An ester-functionalized diketopyrrolopyrrole molecule with appropriate energy levels for application in solution-processed organic solar cells†
Received
31st August 2012
, Accepted 26th September 2012
First published on 27th September 2012
Abstract
For highly efficient organic solar cells (OSCs), the electron donor should possess not only a narrow band gap (Eg) but also a low highest occupied molecular orbital (HOMO) energy level. To achieve it, in this paper, we designed and synthesized a diketopyrrolopyrrole (DPP) derivative end capped with an ethyl thiophene-2-carboxylate moiety, 3,6-bis{5-[(ethyl thiophene-2-carboxylate)-2-yl]thiophene-2-yl}-2,5-bis(2-ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-dione (DPP(CT)2). Through UV-vis absorption and cyclic voltammetry (CV) measurements, we demonstrated that the resulting molecule exhibits both a low optical Eg of 1.65 eV and a lower-lying HOMO energy level of −5.33 eV owing to the electronegativity of the ester group and the conjugation effect of the thiophene ring. Therefore, when DPP(CT)2 is used as the electron donor to blend with [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) for solution processable OSCs, a power conversion efficiency (PCE) of 4.02% combined with an open-circuit voltage (VOC) as high as 0.94 V and a broad photovoltaic response range extending to around 750 nm is obtained.
Introduction
Recently, organic solar cells (OSCs) have been evolving into promising clean and renewable energy sources because they can be fabricated into large-area and flexible devices by low-cost solution-processing.1–3 In general, OSCs feature a bulk heterojunction structure, where a p-type organic semiconductor (either conjugated polymer or small molecule) and a fullerene derivative (e.g. [6,6]-phenyl-C71-butyric acid methyl ester, abbreviated as PC71BM) function as the electron donor and acceptor, respectively. To achieve a higher power conversion efficiency (PCE) of OSCs, an ideal p-type organic semiconductor should possess both a low band gap (Eg) and a low highest occupied molecular orbital (HOMO) energy level: a low Eg will favor the efficient harvest of solar photons at longer wavelengths, giving a larger short-circuit current density (JSC) of OSCs; and a low HOMO energy level can increase the open-circuit voltage (VOC) of OSCs since VOC is proportional to the difference between the HOMO of the donor and the lowest unoccupied molecular orbital (LUMO) of the acceptor.4 To meet the above requirements in the past five years a lot of new p-type conjugated polymers have been reported, and the best OSCs based on conjugated polymers show PCEs of more than 8%.5–7 In addition to the conjugated polymers, small molecule donors have also been widely examined with significant progress having been made.8–14 However, the development of the OSCs based on small molecule donors still lags behind their polymer-based counterparts.
Small molecule donors possess particular advantages over conjugated polymers, such as well-defined molecular structures, definite molecular weights, and high purity without batch to batch variations.8–14 Diketopyrrolopyrrole (DPP) is one of the most attractive units for the design of high-performance p-type organic photovoltaic materials since DPP contains a well-conjugated core including two electron-deficient carbonyl groups.8–11,15,16 When it is copolymerized or linked with an electron-rich moiety (e.g. thiophene), low Eg polymers or small molecules can be obtained, but their HOMOs are relatively high.9,16 Instead, if DPP is attached to an electron-withdrawing unit or an aromatic unit (e.g. benzofuran and pyrene),8,10 the HOMO of the resulting molecule decreases in favor of a higher VOC, while the Eg becomes undesirably wider. To solve this problem, in this paper, we designed and prepared an ester-functionalized DPP derivative, 3,6-bis{5-[(ethyl thiophene-2-carboxylate)-2-yl]thiophene-2-yl}-2,5-bis(2-ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-dione (DPP(CT)2). It is expected that the attachment of two thiophene-2-carboxylate units to the DPP core can deepen the HOMO of DPP(CT)2 since the ester group is a weak electronegative substituent and, due to its small amount of steric hindrance, the ester group hardly influences on the conjugation between the thiophene and DPP core, achieving a narrowed Eg.17–22 Then, the optical, electrochemical and photovoltaic properties of this new molecule were investigated, in order to verify the feasibility of the above molecular design.
Results and discussion
Synthesis and thermal property of DPP(CT)2
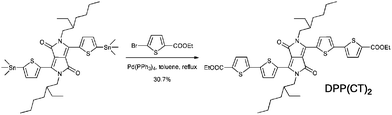
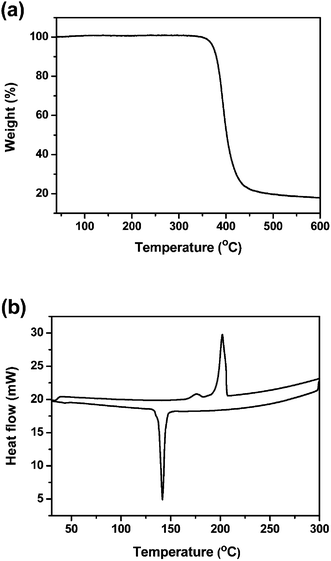
The target molecule DPP(CT)2 was synthesized via a traditional Pd(0)-catalyzed Stille coupling reaction, shown in Scheme 1. The thermal properties of DPP(CT)2 were investigated with thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) in a nitrogen atmosphere, as outlined in Fig. 1. It was found that DPP(CT)2 exhibits a thermal decomposition temperature (Td), with a 5% weight loss, of 359 °C and a melting point (Tm) of more than 100 °C, indicating that the obtained DPP molecule modified with ester groups is still thermally stable enough for its application in small molecule-based solar cells.
 |
| | Scheme 1 Synthetic route to DPP(CT)2. | |
 |
| | Fig. 1 Thermal properties of DPP(CT)2: (a) TGA curve; (b) DSC curve. The heating rate was 20 °C min−1. | |
Energy levels of DPP(CT)2
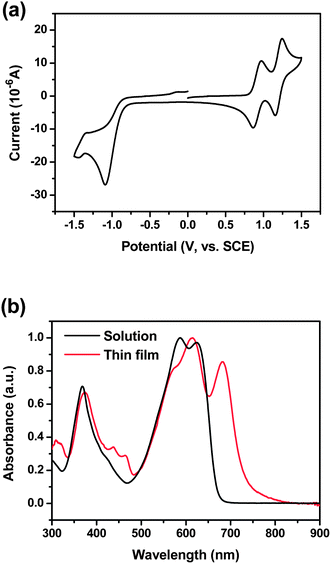
Cyclic voltammetry (CV) measurements were done to characterize the energy level structure of DPP(CT)2. From the onset oxidation potential (0.93 V versus SCE) and the onset reduction potential (−0.80 V versus SCE), presented in Fig. 2a, the HOMO and LUMO energy levels of DPP(CT)2 are obtained as −5.33 and −3.60 eV, respectively. Fig. 2b depicts the UV-visible absorption spectra of DPP(CT)2 in CH2Cl2 solution and as a thin film. It is observed that, compared to the solution, the thin film exhibits an additional absorption peak centered at 682 nm, indicating strong intermolecular π–π overlapping in the condensed state.10 It is exciting to observe that this absorption peak locates exactly at the wavelength of the maximum photon flux of solar irradiation (∼680 nm),23 and from its absorption band-edge (λonset, 752 nm), the optical band gap (Egopt) of DPP(CT)2 is calculated as 1.65 eV. Compared with two previously reported DPP derivatives with benzofuran and pyrene as end-groups that have the same HOMO levels at −5.20 eV and different optical band gaps of 1.80 and 1.70 eV, respectively,8,10 DPP(CT)2 possesses a smaller Egopt and a lower HOMO level, demonstrating the rationality of our molecular design.
Geometry and front orbitals of DPP(CT)2
To expose the effect of the thiophene-2-carboxylate moiety on the energy levels of DPP(CT)2, the molecular conformations and front orbitals of DPP(CT)2 and the two DPP derivatives with benzofuran and pyrene as the end-groups, respectively, in the free states, were simulated by theoretical calculations. The simulated front orbitals, energy levels and dihedral angles between the end-groups and DPP core are presented in Fig. 3 and Table 1. As shown in Fig. 3 and Table 1, for DPP(CT)2, due to the negligible steric repulsion of the ester group, the two thiophene rings of the end-groups exhibit excellent coplanarity and conjugation with the DPP core, thus, the Π-electrons are delocalized throughout the whole molecule, giving the narrowest Eg among the three DPP derivatives. In the mean time, the ester groups attached to the two thiophene rings endow DPP(CT)2 with the lowest HOMO energy level among the three DPP derivatives due to its electron-withdrawing ability. For the DPP derivative with benzofuran as the end-group, benzofuran rings also show good coplanarity but relatively worse conjugation with the DPP core, which can be ascribed to the aromaticity of the fused benzene rings, therefore, it provides a bigger Eg than DPP(CT)2. For the DPP derivative with pyrene as the end-group, pyrene rings show the worst coplanarity and conjugation with the DPP core, consequently, it presents the largest Eg among the three DPP derivatives. Because neither of the two DPP derivatives with benzofuran and pyrene as end-groups are end capped by electronegative substituents, both of them possess relatively higher HOMO energy levels compared to that of DPP(CT)2. The above results are in good agreement with our experimental data, suggesting that the introduction of the thiophene carboxylate moiety is an effective strategy to achieve p-type organic semiconductors with favorable energy levels for photovoltaic applications.
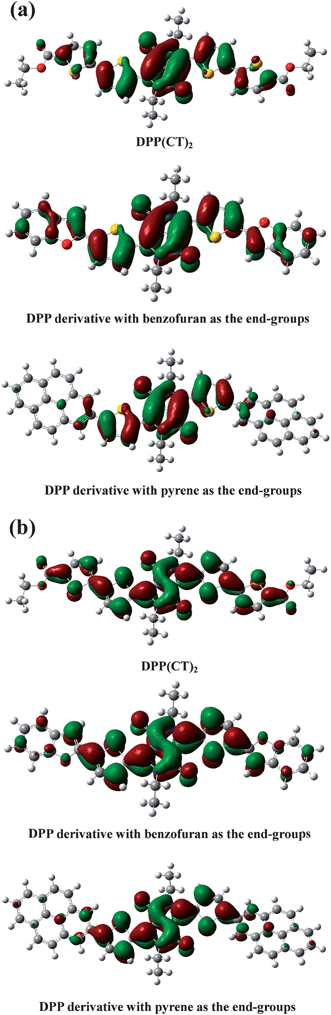 |
| | Fig. 3 HOMO (a) and LUMO (b) of DPP(CT)2 and the two DPP derivatives with benzofuran and pyrene as the end-groups. | |
Table 1 Energy levels and dihedral angles of three DPP derivatives obtained by theoretical simulations
| Molecule |
HOMO (eV) |
LUMO (eV) |
E
g
(eV) |
Dihedral angles between the two end-groups and DPP core (°) |
| DPP(CT)2 |
−5.35 |
−3.24 |
2.11 |
5.27; 5.27 |
| DPP derivative with benzofuran as the end-groups |
−5.19 |
−3.06 |
2.13 |
9.45; 9.47 |
| DPP derivative with pyrene as the end-groups |
−5.14 |
−2.86 |
2.28 |
13.25; 36.55 |
Photovoltaic properties of DPP(CT)2
Because DPP(CT)2 has the appropriate energy levels for application in organic photovoltaics, OSCs with a device structure of ITO/PEDOT:PSS/DPP(CT)2:PC71BM/Al were fabricated to study the photovoltaic property of DPP(CT)2. Shown in Fig. 4a are the J–V characteristics of the devices under AM 1.5G illumination at an intensity of 100 mW cm−2, and all the photovoltaic data is summarized in Table 2. From Fig. 4a, it is found that, the as-cast device with a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 DPP(CT)2:PC71BM blend weight ratio gives an outstanding VOC of 1.02 V, but a small JSC of 3.31 mA cm−2 and a low fill factor (FF) of 0.34, and thereby a poor PCE of 1.14%. After thermal annealing at 90 °C for 10 min, the photovoltaic performance of the device with the same DPP(CT)2:PC71BM blend ratio improves tremendously: the JSC increases by 1.5 times to 8.55 mA cm−2, the FF reaches a relatively high value of 0.50, and the VOC drops slightly to 0.94 V, thus, a maximum PCE of 4.02% was achieved. When the concentration of DPP(CT)2 in the active layer was changed to 50 and 70 wt%, it was observed that, although the VOC remains unchanged, both the JSC and FF of the devices decrease to some extent, providing the reduced PCEs of 3.11% and 3.36%, respectively. Fig. 4b presents external quantum efficiency (EQE) curves of the OSCs. It is apparent that the shape of the EQE spectra resembles that of DPP(CT)2's absorptions, except that the photovoltaic response in the region of 300–550 nm was elevated, which is attributed to the absorptions of PC71BM.8 It was also found that thermal annealing results in a remarkable increase of EQE values covering the whole absorption range, displaying a moderate value of 45% for a 50:50 ratio, a maximum value of 55% for a 60:40 ratio, and a slightly lower value of 50% for the 70:30 ratio at around 630 nm. These results are consistent with the varying trend of the JSC illustrated in Fig. 4a.
:40 DPP(CT)2:PC71BM blend weight ratio gives an outstanding VOC of 1.02 V, but a small JSC of 3.31 mA cm−2 and a low fill factor (FF) of 0.34, and thereby a poor PCE of 1.14%. After thermal annealing at 90 °C for 10 min, the photovoltaic performance of the device with the same DPP(CT)2:PC71BM blend ratio improves tremendously: the JSC increases by 1.5 times to 8.55 mA cm−2, the FF reaches a relatively high value of 0.50, and the VOC drops slightly to 0.94 V, thus, a maximum PCE of 4.02% was achieved. When the concentration of DPP(CT)2 in the active layer was changed to 50 and 70 wt%, it was observed that, although the VOC remains unchanged, both the JSC and FF of the devices decrease to some extent, providing the reduced PCEs of 3.11% and 3.36%, respectively. Fig. 4b presents external quantum efficiency (EQE) curves of the OSCs. It is apparent that the shape of the EQE spectra resembles that of DPP(CT)2's absorptions, except that the photovoltaic response in the region of 300–550 nm was elevated, which is attributed to the absorptions of PC71BM.8 It was also found that thermal annealing results in a remarkable increase of EQE values covering the whole absorption range, displaying a moderate value of 45% for a 50:50 ratio, a maximum value of 55% for a 60:40 ratio, and a slightly lower value of 50% for the 70:30 ratio at around 630 nm. These results are consistent with the varying trend of the JSC illustrated in Fig. 4a.
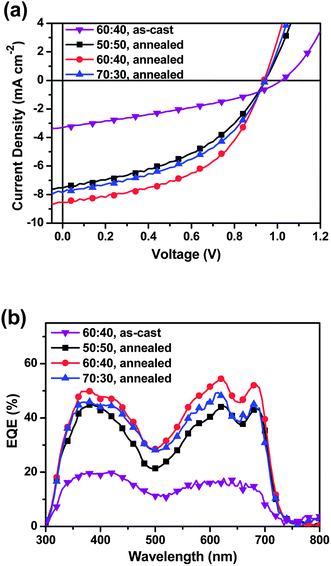 |
| | Fig. 4 (a) J–V characteristics and (b) external quantum efficiency spectra of DPP(CT)2:PC71BM based OSCs under illumination of AM 1.5G, 100 mW cm−2. | |
Table 2 Photovoltaic performances of the OSCs based on DPP(CT)2
| Active layer |
Blend ratio |
V
OC (V) |
J
SC (mA cm−2) |
FF |
PCE (%) |
|
These devices are annealed at 90 °C for 10 min.
As-cast.
|
| DPP(CT)2:PC71BMa |
50:50 |
0.94 |
7.53 |
0.44 |
3.11 |
| DPP(CT)2:PC71BMa |
60:40 |
0.94 |
8.55 |
0.50 |
4.02 |
| DPP(CT)2:PC71BMa |
70:30 |
0.94 |
7.84 |
0.46 |
3.36 |
| DPP(CT)2:PC71BMb |
60:40 |
1.02 |
3.31 |
0.34 |
1.14 |
Charge carrier mobilities of the active layers
To interpret the dependence of the photovoltaic performances on thermal annealing and the DPP(CT)2:PC71BM blend ratio, the space charge limited current (SCLC) method was employed to determine the charge carrier mobilities of the active layers prepared under different conditions. Devices with a structure of ITO/PEDOT:PSS/DPP(CT)2:PC71BM/MoO3/Al were fabricated to measure the hole mobilities while other devices with a structure of ITO/Al/DPP(CT)2:PC71BM/Al were fabricated for measuring the electron mobilities. Fig. 5a and b show the J0.5–V characteristics of the hole-only and electron-only devices, respectively,24,25 and the mobility values are summarized in Table 3. From Fig. 5a, the hole mobility of the as-cast 60:40 film was calculated as 7.44 × 10−6 cm2 V−1 s−1. Upon annealing, the hole mobilities do not change notably and almost remained constant with an increasing donor concentration: the hole mobilities of the annealed 50:50, 60:40 and 70:30 films were 8.68 × 10−6, 9.64 × 10−6 and 9.57 × 10−6 cm2 V−1 s−1, respectively. The above results suggest that the good hole-transporting ability of the blended films originates from the strong intermolecular π–π stacking of DPP(CT)2, and it is basically independent on blend ratio and thermal annealing, because the annealing temperature of 90 °C is much lower than the melting point (Tm) of DPP(CT)2 (refer to Fig. 1b). However, from Fig. 5b, it is observed that thermal annealing and the blend ratio can greatly affect the electron mobilities of the active layers. After thermal annealing, the electron mobility of the 60:40 film increased by an order of magnitude from 1.66 × 10−7 to 2.19 × 10−6 cm2 V−1 s−1. With increasing the fraction of PC71BM in the blended films, the electron mobility was enhanced significantly: the electron mobilities of the annealed 50:50 and 70:30 films were 1.91 × 10−5 and 1.69 × 10−6 cm2 V−1 s−1, respectively. These phenomena are reasonable. As disclosed by previous studies, thermal annealing would lead to larger fullerene domains in the active layer of OSCs, facilitating efficient electron transportation.26,27 It is also natural that a higher PC71BM concentration is more beneficial to form percolated electron transport pathways. Therefore, based on the above data, the better photovoltaic properties of the annealed blended films may be ascribed to their better balanced hole and electron mobilities.
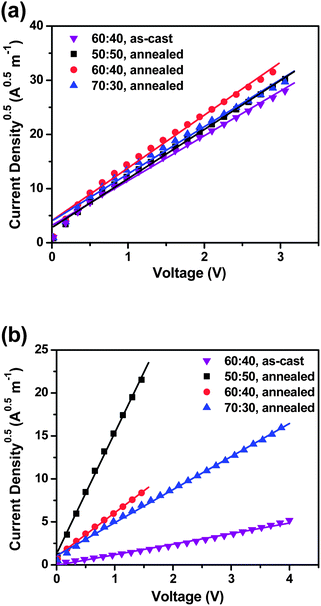 |
| | Fig. 5
J
0.5–V characteristics of (a) the hole-only and (b) electron-only devices based on DPP(CT)2:PC71BM blended films. The symbols represent the experimental data and the solid lines were fitted according to the Mott–Gurney Law. | |
Table 3 Charge carrier mobilities of the DPP(CT)2:PC71BM blended films
| Blend ratio |
μ
h (cm2 V−1 s−1) |
μ
e (cm2 V−1 s−1) |
|
The films were annealed at 90 °C for 10 min.
As-cast.
|
| 50:50a |
8.68 × 10−6 |
1.91 × 10−5 |
| 60:40a |
9.64 × 10−6 |
2.19 × 10−6 |
| 70:30a |
9.57 × 10−6 |
1.69 × 10−6 |
| 60:40b |
7.44 × 10−6 |
1.66 × 10−7 |
Morphologies of the active layers
This hypothesis is also supported by the morphologies of the DPP(CT)2:PC71BM films probed with atomic force microscopy (AFM) (Fig. 6). It can be seen from Fig. 6a that the as-cast 60:40 film displays an obvious donor–acceptor phase separation with large DPP(CT)2 domains in the scale of 45–70 nm, and the PC71BM domains are not well interconnected. After annealing, with a slightly bigger DPP(CT)2 domains (50–80 nm), a percolated acceptor network forms in the annealing 60:40 film because the size of the PC71BM domains became larger (Fig. 6c), which agrees well with the significant improvement of the electron mobility. For the annealing 50:50 film, the biggest phase separation domains (80–110 nm) were observed (Fig. 6b), which were responsible for the poorest photovoltaic property among the three annealed films. When the content of DPP(CT)2 in the blended film was increased to 70 wt%, the DPP(CT)2 domains isolate the PC71BM domains again (Fig. 6d), which led to the lowest electron mobility among the three annealed films, as demonstrated in the above section. Therefore, the best photovoltaic performance of the annealed 60:40 film could be attributed to more efficient exciton dissociation and charge transportation in the active layer. It should be noted that, all DPP(CT)2:PC71BM films, regardless of thermal annealing and blend ratio, present continuous hole pathways composed of large DPP(CT)2 domains in the range of 45–110 nm, which grants good hole-transporting ability, but inevitably disrupts efficient exciton dissociation in the active layer because the exciton diffusion length is typically 10–20 nm. It is thought that the large DPP(CT)2 domains result from the bad miscibility between DPP(CT)2 and PC71BM since the end-group of ethyl ester in DPP(CT)2 is linear and short. Further molecular modification of DPP(CT)2 is necessary to achieve a much finer phase separation in the active layer.
 |
| | Fig. 6 AFM images of DPP(CT)2:PC71BM blended films: (a) as-cast 60:40 film, (b) annealed 50:50 film, (c) annealed 60:40 film and (d) annealed 70:30 film. | |
Conclusions
In summary, an ester group end-capped DPP molecule, DPP(CT)2, was successfully synthesized and demonstrated to be a promising donor for solution processable OSCs. Owing to its narrow band gap as well as lower-lying HOMO, the DPP(CT)2-based OSCs exhibited a broad photovoltaic response range extending to around 750 nm and a very high VOC of 0.94 V, providing a PCE of 4.02%. The results also imply that the molecular structure of DPP(CT)2 should be optimized to improve PCE through tuning the morphology of the active layer. Such work is underway.
Experimental section
Instrument
1H-NMR and 13C-NMR spectra were measured using a Bruker Advance DMX 300 (300 MHz) nuclear magnetic resonance spectroscope. UV-visible absorption spectra were taken on a Shimadzu UV-2450 spectrophotometer. Elemental analyses were conducted on a Flash EA 1112 elemental analyzer. Thermogravimetric analysis (TGA) was carried out on a WCT-2 thermal balance under protection of nitrogen at a heating rate of 20 °C min−1. Differential scanning calorimetry (DSC) was recorded on a Perkin-Elmer Pyris 1 differential scanning calorimeter. Cyclic voltammetry (CV) was done on a CHI 660C electrochemical workstation with a Pt disk, Pt plate and standard calomel electrode (SCE) as the working electrode, counter electrode and reference electrode, respectively, in a 0.1 mol L−1 tetrabutylammonium hexafluorophosphate (Bu4NPF6) CH2Cl2 solution. Topographic images of the films were obtained on a Veeco MultiMode atomic force microscopy (AFM) in the tapping mode using an etched silicon cantilever at a nominal load of ∼2 nN, and the scanning rate for a 5 μm × 5 μm image size was 1.0 Hz.
Materials
All the chemicals were purchased from Aldrich, Acros and TCI Chemical Co. and used as received if not specified otherwise. Toluene was dried over Na/benzophenone and freshly distilled prior to use. Ethyl 5-bromothiophene-2-carboxylate was purchased from Adamas Chemical Co. and used without further purification. 3,6-bis[(5-(trimethylstannyl)thiophen-2-yl)]-2,5-bis(2-ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-dione were synthesized according to the published procedures.17
Theoretical calculations
Geometry optimizations were carried out by the density functional theory (DFT) method at the B3LYP/6-31G(d) level. Energies of front orbitals (HOMOs and LUMOs) were evaluated with the basis set of 6-311+G(d,p). All the calculations were performed using the Gaussian 03 program. Model compounds of the three DPP derivatives were simplified by replacing pendant alkyl side chains with ethyl groups to save computational time due to the fact that the geometries and energies negligibly depended on the pendant alkyl groups.28
Device fabrication and characterization
Organic solar cells were fabricated on commercially available glass substrates with a layer of indium tin oxide (ITO) as the anode, aluminum (Al) metal as the cathode and a blended film of DPP(CT)2:PC71BM sandwiched between the two electrodes as the active layer. Prior to device fabrication, the ITO-coated glass substrates were cleaned using detergent, deionized water, acetone and isopropanol consecutively every 15 min, and then treatment in an ultraviolet ozone generator for 15 min before being spin-coated with aqueous poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) dispersions (Baytron P AI4083). After baking the PEDOT:PSS in air at 140 °C for 15 min, the substrates were transferred to a glovebox. The active layer was spin-cast at 1500 rpm from a solution of DPP(CT)2 and PC71BM in chlorobenzene at a total solid concentration of 20 mg mL−1. Then the sample was annealed at 90 °C for 10 min. Subsequently, samples were loaded into a vacuum deposition chamber (background pressure ≈ 5 × 10−4 Pa) to deposit a 100 nm thick aluminum cathode with a shadow mask (the device area was 9 mm2). The current density–voltage (J–V) curves under illumination were measured with Keithley 236 measurement source units at room temperature in air. The photocurrent was measured under a calibrated solar simulator (Abet 300 W) at 100 mW cm−2 and the light intensity was calibrated with a standard photovoltaic (PV) reference cell. External quantum efficiency (EQE) spectrum was measured with a Stanford lock-in amplifier 8300 unit.
The charge carrier mobility of the blends was measured using the space-charge-limited current (SCLC) method. Hole-only devices were fabricated in a structure of ITO/PEDOT:PSS/DPP(CT)2:PC71BM/MoO3(10 nm)/Al(100 nm) and electron-only devices were fabricated in a structure of Al(120 nm)/DPP(CT)2:PC71BM/Al(100 nm). The device characteristics were extracted by modelling the dark current under forward bias using the SCLC expression described by the Mott–Gurney law:
| | 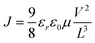 | (1) |
Here,
εr ≈ 3 is the average dielectric constant of the blended film,
ε0 is the permittivity of the free space,
μ is the carrier mobility,
L ≈ 70 nm is the thickness of the film and
V is the applied voltage.
Synthesis
The synthetic route to DPP(CT)2 is shown in Scheme 1. The detailed synthetic process is as follows.
Synthesis of 3,6-bis{5-[(ethyl thiophene-2-carboxylate)-2-yl]thiophene-2-yl}-2,5-bis(2-ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-dione (DPP(CT)2).
Compound 3,6-bis[(5-(trimethylstannyl)thiophen-2-yl)]-2,5-bis(2-ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-dione (1.37 g, 1.6 mmol) and ethyl 5-bromothiophene-2-carboxylate (0.933 g, 4.0 mmol) were put into a two-neck 100 mL round flask and were purged three times with successive vacuum and nitrogen fill cycles. Then 30 mL of dry degassed toluene was added under the protection of nitrogen and the solution was flushed with nitrogen before the addition of 70 mg of tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4). The reaction mixture was further degassed and subsequently sealed before being heated to 110 °C for 48 h. Removal of the solvent on a rotary evaporator afforded the crude product, which was purified using column chromatography (silica gel) with dichloromethane (CH2Cl2) and petroleum mixture (6:1, v/v) to give a dark blue powder (0.40 g, yield 30.7%). δH (300 MHz; CDCl3; Me4Si): 8.91 (d, 2H, J = 4.1 Hz), 7.72 (d, 2H, J = 3.9 Hz), 7.39 (d, 2H, J = 3.9 Hz), 7.26 (d, 2H, J = 4.1 Hz), 4.37 (q, 4H, J = 7.1 Hz), 3.95–4.10 (m, 4H), 1.82–1.97 (m, 2H), 1.21–1.46 (m, 22H), 0.82–0.98 (m, 12H). δC (300 MHz; CDCl3; Me4Si): 161.99, 161.73, 142.49, 141.57, 139.65, 136.87, 134.38, 133.73, 129.76, 126.34, 125.36, 109.09, 61.70, 46.12, 39.52, 30.58, 28.77, 23.92, 23.33, 14.56, 14.30, 10.79. Anal. calcd for C44H52N2O6S4: C, 63.43; H, 6.29; N, 3.36. Found: C, 63.54; H, 6.27; N, 3.21%.
Acknowledgements
The authors would like to gratefully acknowledge the financial support from the National Natural Science Foundation of China (nos. 50990063, 51073135, 51011130028). The work was also partly supported by National High Technology Research and Development Program of China (863 Program) (no. 2011AA050520).
Notes and references
- R. F. Service, Science, 2011, 332, 293 CrossRef CAS.
- B. Walker, C. Kim and T. Q. Nguyen, Chem. Mater., 2011, 23, 470–482 CrossRef CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- H. Y. Chen, J. H. Hou, S. Q. Zhang, Y. Y. Liang, G. W. Yang, Y. Yang, L. P. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- Z. C. He, C. M. Zhong, X. Huang, W. Y. Wong, H. B. Wu, L. W. Chen, S. J. Su and Y. Cao, Adv. Mater., 2011, 23, 4636–4643 CrossRef CAS.
- X. H. Li, W. C. H. Choy, L. J. Huo, F. X. Xie, W. E. I. Sha, B. F. Ding, X. Guo, Y. F. Li, J. H. Hou, J. B. You and Y. Yang, Adv. Mater., 2012, 24, 3046–3052 CrossRef CAS.
- B. Walker, A. B. Tamayo, X. D. Dang, P. Zalar, J. H. Seo, A. Garcia, M. Tantiwiwat and T. Q. Nguyen, Adv. Funct. Mater., 2009, 19, 3063–3069 CrossRef CAS.
- A. B. Tamayo, X. D. Dang, B. Walker, J. Seo, T. Kent and T. Q. Nguyen, Appl. Phys. Lett., 2009, 94, 103301 CrossRef.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. J. Fréchet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS.
- S. Loser, C. J. Bruns, H. Miyauchi, R. P. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 8142–8145 CrossRef CAS.
- Z. Li, G. R. He, X. J. Wan, Y. S. Liu, J. Y. Zhou, G. K. Long, Y. Zuo, M. T. Zhang and Y. S. Chen, Adv. Energy Mater., 2012, 2, 74–77 Search PubMed.
- Y. M. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44–48 CAS.
- T. S. van der Poll, J. A. Love, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646–3649 CrossRef CAS.
- E. J. Zhou, Q. S. Wei and K. Hashimoto, Macromolecules, 2010, 43, 821–826 CrossRef CAS.
- P. M. Beaujuge, H. N. Tsao, M. R. Hansen, C. M. Amb, C. Risko, J. Subbiah, K. R. Choudhury, A. Mavrinskiy, W. Pisula, J. L. Brédas, F. So, K. Müllen and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 8944–8957 Search PubMed.
- X. L. Hu, L. J. Zuo, W. F. Fu, T. T. Larsen-Olsen, M. Helgesen, E. Bundgaard, O. Hagemann, M. M. Shi, F. C. Krebs and H. Z. Chen, J. Mater. Chem., 2012, 22, 15710–15716 RSC.
- Z. G. Zhang and J. Z. Wang, J. Mater. Chem., 2012, 22, 4178–4187 RSC.
- L. J. Huo, T. L. Chen, Y. Zhou, J. H. Hou, H. Y. Chen, Y. Yang and Y. F. Li, Macromolecules, 2009, 42, 4377–4380 CrossRef CAS.
- X. L. Hu, M. M. Shi, J. Chen, L. J. Zuo, L. Fu, Y. J. Liu and H. Z. Chen, Macromol. Rapid Commun., 2011, 32, 506–511 CAS.
- M. M. Shi, L. Fu, X. L. Hu, L. J. Zuo, D. Deng, J. Chen and H. Z. Chen, Polym. Bull., 2012, 68, 1867–1877 CrossRef CAS.
- M. Pomerantz, Y. Cheng, R. K. Kasim and R. L. Elsenbaumer, J. Mater. Chem., 1999, 9, 2155–2163 RSC.
- J. H. Hou, M. H. Park, S. Q. Zhang, Y. Yao, L. M. Chen, J. H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- L. T. Dou, J. B. You, J. Yang, C. C. Chen, Y. J. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS.
- C. Shim, M. Kim, S. G. Ihn, Y. S. Choi, Y. Kim and K. Cho, Chem. Commun., 2012, 48, 7206–7208 RSC.
- W. L. Ma, C. Y. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- X. N. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579–583 CrossRef CAS.
- M. M. Shi, D. Deng, L. Chen, J. Ling, L. Fu, X. L. Hu and H. Z. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1453–1461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ta00148a |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.