In situ growth of oriented polyaniline nanowires array for efficient cathode of Co(III)/Co(II) mediated dye-sensitized solar cell†
Hong
Wang
,
Quanyou
Feng
,
Feng
Gong
,
Yan
Li
,
Gang
Zhou
and
Zhong-Sheng
Wang
*
Department of Chemistry, Lab of Advanced Materials, Fudan University, 2205 Songhu Road, Shanghai 200438, P. R. China. E-mail: zs.wang@fudan.edu.cn
First published on 5th November 2012
Abstract
To improve the electrocatalytic performance of polyaniline thin films, an oriented polyaniline nanowires array has been successfully grown in situ on conductive glass substrates without templates and applied as the cathode of dye-sensitized solar cells (DSSCs) mediated with a Co(bpy)33+/2+ (bpy = 2,2′-bipyridine) redox couple. Compared to the polyaniline film with a random network, the oriented polyaniline nanowires array exhibits much better electrocatalytic performance, and even outperforms the typical Pt electrode in both electrocatalytic performance and electrochemical stability when exposed to the acetonitrile solution of the Co(bpy)33+/2+ redox couple. Owing to the higher electrocatalytic performance, the DSSC with the oriented nanowires array produces a higher short-circuit photocurrent and fill factor than the DSSCs with the random polyaniline network or Pt cathodes. Consequently, the power conversion efficiency of DSSCs based on a typical D–π–A organic dye sensitizer increases from 5.97% for the polyaniline random network cathode to 8.24% for the oriented polyaniline nanowires array cathode, which is also higher than the efficiency (6.78%) of the DSSC with the Pt cathode.
Introduction
Dye-sensitized solar cells (DSSCs) have been attracting increasing attention over the last two decades due to their advantages over silicon based solar cells such as low production costs and environmental friendliness.1 Solar cell performance, including efficiency and long-term stability, has been improved steadily through considerable efforts devoted to the development of new materials such as sensitizers,2–5 electrolytes6–10 and anodes,11–13 and improvement in fundamental understanding.14,15 The typical redox couple often used in DSSCs is I3−/I−, but its redox potential is not positive enough for the generation of higher photovoltage and hence higher power conversion efficiency.16 Other redox couples with more positive redox potentials are favorable for increasing the photovoltage.17–19 Recently, the highest efficiency (12.3%) for DSSCs was achieved using the Co(bpy)33+/Co(bpy)32+ (bpy = 2,2′-bipyridine) redox couple in conjunction with a combination of dyes as sensitizers and Pt as the cathode.18 It has been demonstrated that the use of polypyridine complexes of Co(III)/Co(II) in conjunction with donor (D)–π-bridge–acceptor (A) organic dyes is a promising route to achieve high photovoltage and hence power conversion efficiency19 as compared to the combination of the famous N719 dye20 and the I3−/I− redox couple.Although Pt is a preferred material for the counter electrode in DSSCs due to its outstanding conductivity, electrocatalytic activity and stability, its low abundance ratio and high cost prevent Pt from being used for the large-scale manufacturing of DSSCs, which has stimulated great efforts to develop substitutes for Pt in order to reduce the overall cost and simultaneously maintain or improve the performance of DSSCs. For this reason, various substitutes for expensive Pt have been developed for the purpose of reducing cost. Hitherto, a variety of low-cost cathode materials such as inorganic compounds,21 carbon materials22–28 and conducting polymers29,30 have been developed for use in DSSCs, and good performance has been achieved with some of them.
Among the Pt-free cathode materials, conducting polymers such as polyaniline (PANI) are also promising because of their low cost, high conductivity, good catalytic activity, and simple solution processing at low temperature. However, the current reported conducting polymer cathodes, usually prepared by a drop-casting or electro-polymerization method, show a random network,31 which limits the electrocatalytic activity on the reduction of oxidized species because a lot of boundaries are present in a random network. Thus, the photocurrent generation and hence power conversion efficiency of DSSCs are limited by using a random polymer film as the cathode. To overcome this problem, an array of oriented conducting polymer nanowires as the cathode is desirable since the electron transport along the polymer nanowires is expected to be faster than percolation in a random polymer network. With a sufficiently dense array of oriented polymer nanowires as the cathode in DSSCs, all the exposed polymer parts should be effective for the catalytic reduction of the oxidized species in the electrolyte due to the fast electron transport along the wire. Therefore, enhancement of the photocurrent and the fill factor are anticipated with an oriented polymer nanowires array as the cathode of DSSCs.
Although oriented PANI nanowires arrays have been prepared via template synthesis methods,32,33 complex post-processing is needed to remove the template. The intrinsic electronic properties could be affected, and the aligned nanostructure could also be destroyed to some extent. Therefore, the development of an easy, template-free and scalable one step method to generate aligned PANI nanowires on substrates is desired, considering their fascinating electronic and electrochemical applications.34 In this study, we first present an array of oriented PANI nanowires grown in situ on a conductive glass (fluorine-doped tin oxide, FTO) substrate as an efficient cathode for DSSCs, and illustrate how this oriented array improves the electrocatalytic activity and the performance of DSSCs with a D–π–A structured metal-free organic dye (FNE29, Fig. S1†) as the sensitizer and the Co(III)/Co(II) redox couple (Fig. S2†) in acetonitrile as the electrolyte. The PANI nanowires array demonstrates high catalytic performance for the reduction of the Co(III) species compared to the drop-cast PANI film with a random network and Pt. Using the PANI nanowire array as the cathode, a power conversion efficiency of 8.24% has been achieved under irradiation of simulated AM 1.5G solar light (100 mW cm−2). This efficiency is much higher than that obtained with a drop-cast PANI film with a random network (5.97%) and that obtained with the Pt cathode (6.78%) under the same conditions.
Results and discussion
PANI nanowires can be grown in situ on FTO glass, as detailed in the Experimental section (see ESI†). The surface morphologies of the PANI films grown in situ on FTO with a fixed growth time of 24 h are sensitive to the initial concentration of aniline, as revealed by the field emitting scanning electronic microscopy (FESEM) images (Fig. 1). Vertical nanowires were formed when the initial concentration of aniline increased from 2 to 11 mM (Fig. 1(a)–(c)). However, when the initial concentration of aniline was 50 mM or higher, the nanowires agglomerated into an interconnected network (Fig. 1(d) and (e)). | ||
| Fig. 1 Tilted views of FESEM images of PANI films obtained from (a) 2 mM, (b) 6 mM, (c) 11 mM, (d) 50 mM, and (e) 200 mM aniline with a growth time of 24 h. | ||
The different surface morphologies of the resultant PANI films can be explained by the heterogeneous nucleation mechanism, as discussed elsewhere.34,35 Generally, there are two possible nucleation sites for the growth of polyaniline in chemical oxidative polymerization, the bulk solution and the solid substrate. For polymerization in concentrated aniline solution, homogeneous nucleation in the bulk solution is dominant. Consequently, individual PANI nanowires pack very densely and merge with each other. In contrast, for polymerization in dilute solution, heterogeneous nucleation occurs first on the solid substrate. As a consequence, most active nucleation centers are generated on the solid substrate at the beginning of the polymerization. These active sites would minimize the interfacial energy barrier for the subsequent vertical growth of polyaniline on the solid substrate. When the initial concentration of aniline increases from 2 to 11 mM, more active nuclei are generated on the FTO substrate so that more nanowires are formed, resulting in a dense nanowires array. However, when the initial concentration of aniline is 50 mM or higher, the nanowires are so dense that they tend to agglomerate into an interconnected network.
At an initial concentration of 11 mM of aniline, the time dependent growth of PANI nanowires on FTO was also investigated. While the length of nanowires was hardly dependent on the growth time, the nanowires became denser with increasing growth time up to 24 h, as revealed by the FESEM images shown in Fig. 2. According to the above observations, the densest PANI nanowires array is obtained at the initial concentration of 11 mM of aniline with a growth time of 24 h. The length of the PANI nanowires was determined to be ∼340 nm, and the diameter was around 40 nm. For comparison, the FESEM image of the drop-cast PANI film is shown in Fig. S3,† where a random compact network is observed.
 | ||
| Fig. 2 Cross-sectional views of FESEM images of PANI films obtained from 11 mM aniline after (a) 3 h, (b) 8 h, (c) 14, (d) 18 h, and (e) 24 h. | ||
The in situ grown oriented PANI nanowires film was highly uniform and translucent with a green colour (Fig. S4†), which was caused by the doping of hydrochloric acid for the purpose of achieving high conductivity. The Fourier Transform Infrared (FTIR) spectrum for the oriented PANI nanowire array is shown in Fig. S5.† The bands at 1622 and 1520 cm−1 are due to quinoid and benzenoid ring deformation, respectively.36 The peak at 1358 cm−1 is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching near a quinoid ring, while the peak at 1193 cm−1 is assigned to the C–N stretching in a secondary aromatic amine.36 The C–H out-of-plane bending located at 845 cm−1 is indicative of para-substitution.36 The FTIR data confirms the successful formation of polyaniline. The crystal structure of the formed PANI nanowires powder was characterized by X-ray diffraction (XRD), as shown in Fig. S6.† Two characteristic XRD peaks located at 19.3° and 25.0° were observed. The XRD peak at 25.0° is attributed to the plane-to-plane stacking of benzene rings in PANI.37 The high peak intensity indicates that the formed PANI nanowires have good crystallinity, which is crucial to achieving high conductivity along the wire.
N stretching near a quinoid ring, while the peak at 1193 cm−1 is assigned to the C–N stretching in a secondary aromatic amine.36 The C–H out-of-plane bending located at 845 cm−1 is indicative of para-substitution.36 The FTIR data confirms the successful formation of polyaniline. The crystal structure of the formed PANI nanowires powder was characterized by X-ray diffraction (XRD), as shown in Fig. S6.† Two characteristic XRD peaks located at 19.3° and 25.0° were observed. The XRD peak at 25.0° is attributed to the plane-to-plane stacking of benzene rings in PANI.37 The high peak intensity indicates that the formed PANI nanowires have good crystallinity, which is crucial to achieving high conductivity along the wire.
The above PANI films were employed as cathodes to construct DSSCs. It was found that the photovoltaic performance was largely influenced by the film morphology. Table 1 summarizes the photovoltaic performance of DSSCs based on PANI films obtained from different initial concentrations of aniline. The change in short-circuit photocurrent density (Jsc) that accompanied the change in initial concentration was more remarkable than the changes in open-circuit photovoltage (Voc) or fill factor (FF). Jsc and FF increased gradually as the initial concentration of aniline increased from 2 to 11 mM, and then decreased gradually with further increase in the concentration. As a consequence, the power conversion efficiency (η) increased upon increasing the initial concentration of aniline up to 11 mM, and then decreased with further increase in the concentration.
| Cathodesa | V oc (V) | J sc (mA cm−2) | FF | η (%) | R s (Ω) | R ct (Ω) |
|---|---|---|---|---|---|---|
| a The cathodes were obtained from different initial concentrations of aniline as listed in the column, with a growth time of 24 h. The relative deviations of the photovoltaic parameters were less than 5%. The electrolyte for the DSSCs was 0.22 M [Co2+(bpy)3](PF6)2, 0.05 M [Co3+(bpy)3](PF6)3, 0.1 M LiClO4, and 0.2 M tert-butylpyridine in acetonitrile. The electrolyte for the dummy cells was 0.22 M [Co2+(bpy)3](PF6)2, and 0.05 M [Co3+(bpy)3](PF6)3 in acetonitrile. | ||||||
| 2 mM | 0.75 | 13.22 | 0.64 | 6.35 | 5.31 | 1.12 |
| 6 mM | 0.77 | 14.32 | 0.68 | 7.50 | 5.32 | 0.69 |
| 11 mM | 0.77 | 14.89 | 0.71 | 8.14 | 5.42 | 0.59 |
| 50 mM | 0.76 | 13.89 | 0.67 | 7.07 | 5.54 | 0.91 |
| 200 mM | 0.75 | 13.29 | 0.66 | 6.58 | 5.69 | 1.01 |
The photovoltaic performance also depended on the growth time of the PANI film. Table 2 summarizes the photovoltaic performances of DSSCs with PANI cathodes obtained with different growth times. Jsc and FF increased with growth time up to 24 h (Table 2) and then decreased a little (Table S1†) with further increase in the growth time. Accompanying the increase in Jsc, Voc also increased with growth time up to 24 h because of the increased Jsc. The highest power conversion efficiency was thus obtained at a growth time of 24 h.
| Cathodesa | V oc (V) | J sc (mA cm−2) | FF | η (%) | R s (Ω) | R ct (Ω) |
|---|---|---|---|---|---|---|
| a PANI cathodes were obtained after different growth times with an initial aniline concentration of 11 mM. The relative deviations of the photovoltaic parameters were less than 4%. | ||||||
| 3 h | 0.74 | 12.43 | 0.64 | 5.89 | 5.54 | 0.91 |
| 8 h | 0.75 | 14.31 | 0.66 | 7.08 | 5.41 | 0.83 |
| 14 h | 0.76 | 14.54 | 0.68 | 7.51 | 5.41 | 0.79 |
| 18 h | 0.77 | 14.96 | 0.69 | 7.95 | 5.42 | 0.66 |
| 24 h | 0.78 | 15.09 | 0.70 | 8.24 | 5.42 | 0.59 |
To clarify the reason why Jsc and FF were influenced by the surface morphology of the cathode, the electrochemical catalytic performance of various cathodes was investigated on symmetrical dummy cells, in which a thin layer of the redox electrolyte solution (the same as the electrolyte used in DSSCs, ESI†) was sandwiched between two identical electrodes to be used as cathodes in DSSCs. Fig. 3 shows the cyclic voltammetry (CV) curves of the symmetrical dummy cells with various PANI films grown from different initial concentrations of aniline or with different growth times. The CV curve exhibits a plateau of limiting current density (jL) in all cases, which is controlled by the mass transport of the redox species and the electrode property. For a redox electrolyte with fixed concentration and layer thickness, the limiting current depends on the electron transfer rate at the electrode–electrolyte interface. Therefore, a cathode with a higher catalytic activity can produce a higher limiting current. It can be seen from Fig. 3(a) that the catalytic activity increases with increasing the initial concentration of aniline up to 11 mM, and then decreases with further increase in the initial concentration of aniline. As the limiting current increases in the order of 2 mM < 200 mM < 50 mM < 6 mM < 11 mM (Fig. 3(a)) for PANI films obtained from different initial concentrations of aniline, the electrocatalytic activity of the PANI nanowire arrays increases in the same order. The PANI nanowire array obtained at 11 mM has the highest limiting current and hence the highest catalytic perfromance. For the PANI nanowire arrays obtained with different growth times, the limiting current increases with growth time up to 24 h (Fig. 3(b)), indicating that the catalytic performance becomes higher with growth time up to 24 h.
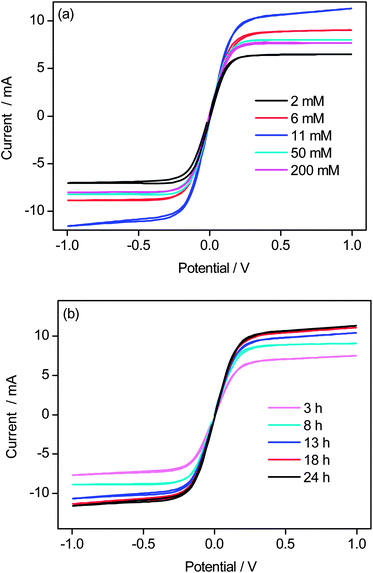 | ||
| Fig. 3 Cyclic voltammograms of dummy cells with Co(bpy)33+/2+ in acetonitrile as the electrolyte solution based on PANI films obtained from (a) different initial concentrations of aniline with a growth time of 24 h, and (b) different growth times at 11 mM initial concentration of aniline. Scan rate: 50 mV s−1. | ||
The electrocatalytic performance of various cathodes in the above dummy cells was also evaluated by electrochemical impedance spectroscopy (EIS). The impedance of a dummy cell can be fitted to the equivalent circuit38 shown in the inset of Fig. 4(a), where Rs is the ohmic series resistance, Rct is the charge transfer resistance, ZN is the Nernst diffusion impedance of the bulk electrolyte solution, and CPE is a constant phase element describing the deviation from the ideal capacitance due to the roughness of the electrodes. Two semicircles located in the higher (left) and lower (right) frequency regions are observed in the Nyquist plots (Fig. 4(a) and (c)). The left arc arises from the charge transfer at the cathode–electrolyte interface, and the right one stems from the diffusion of the electrolyte. Rs is estimated by the high-frequency intercept of the left arc on the real axis, while the Rct value is estimated by the real component radius of the fitted left arc. The estimated values of Rs and Rct are listed in Tables 1 and 2.
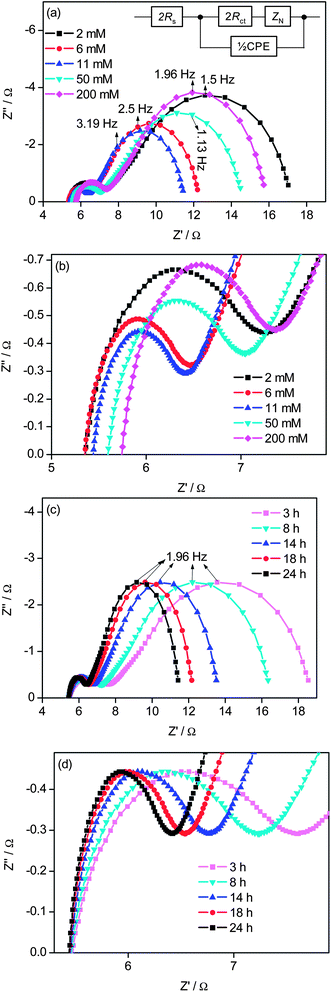 | ||
| Fig. 4 Electrochemical impedance spectra of dummy cells based on PANI films obtained from (a and b) different initial concentrations with growth time of 24 h, and (c and d) different growth times at 11 mM initial concentration of aniline. The inset in (a) is the equivalent circuit diagram for fitting the electrochemical impedance spectra. The Nyquist plots of the electrochemical impedance spectra were measured from 1 MHz to 0.1 Hz. | ||
R ct is inversely correlated with the electrocatalytic performance of the cathode on the reduction of the Co(III) species. For the PANI films obtained from different initial concentrations of aniline, Rct decreases from 2 to 11 mM and then increases with further increase in the initial concentration (Fig. 4(b), Table 1). These results indicate that the PANI film obtained from 11 mM aniline has the highest electrocatalytic activity in the investigated concentration range. For the PANI films obtained with different growth times, Rct decreases and electrocatalytic activity increases with growth time (Fig. 4(d), Table 2).
The CV and EIS results are consistent with the electrocatalytic performance of various PANI films. For the PANI nanowires arrays with a similar thickness, the difference in electrocatalytic performance should be explained by the different surface areas, but the specific surface area of the PANI samples could not be determined using the nitrogen sorption technique due to their decomposition above 150 °C.
For DSSCs with given dye-sensitized TiO2 and redox couple, Jsc depends on the electron collection efficiency, which is determined by the catalytic performance of the cathode. As the exchange current density (J0) of a cathode is related to the photocurrent density of a DSSC under illumination, a higher J0 signifies a higher Jsc for a DSSC.39 The J0 of a cathode can be calculated using the equation J0 = RT/nFRct, where n (= 1 here) is the number of electrons, R is the gas constant, T is the absolute temperature (here 298 K), and F is the Faraday constant. J0 is thus inversely proportional to Rct. Evidently, the changing tendency of J0 (or 1/Rct) is consistent with the observed Jsc trend on changing the initial concentration of aniline or the growth time. Therefore, the difference in Jsc can be well explained by the catalytic performance of the various films.
For DSSCs, FF depends on the Rs and Rct values of the cathode. As the Rs values are similar for these films, the difference in FF for the different DSSCs mainly stems from the difference in Rct; a higher catalytic performance (or a lower Rct) corresponds to a higher FF. The trend of catalytic performance (or the reciprocal of Rct) is in good agreement with the trend of FF for the DSSCs with the PANI cathodes obtained from different initial concentrations of aniline or with different growth times, as shown in Tables 1 and 2.
Among the conditions tested above, the PANI nanowires array obtained from 11 mM aniline with a growth time of 24 h is the best, not only in electrocatalytic activity but also in solar cell performance. To emphasize the advantage of the PANI nanowires array as the cathode of DSSCs, we compared its catalytic performance with that of the drop-cast PANI film (the preparation method is shown in the Experiment section in the ESI†) having a random network and with that of Pt by recording the CV curves (Fig. 5(a)) and EIS spectra (Fig. 5(b)) of the corresponding dummy cells. The limiting current (Fig. 5(a)) increased in the order of drop-cast PANI film < Pt < PANI nanowires array, indicating that the catalytic activity increased in the same order. The Rs and Rct values were derived from Fig. 5(b) and are listed in Table 3. Rct increased in the order of PANI nanowires array < Pt < drop-cast PANI film (Table 3), further confirming that the catalytic performance increased in the order of drop-cast PANI film < Pt < PANI nanowires array. As expected, the catalytic performance of the PANI film was improved significantly when the morphology changed from a random network to an oriented nanowires array. Impressively, the oriented PANI nanowires array also showed higher catalytic performance than the typical Pt, indicating that the low-cost PANI nanowires array is very promising to replace the expensive Pt as the cathode of DSSCs.
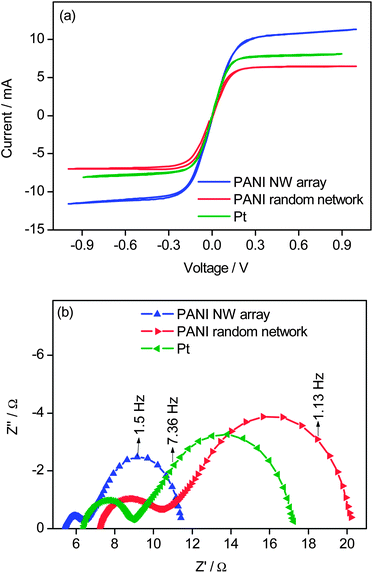 | ||
| Fig. 5 (a) Cyclic voltammograms of dummy cells based on a PANI nanowires array, a PANI random network, and Pt, with a scan rate of 50 mV S−1, and (b) EIS spectra of dummy cells with Co(bpy)33+/2+ in acetonitrile as the electrolyte solution based on a PANI nanowires array, a PANI random network, and Pt. The Nyquist plots of electrochemical impedance spectra were measured from 1 MHz to 0.1 Hz. The PANI nanowires array and random films had similar thicknesses. | ||
| Cathodes | V oc (V) | J sc (mA cm−2) | FF | η (%) | R s (Ω) | R ct (Ω) |
|---|---|---|---|---|---|---|
| a The PANI nanowires array was obtained from 11 mM initial concentration of aniline with growth time of 24 h. The relative deviations of the photovoltaic parameters were less than 5%. | ||||||
| Drop-cast PANI film | 0.72 | 12.76 | 0.65 | 5.97 | 7.23 | 1.68 |
| Pt | 0.76 | 13.11 | 0.68 | 6.78 | 6.37 | 1.29 |
| PANI nanowires array | 0.78 | 15.09 | 0.70 | 8.24 | 5.42 | 0.59 |
The electrochemical stability of the oriented PANI nanowires array and Pt cathodes were examined by repeated EIS measurements of the dummy cells, which were pre-treated by potential cycling before each EIS measurement, as displayed in Fig. 6. When this sequential test was repeated 10 times, Rct hardly changed for the PANI nanowires array (Fig. 6(a)), while Rct increased from 1.29 Ω to 1.70 Ω for the Pt cathode (Fig. 6(b)). Therefore, the PANI nanowires array had better electrochemical stability than the Pt cathode against potential cycling. For both cathodes, the series ohmic resistance and mass transportation in the redox electrolyte solution were hardly affected by the potential cycling, as revealed by the almost unchanged Rs and ZN.
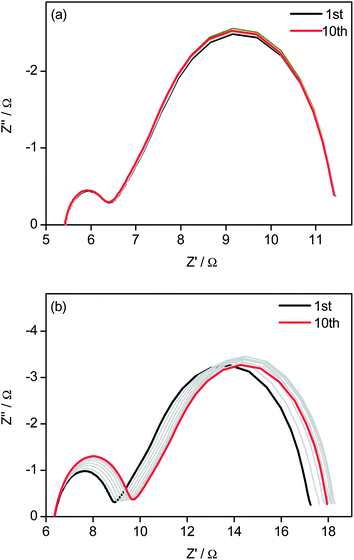 | ||
| Fig. 6 Potential cycling stability of the symmetric cells with (a) oriented PANI nanowires and (b) Pt electrodes. The dummy cells were first subjected to cyclic voltammetry scanning from 0 to 1 V and then from −1 to 0 V with a scan rate of 50 mV s−1, followed by 20 s relaxation at 0 V, and then EIS measurement at 0 V from 1 MHz to 0.05 Hz was performed. This sequence of electrochemical tests was repeated 10 times. | ||
Finally, it is of interest to compare the solar cell performance of the DSSCs based on the PANI nanowires array, drop-cast PANI with a random network and Pt cathodes, respectively. Fig. 7(a) shows the J–V curves, and the corresponding data is summarized in Table 3. With the PANI nanowire array grown from 11 mM aniline for 24 h as the cathode, the DSSC produced Jsc of 15.09 mA cm−2, Voc of 0.78 V, and FF of 0.70, corresponding to η of 8.24%. However, the drop-cast PANI film as the cathode produced a lower η of 5.97% (Jsc = 12.76 mA cm−2, Voc = 0.72 V, FF = 0.65). When the cathode was changed from the drop-cast PANI film to the oriented nanowires array, the Jsc increased from 12.76 to 15.09 mA cm−2 by 18%, Voc increased from 0.72 to 0.78 V, and FF increased from 0.65 to 0.70, resulting in a remarkable 38% improvement in η. The formation of the nanowires array should be responsible for the remarkable enhancement of Jsc and FF. As Pt is a typical cathode in DSSCs, the photovoltaic performance of a Pt based DSSC under the same conditions was also tested (Fig. 7(a)) for comparison. The cell with the Pt cathode produced η of 6.78% (Jsc = 13.11 mA cm−2, Voc = 0.76 V, FF = 0.68), which is better than that of the drop-cast PANI cathode but inferior to that of the PANI nanowires array. Evidently, the cell with the oriented PANI nanowire array cathode outperformed that with the Pt cathode in solar cell performance. As the apparent area of DSSCs is small (here 0.2304 cm2), the current passing through the cell is small, and therefore the observed series resistance below 20 Ω does not have a significant influence on solar cell performance.40 Therefore, the Jsc and FF are mainly influenced by Rct; higher catalytic performance (or lower Rct) results in higher Jsc and FF, and vice versa. For the three cathodes, the trend of catalytic performance is consistent with the trends of Jsc and FF.
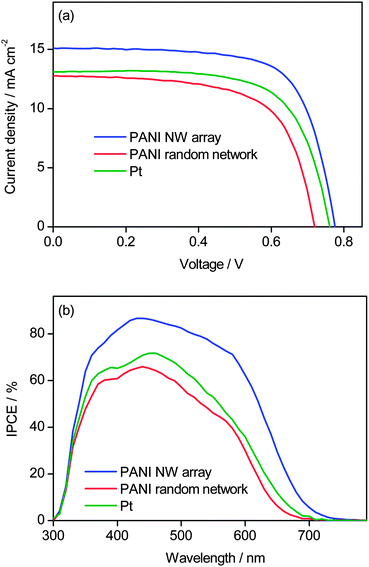 | ||
| Fig. 7 (a) Current density–voltage characteristics of DSSCs based on the PANI nanowires array, drop-cast PANI film and Pt cathodes, respectively, under simulated AM 1.5G solar light (100 mW cm−2), and (b) IPCE spectra of the above cells. | ||
As this work focuses on cathodes with novel morphology, we did not optimize the efficiency by screening dye sensitizers, which is very important for light harvesting and hence photocurrent generation. We believe that the efficiency can be improved when a better dye with a broader absorption spectrum is used in this system.
Fig. 7(b) shows the incident monochromatic photon-to-electron conversion efficiency (IPCE) measured at different wavelengths. The IPCE values in the visible range increase in the order of PANI film with a random network < Pt < oriented PANI nanowires array, which is consistent with the trend of Jsc. For the PANI nanowires array, the maximum IPCE reaches 87% at 440 nm, owing to its outstanding catalytic performance. As compared to the IPCE spectra for random PANI or Pt, the PANI array shows a red shift of the IPCE cut edge, which is likely attributed to the optical trapping effect of the nanostructured PANI array.41 The integrated photocurrents calculated from the overlap integral of the IPCE spectra with the standard AM 1.5G solar emission spectrum are 15, 10 and 11 mA cm−2 for the PANI array, random PANI and Pt, respectively. The calculated photocurrents for the devices based on random PANI or Pt are lower than the measured Jsc values shown in Fig. 7(a), while the calculated photocurrent is in good agreement with the measured Jsc for the PANI array. At present we do not know the reason for this.
Conclusions
In summary, an oriented PANI nanowire array has been successfully prepared in situ on FTO substrates and applied as a low-cost and efficient cathode material for DSSCs. The PANI nanowires array has a much higher electrocatalytic activity on the Co(III)/Co(II) redox couple than the PANI film with a random network, and even outperforms the often used Pt electrode. Using the oriented PANI nanowires array as the cathode, a Co(III)/Co(II) redox couple mediated DSSC with the FNE29 dye has achieved a power conversion efficiency of 8.24% under simulated AM 1.5G solar illumination (100 mW cm−2), which is higher than the efficiencies obtained using the drop-cast PANI film (5.97%) and Pt cathode (6.78%). The oriented PANI nanowires array can also be grown on flexible substrates such as flexible plastics and fibers, providing straightforward applications in flexible and fiber DSSCs.42 Our discovery expands the architectures of cathodes through the introduction of aligned one-dimensional nanostructures, and provides a paradigm for the fabrication of high-performance solar cells.Acknowledgements
This work was financially supported by the National Basic Research Program (no. 2011CB933302) of China, the National Natural Science Foundation of China (20971025, 90922004, and 50903020), STCSM(12JC1401500), the Shanghai Pujiang Program (11PJ1401700), the Jiangsu Major Program (BY2010147), the Shanghai Leading Academic Discipline Project (B108), the Project sponsored by SRF for ROCS, SEM, and the Jiangsu Major Program (BY2010147).Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- H. Choi, C. Baik, S. O. Kang, J. Ko, M.-S. Kang, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 327–330 CrossRef CAS.
- T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218–12219 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- K.-L. Wu, C.-H. Li, Y. Chi, J. N. Clifford, L. Cabau, E. Palomares, Y.-M. Cheng, H.-A. Pan and P.-T. Chou, J. Am. Chem. Soc., 2012, 134, 7488–7496 CrossRef CAS.
- H. X. Wang, H. Li, B. F. Xue, Z. X. Wang, Q. B. Meng and L. Q. Chen, J. Am. Chem. Soc., 2005, 127, 6394–6401 CrossRef CAS.
- Y. Bai, Y. M. Cao, J. Zhang, M. K. Wang, R. Z. Li, P. Wang, S. Zakeeruddin and M. Grätzel, Nat. Mater., 2008, 7, 626–630 CrossRef CAS.
- X. Liu, W. Zhang, S. Uchida, L. Cai, B. Liu and S. Ramakrishna, Adv. Energy Mater., 2010, 22, 150–155 Search PubMed.
- M. K. Wang, N. Chamberland, L. Breau, J.-E. Moser, R. H. Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389 CrossRef CAS.
- A. M. Spokoyny, T. C. Li, O. K. Farha, C. W. Machan, C. She, C. L. Stern, T. J. Marks, J. T. Hupp and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 5339–5341 CrossRef CAS.
- H. Zhang, Y. Wang, D. Yang, Y. Li, H. Liu, P. Liu, B. J. Wood and H. Zhao, Adv. Mater., 2012, 24, 1598–1603 CrossRef CAS.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS.
- O. K. Varghese, M. Paulose and C. A. Grimes, Nat. Nanotechnol., 2009, 4, 592–597 CrossRef CAS.
- T. L. Bahers, F. Labat, T. Pauporté, P. P. Lainé and I. Ciofini, J. Am. Chem. Soc., 2011, 133, 8005–8013 CrossRef.
- S. Y. Huang, G. Schlichthörl, A. J. Nozik, M. Grätzel and A. J. Frank, J. Phys. Chem. B, 1997, 101, 2576–2582 CrossRef CAS.
- T. W. Hamann and J. W. Ondersma, Energy Environ. Sci., 2011, 4, 370–381 CAS.
- J. H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J. E. Moser, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2012, 3, 631 CrossRef.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS.
- F. Gong, H. Wang, X. Xu, G. Zhou and Z.-S. Wang, J. Am. Chem. Soc., 2012, 134, 10953–10958 CrossRef CAS.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS.
- J. Han, H. Kim, D. Y. Kim, S. M. Jo and S.-Y. Jang, ACS Nano, 2010, 4, 3503–3509 CrossRef CAS.
- K. Suzuki, M. Yamaguchi, M. Kumagai and S. Yanagida, Chem. Lett., 2003, 32, 28–29 CrossRef CAS.
- J. E. Trancik, S. C. Barton and J. Hone, Nano Lett., 2008, 8, 982–987 CrossRef CAS.
- J. D. Roy-Mayhew, D. J. Bozym, C. Punckt and I. A. Aksay, ACS Nano, 2010, 4, 6203–6211 CrossRef CAS.
- L. Kavan, J. H. Yum, M. K. Nazeeruddin and M. Grätzel, ACS Nano, 2011, 5, 9171–9178 CrossRef CAS.
- F. Gong, Z. Li, H. Wang and Z.-S. Wang, J. Mater. Chem., 2012, 22, 17321–17327 RSC.
- S. Ahmad, J.-H. Yum, Z. X. Xi, M. Grätzel and H.-J. Butta, J. Mater. Chem., 2010, 20, 1654–1658 RSC.
- Q. Tai, B. Chen, F. Guo, S. Xu, H. Hu, B. Sebo and X.-Z. Zhao, ACS Nano, 2011, 5, 3795–3799 CrossRef CAS.
- Q. Li, J. Wu, Q. Tang, Z. Lan, P. Li, J. Lin and L. Fan, Electrochem. Commun., 2008, 10, 1299–1302 CrossRef CAS.
- L. Liang, J. Liu, C. F. Windisch, G. J. Exarhos and Y. Lin, Angew. Chem., Int. Ed., 2002, 41, 3665–3668 CrossRef CAS.
- B. K. Kuila and M. Stamm, J. Mater. Chem., 2010, 20, 6086–6094 RSC.
- N. R. Chiou, C. Lu, J. Guan, L. J. Lee and A. J. Epstein, Nat. Nanotechnol., 2007, 2, 354–357 CrossRef CAS.
- N. R. Chiou and A. J. Epstein, Adv. Mater., 2005, 17, 1679–1683 CrossRef CAS.
- Y. Furukawa, F. Ueda, Y. Uyodo, I. Harada, T. Nakajima and T. Kawagoe, Macromolecules, 1988, 21, 1297–1305 CrossRef CAS.
- S. K. Pillalamarri, F. D. Blum, A. T. Tokuhiro and M. F. Bertino, Chem. Mater., 2005, 17, 5941–5944 CrossRef CAS.
- A. Hauch and A. Georg, Electrochim. Acta, 2001, 46, 3457–3466 CrossRef CAS.
- L. Kavan, J. H. Yum and M. Grätzel, Nano Lett., 2011, 11, 5501–5506 CrossRef CAS.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Seró and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118 RSC.
- E. Garnett and P. Yang, Nano Lett., 2010, 10, 1082–1087 CrossRef CAS.
- X. Fan, Z. Chu, F. Wang, C. Zhang, L. Chen, Y. Tang and D. Zou, Adv. Mater., 2008, 20, 592–595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2ta00705c |
| This journal is © The Royal Society of Chemistry 2013 |