH, 3Si) or Si(2OH, 2Si).
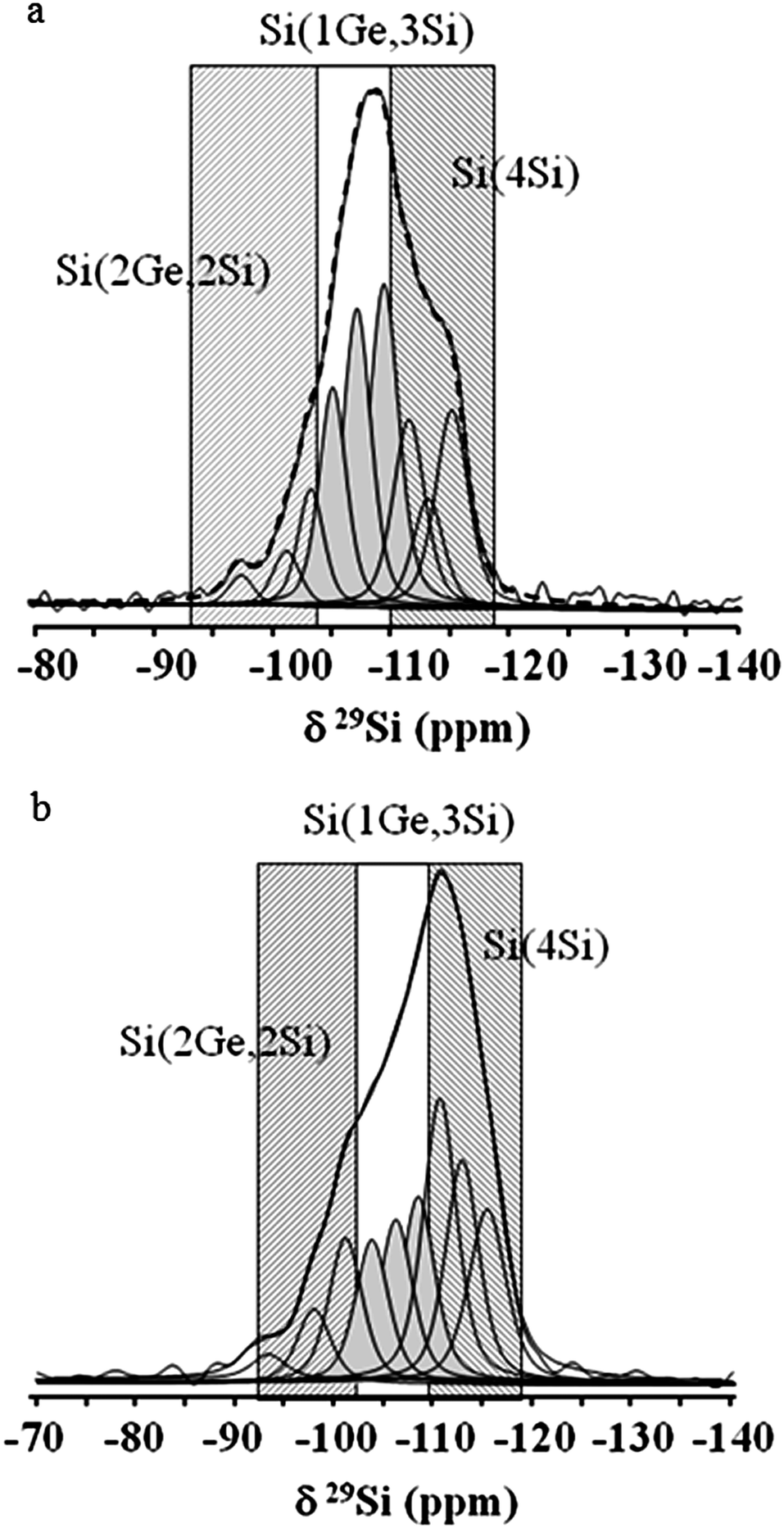 | ||
| Fig. 6 29Si MAS NMR decomposition spectra of the calcined and exposed to inert atmosphere initial (a) and Si-substituted (b) BEC-type materials. | ||
The samples containing Ti were much more complex due to the fact that there are two chemically different replacing atoms in tetrahedral coordination. Consequently, it is impossible to successfully decompose the overlapping peaks corresponding to Ti species interacting with the zeolite framework. Thus a simplified decomposition of the three main peaks was performed in order to distinguish the main tetrahedral environments after post-synthesis modification with Ti (Fig. 7). Therefore, based on this decomposition we could only state that the replacing cations interact with the framework of BEC-type Ge-silicate.
 | ||
| Fig. 7 29Si MAS NMR decomposition spectra of (a) BEC-calcined under dry air, (b) Ti-BEC, (c) Ti,Si-BEC and (d) Ti,Si-BEC calcined. | ||
The 29Si MAS NMR of a zeolite can be used to estimate the ratio between framework cations.35 However, in the current study the complex nature of the interaction between Si, Ge and Ti makes it difficult to obtain meaningful Si/Ge and Si/Ti ratios (Fig. 7). Nevertheless, the increase of the area of the peaks at −109 and −104 ppm, revealed by the decomposition of the 29Si NMR spectrum of TiCl4-plasma modified sample, can be explained by the incorporation of some Ti atoms in the framework (Fig. 7b). Actually, a similar result has been reported in the case of an MFI-type titanium silicalite (TS-1) sample, where the presence of framework Ti was unambiguously proved.36 This effect is less pronounced in the sample subjected to consecutive TiCl4 and SiCl4 plasma treatments, which is not surprising taking into account the competition of Ti and Si for the available tetrahedral positions. It is important to note the changes in the ratio between different peaks after the calcination of the sample (Fig. 7d), which suggest that significant changes in zeolite framework take place during high-temperature calcination, which was confirmed by XRD study.
IR results
The interaction of the SiCl4/TiCl4 plasma with zeolite has been examined by IR spectroscopy (Fig. 8). Fig. 8 presents the evolution of the IR spectra upon: (a) introduction of SiCl4, (b) SiCl4-plasma and (c) O2-plasma. A simple introduction of SiCl4 in the reaction cell led to a shift of the hydrogen-bonded Si–OH/Ge–OH band from 3310 to 3150 cm−1. The latter was assigned to the reaction between SiCl4 and water adsorbed on SiOH and/or GeOH sites (SiOH⋯H2O + SiCl4 → SiOH⋯(HO)SiCl3 + HCl). This band disappeared completely after the SiCl4-plasma treatment, while an important decrease of CH band at 3070–2730 cm−1 was observed. Another important change during plasma treatment is the decrease of the broad band at 3660–3070 cm−1 that was assigned to desorption and/or condensation of adsorbed (HO)SiCl3 species.37 | ||
| Fig. 8 (A) Evolution of IR spectra of BEC-type material vs. treatment time during SiCl4 introduction, SiCl4-plasma and O2-plasma treatments. (B) IR spectra of BEC zeolite before treatment (a), after SiCl4 introduction (b), after SiCl4-plasma treatment (c), and after SiCl4-plasma followed by O2-plasma treatment (d). | ||
Such a result confirms the formation of highly active chlorine-containing compounds (radicals and ions), which led to a substantial degradation of the organic templates. These species could also be related to an extraction and transport of GeCln species through the channel system as well as a consequent incorporation of Si in BEC-type structure. CH bands disappear after oxygen plasma treatment, suggesting the removal of the structure directing agent. This means that the residual template observed by TG analysis of the plasma treated BEC (∼20%) could be assigned to coke formation or to template remaining at the edges of the pellet. It should be noted that the IR beam is focused on the center of pellet and the signal from the peripheral part is negligible.
The preparation of Si-BEC by method II has also been monitored by IR. This method includes preliminary treatment with O2-plasma followed by SiCl4 plasma (Fig. S5†). The application of oxygen plasma resulted in complete removal of the template. However, this approach did not change substantially the characteristics of the final product. As can be seen, the materials prepared by methods I and II show similar total surface area and micropore volume (Table 2). Therefore, the debris from the template that were detected after the application of method I do not affect substantially the diffusion of reactive species and their reaction with the zeolite framework.
The Ti-BEC formation has also been studied by IR spectroscopy during plasma treatment. The evolution of IR spectra while applying method II was similar to that of the SiCl4 treated sample described above. According to XRD analysis Si-BEC and Ti-BEC samples synthesized according to method II exhibit a similar level of crystallinity as samples prepared according to method I (Fig. S6†).
DR-UV results
UV-visible technique provides information about the nature of Ti species in silica networks. Fig. 9 includes the UV spectra of BEC-type material after calcination, Ti-BEC sample, TiO2-anatase and TiO2-P25. The initial Ge-containing BEC-type material exhibits a very low UV absorption. Titanium-modified BEC material showed substantially different UV absorption characteristics, namely a blue shift of the UV spectra as compared to anatase and rutile forms of TiO2. A broad band is observed between 200 and 350 nm, with a maximum centered at ca. 270 nm (Fig. 9). It was assigned to a charge transfer between oxygen and Ti atoms, simultaneously present in tetra-, penta- and hexacoordinated structures. This band is broader than that observed in TS-1 zeolite containing only framework Ti.38 Similar spectrum has been recorded for very nanometric titanium oxide clusters on silica.39 Therefore, the UV spectra reveals the presence of extremely small Ti (TiOx) species in Ti-BEC zeolite.38–42 This result shows also the presence of species with different, including tetrahedral coordination of titanium, which could be attributed to Ti atoms in the zeolite framework.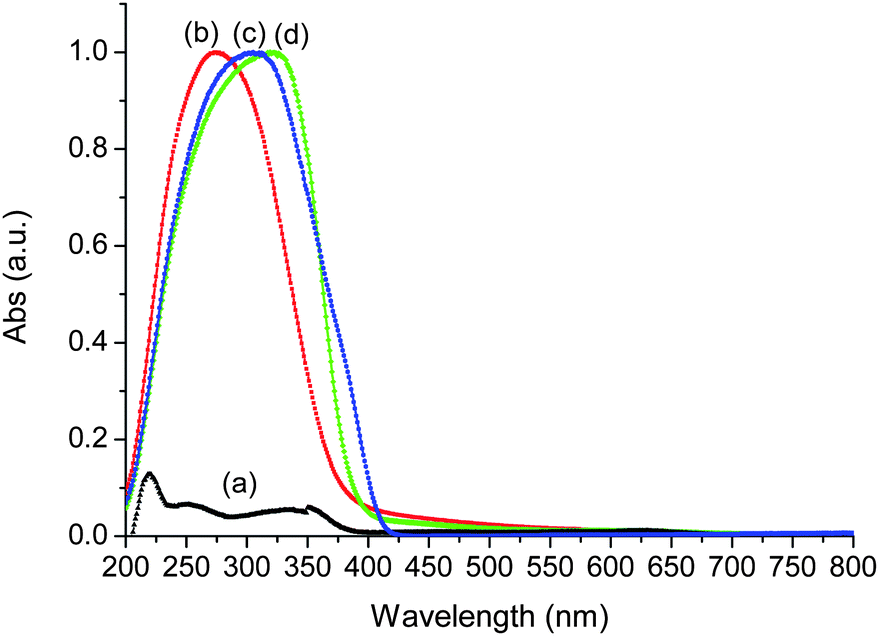 | ||
| Fig. 9 DR UV-visible spectra of BEC-type material (a), Ti-BEC (b), TiO2-P25 (c) and TiO2 anatase (d). | ||
Photocatalytic activity of Ti-BEC
Photocatalytic activities of Beta, HZSM-5 and FAU zeolite types supported TiO2 materials introduced by sol–gel synthesis or by solid-state dispersion have already been discussed in the literature.43–45 In general an enhancement of the photocatalytic activity in comparison with bulk TiO2 photocatalyst could be observed. Therefore the presence of highly dispersed (framework Ti and/or extraframework TiO2) in the Ti-BEC and Ti,Si-BEC samples is expected to induce photocatalytic activity. Methanol photooxidation using Ti-BEC has been performed using a new IR operando technique recently developed in our laboratory.46,47 For comparison, a commercial TiO2 material (TiO2-P25 from Degussa) has been used as reference.The IR spectra of Ti-BEC during methanol adsorption are reported in Fig. 10A. A decrease of the band at 3660 cm−1 assigned to ν(Ti(IV)OH) vibration46 and/or to ν(GeOH) vibration48 modes was observed. This decrease was coupled with an increase of the broad band in the range 3660–3000 cm−1. The latter was assigned to the vibration band of H-bonded TiOH sites, in other words to the methanol adsorption on TiOH sites (Fig. 10A and B). The band observed at 3740 cm−1 was assigned to ν(SiOH) or/and ν(GeOH) vibration modes (isolated SiOH and GeOH sites). In contrast to the band at 3660 cm−1 that has dramatically decreased during methanol adsorption, the one at 3740 cm−1 was still present. This is probably due to the presence of inaccessible germanium oxy–hydroxide species (Ge(O)OH) in zeolite channels. Fig. 10B shows the IR spectra of TiO2-P25 and Ti-BEC samples monitored at RT after saturation with methanol (MeOH) prior to the UV irradiation. Four main bands assigned to CH3 (methanol) vibration modes were observed between 3000 and 2700 cm−1. The bands at 2920 and 2820 cm−1 correspond, respectively, to the νs(CH3) and νas(CH3) vibration modes of dissociative chemisorbed methanol on the TiO2 surface.42 The relatively high intensity of these two bands on TiO2-P25 compared to those on Ti-BEC shows lower methanol chemisorption for the treated zeolite material. This is obviously related to the lower amount of defect sites (TiOH) in Ti-BEC.
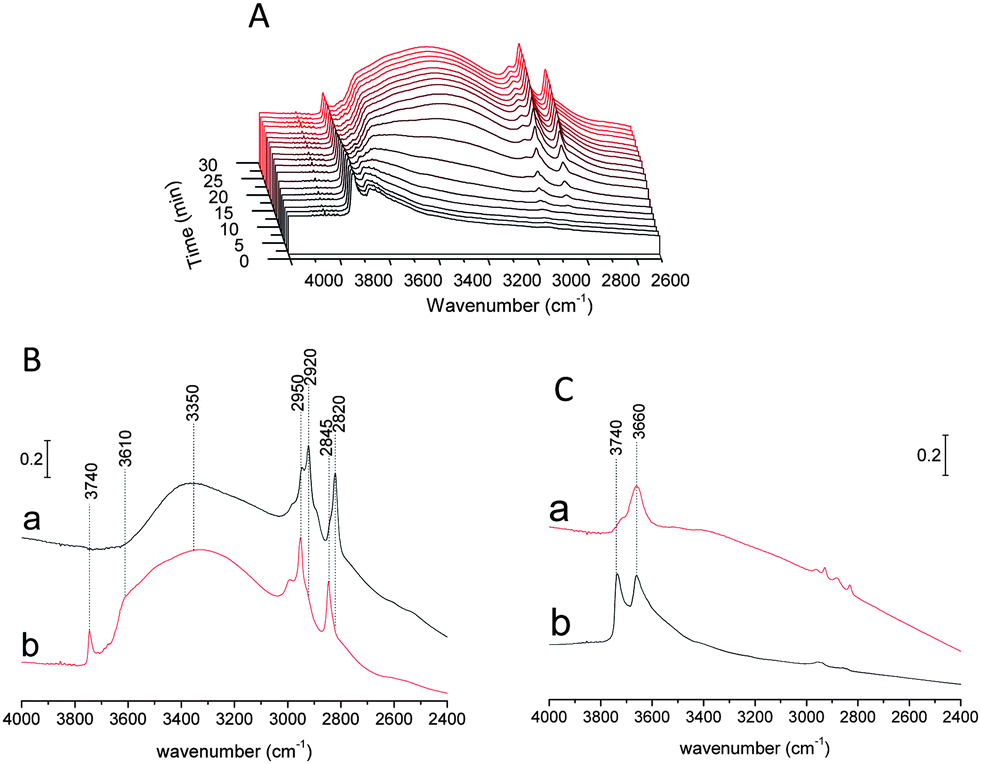 | ||
| Fig. 10 (A) Evolution of IR spectra of Ti-BEC material during methanol saturation (methanol–O2–Ar 0.5:20:79.5 wt%). (B) IR spectra of TiO2-P25 (a), and Ti-BEC (b) materials after methanol saturation. (C) IR spectra of TiO2-P25 (a) and Ti-BEC (b) materials during methanol photooxidation. | ||
The additional bands situated at 2950 and 2845 cm−1 are attributed to νs(CH3) and νs(CH3) vibration modes of methanol molecularly physisorbed on TiO2 surface.46 These results showed a high accessibility of the titanium introduced in BEC-type structure. Fig. 10C presents the IR spectra of TiO2-P25 and Ti-BEC during the methanol photooxidation. The observed high decrease of CH bands is obviously related with methanol desorption and/or photooxidation.
Fig. 11 shows the IR spectra of the gas phase during the photooxidation of methanol using Ti-BEC as photocatalyst. During the first few seconds of irradiation, a high methanol desorption was observed attributed to change in the surface equilibrium. After irradiating Ti-BEC under methanol flow, new peaks appeared in the gas phase at 2395–2290 cm−1 and 1820–1630 cm−1, which were assigned to CO2 and carbonyl vibration bands.
 | ||
| Fig. 11 (A) IR spectra vs. time of the gas phase during the photooxidation of methanol using Ti-BEC. (B) IR spectra of the gas phase before (a) and during methanol photooxidation using Ti-BEC (b) and TiO2-P25 (c) as photocatalysts (methanol–O2–Ar = 0.5:20:79.5 wt%; 25 cm3 min−1). | ||
The effect of the UV light intensity on the photocatalyst reactivity and CO2-selecitivity of methanol photooxidation is presented in Fig. 12 and the results are summarized in Table 3. The photoactivity of Ti-BEC is about 2 to 3.5 times lower than TiO2-P25. Taking into account that the Ti content in Ti-BEC sample is negligible in respect to the bulk P25 sample, this result points out the high photoactivity of the titanium species present in the Ti-BEC sample. The results of methanol photooxidation showed also a different selectivity for Ti-BEC and TiO2-P25 samples. While TiO2-P25 shows a higher selectivity in the conversion of methanol to CO2, Ti-BEC shows a higher selectivity in the conversion of methanol to methyl formate. This molecule is a hightly flexible intermediate for the production of important C1 chemicals such as formic acid and formamide and this result shows the high potential of Ti-BEC in green photochemistry. The difference in the selectivity of TiO2-P25 and Ti-BEC could be assigned to the different nature of active sites and their environment in the two photocatalysts. It could be also assigned to the lower amount of the active sites in Ti-BEC, in comparison with TiO2-P25, which led to a higher covering of Ti-BEC active sites by methanol. An increase of the UV light intensity leads to an increase of the reactivity of the photocatalysts that most probably is a consequence of more abundant formation of active species (electron–hole pairs).
 | ||
| Fig. 12 Evolution of the methanol conversion (a), IR area band of CO2 (b) and carbonyl (c) vs. UV light intensity using Ti-BEC and TiO2-P25 as photocatalyst. | ||
| Samples | UV-intensity/mW cm−2 | Conversion (%) | CO2-selectivity (%) |
|---|---|---|---|
| Ti-BEC | 5 | 20 | 15 |
| 7 | 30 | 18 | |
| 10 | 45 | 21 | |
| TiO2-P25 | 5 | 70 | 58 |
| 7 | 80 | 96 | |
| 10 | 93 | >96 | |
The set of experimental data unambiguously show that low-temperature plasma treatment can be used to stabilize Ge-rich zeolite structures. The mechanism of the process, however, is fairly complex and obviously includes several stages, which were not completely revealed in the present study. It is clear, however, that the plasma treatment allows removal of a substantial part of the organic template and introduces Si and Ti species in zeolite channels. This process is coupled with extraction of Ge from the framework, as shown by the chemical analyses. However, both the Ge extraction and amount of expelled Ge species from the structure is limited due to the short reaction time and low flux of SiCl4/TiCl4 precursors in the plasma unit. Most probably a substantial part of extracted Ge remains in the channels of zeolite, decreasing the pore volume and blocking inter-channel diffusion. Thus, multiple repetition of SiCl4/TiCl4 plasma treatment could contribute to the formation of larger amounts of extra framework species. Partial replacement in the framework of material takes place during high-temperature calcination in air atmosphere when the available Si and/or Ti cations migrate and occupy the T vacancies remaining after the Ge extraction. During this process the cations situated in the channels certainly interact with the zeolite framework. There are two potential consequences of this process. The first one is a partial blocking of micropore space, and this suggestion is supported by relatively low micropore volume of stabilized BEC-type samples. The second one is a possible stabilization of zeolite structure by species situated in the channels, for which we do not have direct proof. It remains an open question as to whether a simple extraction of Ge from T positions could stabilize the zeolite framework. In the present study we did not find such clues. However, our previous investigations, where Ge was extracted from BEC-type framework by means of hydrothermal treatment, showed that the structure collapses if another framework cation does not replace Ge in the zeolite.28
The here presented method is fast and efficient, but there is still a long way to optimize the method and bring it to industrial application. A particularly important issue is the homogeneity of the product and the reproducibility of the material. Our previous study performed on a stable zeolite showed high reproducibility of the results.49–51 It should also be mentioned that Ti-modified zeolite Beta sample obtained by conventional and cold plasma treatments showed similar physicochemical properties.49
The industrial implementation of cold plasma treatment seems feasible. There are no substantive obstacles that would prevent this method from being applied on a larger scale. However, the development of a bigger and automated plasma reactor with higher electrode capacities is an engineering challenge that goes beyond the goals of the present study.
Conclusions
The set of experimental data confirmed partial substitution of Ge for Si and Ti in BEC-type structure. The substitution mechanism is complex, starting with infiltration and attachment of heteroatoms in the zeolite channels, followed by incorporation in the framework through substitution of Ge upon calcination, and expelling Ge mostly in the channels in the form of oxy–hydroxy complexes. Si- and Ti-modified zeolites showed remarkable hydrothermal stability in water-vapor atmosphere.The photocatalytic activity of the samples stabilized by Ti plasma has been investigated in methanol photo-oxidation. The treated samples showed lower activity (2–3.5 times) and higher selectivity in the formation of methyl formate than TiO2-P25 used as reference. This result, however, is promising bearing in mind the much lower Ti content in the zeolite material, and demonstrates also the uniform distribution of Ti in the zeolite matrix.
In conclusion, the applied method allows an efficient and ultrafast stabilization of microporous materials with low thermal/hydrothermal stability. A future study will explore what fraction of framework Ge could be extracted and recycled. The application of this method would make possible the use of microporous zeolite type materials that cannot withstand high-temperature combustion of organic structure directing agent or following exposure of water vapor. The method permits also incorporation of heteroatoms in zeolite structures and thus opens a new route for modification and fine control of their catalytic properties.
Acknowledgements
The project was partially sponsored by the French Research Agency (ANR 2010 BLAN 723 1) project HiZiCOKE (contract no. 723). Louwanda Lakiss acknowledges the financial support of region Lower Normandy for the post-doctoral position.Notes and references
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- A. Corma, J. Catal., 2003, 216, 298–312 CrossRef CAS.
- G. Ferey, Science, 2001, 291, 994–995 CrossRef CAS.
- A. Corma, U. Diaz, M. Arrica, E. Fernandez and I. Ortega, Angew. Chem., Int. Ed., 2009, 48, 6247–6250 CrossRef CAS PubMed.
- R. Fricke, H. Kosslik, G. Lischke and M. Richter, Chem. Rev., 2000, 100, 2303–2405 CrossRef CAS PubMed.
- J. B. Nagy, R. Aiello, G. Giordano, A. Katovic, F. Testa, Z. Konya and I. Kiricsi, Molecular Sieves, 2007, 5, 365–478 CrossRef CAS.
- W. Burton and S. I. Zones, Stud. Surf. Sci. Catal., 2007, 168, 137–179 CrossRef.
- A. Corma, M. J. Diaz-Cabanas, J. Jiang, M. Afework, D. L. Dorset, S. L. Soled and K. G. Strohmaier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13997–14002 CrossRef CAS PubMed.
- C. Cascales, E. Gutierrez-Puebla, M. A. Monge and C. Ruiz-Valero, Angew. Chem., Int. Ed., 1998, 37, 129–131 CrossRef CAS.
- A. Corma, M. T. Navarro, F. Rey, J. Rius and S. Valencia, Angew. Chem., Int. Ed., 2001, 40, 2277–2280 CrossRef CAS.
- J. Su, Y. Wang, Z. Wang and J. Lin, J. Am. Chem. Soc., 2009, 131, 6080–6081 CAS.
- T. Conradsson, M. S. Dadachov and X. D. Zou, Microporous Mesoporous Mater., 2000, 41, 183–191 CrossRef CAS.
- A. Corma, M. J. Diaz-Cabanas, J. Martinez-Triguero, F. Rey and J. Rius, Nature, 2002, 418, 514–517 CrossRef CAS PubMed.
- A. Corma, M. J. Diaz-Cabanas, J. L. Jorda, C. Martinez and M. Moliner, Nature, 2006, 443, 842–845 CrossRef CAS PubMed.
- D. L. Dorset, K. G. Strohmaier, C. E. Kliewer, A. Corma, M. J. Diaz-Cabanas, F. Rey and C. Gilmore, Chem. Mater., 2008, 20, 5325–5331 CrossRef CAS.
- J. Sun, C. Bonneau, A. Cantin, A. Corma, M. J. Diaz-Cabanyas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154–1157 CrossRef CAS PubMed.
- Y. Mathieu, J.-L. Paillaud, P. Caullet and N. Bats, Microporous Mesoporous Mater., 2004, 75, 13–22 CrossRef CAS PubMed.
- J.-L. Paillaud, B. Harbuzaru, J. Patarin and N. Bats, Science, 2004, 304, 990–992 CrossRef CAS PubMed.
- Y. Lorgouilloux, M. Dodin, J.-L. Paillaud, P. Caullet, L. Michelin, L. Josien, O. Ersen and N. Bats, J. Solid State Chem., 2009, 182, 622–629 CrossRef CAS PubMed.
- (a) M. Alvaro, P. Atienzar, A. Corma, B. Ferrer, H. Garcia and M. T. Navarro, J. Phys. Chem. B, 2005, 109, 3696–3700 CrossRef CAS PubMed; (b) R. Castaneda, A. Corma, V. Fornes, F. Rey and J. Rius, J. Am. Chem. Soc., 2003, 125, 7820–7821 CrossRef CAS PubMed; (c) A. Corma, M. J. Diaz-Cabanas and F. Rey, US Pat., 6797254, 2003.
- M. Moliner, M. J. Díaz-Cabañas, V. Fornes, C. Martinez and A. Corma, J. Catal., 2008, 254, 101–109 CrossRef CAS PubMed.
- (a) W. J. Roth, O. V. Scvets, M. Shamzhy, P. Chlubna, M. Kubu, P. Nachtigal and J. Cejka, J. Am. Chem. Soc., 2011, 133, 6130–6133 CrossRef CAS PubMed; (b) E. Verheyen, L. Joos, K. Van Havenberg, E. Breynart, N. Kasian, E. Gobechiya, K. Houthoofd, C. Martineau, M. Hinterstein, F. Taulelle, V. Van Speybroeck, M. Waroquier, S. Bals, G. Van Tandeloo, C. E. A. Kirschhock and J. Martens, Nat. Mater., 2012, 23, 2545–2554 Search PubMed; (c) W. J. Roth, P. Nachtigal, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubna, L. Grajciar, M. Polozij, A. Zukal, O. Schvets and J. Cejka, Nat. Chem., 2013, 5, 628–633 CrossRef CAS PubMed.
- A. Corma and M. E. Davis, ChemPhysChem, 2004, 5, 304–313 CrossRef CAS.
- (a) S. Li, S.-J. Huang, W. Shen, H. Zhang, H. Fang, A. Zheng, S.-B. Liu and F. Deng, J. Phys. Chem. C, 2008, 112, 14486–14494 CrossRef CAS; (b) B. H. Wouters, T. Chen and P. J. Grobet, J. Phys. Chem. B, 2001, 105, 1135–1139 CrossRef CAS; (c) J. A. Martens, H. Geerts, P. J. Grobet and P. A. Jacobs, J. Chem. Soc., Chem. Commun., 1990, 1418–1419 RSC; (d) M. A. Zanjanchi and A. Ebrahimian, Mater. Chem. Phys., 2008, 110, 228–230 CrossRef CAS PubMed; (e) C. S. Triantafillidis, A. G. Vlessidis, L. Nalbandian and N. P. Evmiridis, Microporous Mesoporous Mater., 2001, 47, 369–388 CrossRef CAS.
- (a) C. Yang and Q. J. Xu, J. Chem. Soc., Faraday Trans., 1997, 93, 1675 RSC; (b) C. Yang, J. Wang and Q. Xu, Microporous Mater., 1997, 11, 261–268 CrossRef CAS; (c) A. Omegna, M. Haouas, G. Pirngruber and R. Prins, Stud. Surf. Sci. Catal., 2001, 135, 215 CrossRef; (d) M. W. Anderson, J. Klinowski and L. Xinsheng, J. Chem. Soc., Chem. Commun., 1984, 1596–1597 RSC; (e) S. Namba, K. Yamagishi and T. Yashima, Chem. Lett., 1987, 1109–1112 CrossRef CAS.
- (a) A. Omegna, R. Prins and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 9280–9283 CrossRef CAS PubMed; (b) A. Omegna, J. A. van Bokhoven and R. Prins, J. Phys. Chem. B, 2003, 107, 8854–8860 CrossRef CAS; (c) J. Zhang, L. Zhou and X. Li, Stud. Surf. Sci. Catal., 2001, 135, 213–220 CrossRef.
- M. Moliner, P. Serna, A. Cantín, G. Sastre, M. J. Díaz-Cabanas and A. Corma, J. Phys. Chem. C, 2008, 112, 19547–19554 CAS.
- (a) L. Tosheva, N. Mahe and V. Valtchev, Stud. Surf. Sci. Catal., 2007, 170, 616–621 CrossRef; (b) F. Gao, M. Jaber, K. Bozhilov, A. Vicente, C. Fernandez and V. Valtchev, J. Am. Chem. Soc., 2009, 131, 16580–16586 CrossRef CAS PubMed.
- P. E. Werner, L. Eriksson and M. Westdahl, J. Appl. Crystallogr., 1985, 18, 367–370 CrossRef CAS.
- A. Cantin, A. Corma, M. J. Díaz-Cabañas, J. L. Jorda, M. Moliner and F. Rey, Angew. Chem., Int. Ed., 2006, 45, 8013–8015 CrossRef CAS PubMed.
- S. Pei, G. W. Zajac, J. A. Kaduk, J. Faber, B. I. Boyanov, D. Duck, D. Fazzini, T. I. Morrison and D. S. Yang, Catal. Lett., 1993, 21, 333–344 CrossRef CAS.
- H. K. Beyer and I. Belenykaja, Stud. Surf. Sci. Catal., 1980, 5, 203–210 CrossRef CAS.
- Y. Wang, J. Song and H. Gies, Solid State Sci., 2003, 5, 1421–1433 CrossRef CAS PubMed.
- S. Ganapathy, K. U. Gore, R. Kumar and J. P. Amoureux, Solid State Nucl. Magn. Reson., 2003, 24, 184–195 CrossRef CAS.
- G. Engelhardt and D. Michel, High-Resolution Solid-State NMR of Silicates and Zeolites, John Wiley & Sons, New York, 1987 Search PubMed.
- A. Carati, C. Flego, E. Previde Massara, R. Millini, L. Carluccio, W. O. Parker, Jr and G. Bellussi, Microporous Mesoporous Mater., 1999, 30, 137–144 CrossRef CAS.
- H. Kosslick, V. A. Tuan, R. Fricke and C. Peuker, Ber. Bunsen-Ges. Phys. Chem., 1992, 96, 1761–17655 CrossRef CAS.
- Y. Cheneviere, F. Chieux, V. Caps and A. Tuel, J. Catal., 2010, 269, 161–168 CrossRef CAS PubMed.
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16646–16654 CrossRef CAS.
- A. Tuel and L. G. Hubert-Pfalzgraf, J. Catal., 2003, 217, 343–353 CAS.
- L. Brus, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS.
- O. I. Micic, M. Meglic, D. Lawless, D. K. Sharma and N. Serpone, Langmuir, 1990, 6, 4403–4409 CrossRef.
- M. Lafjah, F. Djafri, A. Bengueddach, N. Keller and V. Keller, J. Hazard. Mater., 2011, 186, 1218–1225 CrossRef CAS PubMed.
- W. Zhanga, K. Wanga, Y. Yua and H. Heb, Chem. Eng. J., 2010, 163, 62–67 CrossRef PubMed.
- Y. Kuwahara, J. Aoyama, K. Miyakubo, T. Eguchi, T. Kamegawa, K. Mori and H. Yamashita, J. Catal., 2012, 285, 223–234 CrossRef CAS PubMed.
- M. El-Roz, P. Cool, M. Kus and F. Thibault-Starzyk, J. Phys. Chem. C, 2012, 116, 13252–13263 CAS.
- M. El-Roz, P. Bazin and F. Thibault-Starzyk, Catal. Today, 2013, 205, 111–119 CrossRef CAS PubMed.
- C.-H. Nicolas and M. Pera-Titus, Microporous Mesoporous Mater., 2012, 153, 254–262 CrossRef CAS PubMed.
- M. El-Roz, L. Lakiss, J. El Fallah, O. I. Lebedev, F. Thibault-Starzyk and V. Valtchev, Phys. Chem. Chem. Phys., 2013, 15, 16198–16207 RSC.
- M. El-Roz, L. Lakiss, V. Valtchev, S. Mintova and F. Thibault-Starzyk, Microporous Mesoporous Mater., 2012, 158, 148–154 CrossRef CAS PubMed.
- K.-L. Wong, M. El Roz, L. Tosheva, J.-M. Goupiland and S. Mintova, Catal. Today, 2013, 204, 66–72 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A scheme of plasma reactor coupled to IR in situ cell; a scheme of “sandwich” reactor-IR cell modified for studying UV photocatalysis; SEM micrographs of Si- and Ti-plasma modified BEC-type crystals; 29Si MAS NMR spectra of the initial and substituted BEC-type materials; IR spectra of BEC-type material modified by method II; XRD patterns of BEC-type material modified by method II. See DOI: 10.1039/c3sc51892b |
| This journal is © The Royal Society of Chemistry 2014 |