Importance of small micropores in CO2 capture by phenolic resin-based activated carbon spheres†
Nilantha P.
Wickramaratne
and
Mietek
Jaroniec
*
Department of Chemistry and Biochemistry, Kent State University, Kent, Ohio 44242, USA. E-mail: jaroniec@kent.edu; Fax: +1-330-672-3816; Tel: +1-330-672-3790
First published on 18th October 2012
Abstract
Phenolic resin-based carbon spheres obtained by a slightly modified Stöber method are shown to be superior CO2 adsorbents. A direct KOH activation of polymeric spheres gave carbons with small micropores (<0.8 nm) and large specific surface area (2400 m2 g−1), which are able to adsorb an unprecedented amount of CO2 (up to 8.9 mmol g−1) at 0 °C and ambient pressure.
1. Introduction
Global warming is one of the most potential phenomena that may cause the extinction of some plants and animals in future. It is well known that global warming is mainly due to greenhouse gases such as water vapor, CO2, N2O and CH4.1 Among them CO2 is probably the main contributor to the greenhouse gas effects. In the last few decades the amount of CO2 in the atmosphere has increased dramatically. The main drivers of the CO2 increment in the atmosphere are vehicular emissions, fossil fuel-fired power plants, deforestation and chemical processes. It has been found that 86% of anthropogenic greenhouse gas emission comes from fossil fuel-fire power plants (coal, petroleum and natural gas), which amounted for ∼26 Gt in 2004.2 About 50% increment of CO2 emission from fossil fuel-fire power plants is expected by 2030.3 At present, most of the commercial CO2 capture is handled via absorption–regeneration technology involving amine-based scrubbing systems and cryogenic coolers.4 The aforementioned methods of reduction of CO2 emissions are energy intensive and not cost-effective. Another main disadvantage of these methods is the oxidative amine degradation leading to the corrosion of process equipment. Therefore, alternative technological solutions for carbon capture and storage (CCS) are urgently needed in order to control the CO2 emission.In the past decade, a variety of adsorbents for CO2 capture including zeolites,5 functionalized porous silica,6,7 metal–organic frameworks (MOFs)8,9 and carbonaceous materials10 have been intensively studied. Among them, nanoporous carbons and MOFs have gained much attention as potential sorbents for CO2 capture. Recently, Furukawa et al. reported that the MOFs studied exhibited exceptional CO2 adsorption capacities at high pressures (50 bar) and 25 °C, reaching in some cases 54.5 mmol g−1.11 However, the highest CO2 adsorption capacities reported for MOFs under ambient conditions did not exceed 8.5 mmol g−1.12 Despite the excellent adsorption capacities of MOFs, their preparation can be more time consuming and expensive in comparison to the synthesis of many carbonaceous adsorbents. Another major drawback of MOFs is their low stability towards water vapor. Küsgens and coworkers reported that MOFs can adsorb a large amount of water and not all water can be desorbed due to chemisorption.13 In contrast, carbonaceous adsorbents have several advantages such as high resistance to water due to their hydrophobicity, high thermal stability, low cost, good chemical resistance to both alkaline and acidic media, easy preparation and control of the pore structure, and low energy requirement for regeneration. Thus, carbonaceous materials can be considered as very promising adsorbents for CO2 capture.
Various types of carbon materials, such as activated carbons, metal–carbon composites, and nitrogen-doped carbons, have been studied for CO2 adsorption.14–19 In order to improve the CO2 adsorption capacity, extensive research has been done with a special emphasis on the development of high surface area microporous carbons and/or the incorporation of basic groups into carbons, mainly nitrogen-containing species. However, until now the later strategy resulted only in moderate enhancement of CO2 adsorption capacity under ambient conditions. For instance, Yang and coworkers reported a maximum CO2 adsorption capacity of 3.2 mmol g−1 under ambient conditions for N-incorporated carbons (with a nitrogen loading of 11.56 wt%).14 Also, recent studies have been focused on the synthesis of N-functionalized high surface area carbons for CO2 adsorption. Sevilla et al. and Xing et al. demonstrated that the high surface area N-containing carbons are suitable to capture larger amounts (3.9–4.0 mmol g−1) of CO2.15 Similarly, Wang et al. reported 4.0 mmol g−1 CO2 adsorption capacities for N-carbons.16 However, there are numerous studies showing that high surface area carbons with appropriate microporous structure can adsorb comparable CO2 amounts under ambient conditions. Recently, Silvestre-Albero and coworkers obtained high surface area carbon molecular sieves by KOH activation of petroleum pitch and achieved CO2 adsorption capacities up to 4.54 mmol g−1.17 Also, Presser et al.18 and Hu et al.19 studied the CO2 uptake on microporous carbons in relation to their pore size. Presser and coworkers18 showed that the CO2 uptake for the carbide-derived carbons (CDCs) at low pressures (<0.1 bar) and room temperature correlates with the volume of micropores smaller or equal to 0.5 nm; while the CO2 uptake under ambient conditions (1 bar) correlates with the volume of micropores smaller than 0.8 nm. Hu and coworkers19 examined activated petroleum coke and pointed out that the micropores smaller than 1 nm are responsible for high CO2 uptake at 1 bar. However, Wang and Yang correlated the CO2 uptake with the BET specific surface area.16
This short appraisal of the existing literature on CO2 adsorption on porous carbon shows that there is no definite consensus on the origin of high CO2 uptake by the aforementioned materials. This experimental work confirms earlier computer modeling studies20 showing that the fine tuning of carbon microporosity is needed to maximize the CO2 adsorption capacity; namely, it is shown that carbons with lower specific surface area but with appropriate microporosity can exhibit very high CO2 adsorption capacities. In contrast, Maxsorb carbon (with the BET surface area larger than 3300 m2 g−1) adsorbed only about 2.7 mmol g−1 of CO2 under ambient conditions.16
Here we show that a slightly modified Stöber recipe afforded phenolic resin-based carbon spheres with high fraction of small micropores (pores below 0.8 nm), which possessed high CO2 adsorption capacity under ambient conditions. Importantly, activation of the as-synthesized polymer spheres with KOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 w/w ratio) enlarged the aforementioned fine microporosity, which resulted in superior CO2 adsorption capacities reaching respectively 4.6 and 8.9 mmol g−1 at 23 and 0 °C under atmospheric pressure (1 bar). The latter value is the highest reported CO2 uptake for a microporous carbon at 0 °C and 1 bar and matches the value reported for KOH activated pitch-based carbons.17 This study provides experimental evidence for the importance of small micropores in CO2 capture; thus, the design of carbon adsorbents with high volume of small micropores is essential for achieving high CO2 uptake under ambient conditions. Note that carbons with larger micropores and much higher surface area such as Maxsorb show lower CO2 adsorption capacity16 than the reported phenolic resin-based carbons.
:4 w/w ratio) enlarged the aforementioned fine microporosity, which resulted in superior CO2 adsorption capacities reaching respectively 4.6 and 8.9 mmol g−1 at 23 and 0 °C under atmospheric pressure (1 bar). The latter value is the highest reported CO2 uptake for a microporous carbon at 0 °C and 1 bar and matches the value reported for KOH activated pitch-based carbons.17 This study provides experimental evidence for the importance of small micropores in CO2 capture; thus, the design of carbon adsorbents with high volume of small micropores is essential for achieving high CO2 uptake under ambient conditions. Note that carbons with larger micropores and much higher surface area such as Maxsorb show lower CO2 adsorption capacity16 than the reported phenolic resin-based carbons.
2. Experimental
2.1 Sample preparation
Monodisperse carbon spheres (CS) were synthesized using a slightly modified recipe reported by Liu et al.21 Two sets of resorcinol–formaldehyde (RF) spheres were synthesized at two different temperatures. The synthesis recipe used was as follows: an aqueous-alcoholic solution was prepared by mixing 16 mL of ethanol and 40 mL of distilled water at room temperature (23 °C; carbon samples denoted as CS series) and 30 °C (samples denoted as CS* series). Subsequently, 0.2 mL of 25 wt% ammonia was added under continuous stirring. Then, 0.4 g of resorcinol was added and stirred until its complete dissolution. Next, 0.6 mL of 37 wt% formaldehyde was added slowly to the solution and stirred for 24 h at respective temperatures. Finally, the reaction mixture was transferred to a 125 mL capacity Teflon container and placed in a sealed metal autoclave vessel. Then, the vessel was placed in an oven at 100 °C for 24 h. The solid product (polymer spheres) was obtained by centrifugation and dried at 100 °C for 12 h.The carbon spheres obtained by carbonization of the aforementioned polymer spheres at 600 °C were prepared as follows: the latter were carbonized under flowing nitrogen in a tube furnace using a heating rate of 1 °C min−1 up to 350 °C, left for 2 h, and resuming the heating rate of 1 °C min−1 up to 600 °C and left for 4 h. Another set of polymer spheres were obtained by thermal treatment at 300 °C, which was performed under flowing nitrogen in a tube furnace using a heating rate of 1 °C min−1 up to 300 °C, and left for 4 h.
The post-synthesis activation of carbon and polymer spheres was performed by impregnation of 0.1 g of sample with KOH solution (0.4 g of KOH dissolved in 1 mL of water) followed by thermal treatment. This treatment was carried out in a ceramic tube furnace under flowing nitrogen by heating the sample at a rate of 2 °C min−1 up to 100 °C and holding for 1 h, and resuming heating up to 700 °C with the rate of 5 °C min−1 and holding for 1 h at 700 °C. To assure a complete removal of KOH, the resulting samples were washed with 0.1 M HCl and deionized water under centrifugation. Finally, the activated samples were dried at 100 °C for 12 h.
The resulting carbon samples are listed in Table 1. The carbon spheres obtained by carbonization of polymer spheres at 600 °C are denoted by CS-6 and CS*-6, where * refers to the synthesis at 30 °C. Accordingly, the KOH activated polymer spheres are denoted by CS-P-A and CS*-P-A. The remaining carbon samples, denoted by CS-T-A and CS*-T-A, were prepared by KOH activation of carbon spheres CS and CS*, respectively, which were obtained at 300 (T = 3) and 600 °C (T = 6). Activation of all samples was performed at 700 °C.
| Sample | S BET (m2 g−1) | V t (cm3 g−1) | W (Å) | n CO2 (mmol g−1) | N% | |
|---|---|---|---|---|---|---|
| 23 °C | 0 °C | |||||
| a Notation: SBET – BET specific surface area; Vt – single-point pore volume, W – pore diameter at the maximum of the PSD curve obtained by the DFT method, nCO2 – CO2 adsorption capacity at 23 and 0 °C and 1 bar, N% – nitrogen percentage obtained by elemental analysis. | ||||||
| CS*-6 | 540 | 0.25 | 5.0 | 3.4 | — | 1.43 |
| CS*-P-A | 2400 | 1.07 | 5.4, 8.0, 12.3 | 4.6 | 8.9 | 0 |
| CS*-3-A | 1960 | 0.84 | 6.0, 8.5, 14.6 | 4.0 | 6.9 | 0 |
| CS-6 | 530 | 0.23 | 4.6 | 3.8 | — | 1.51 |
| CS-P-A | 1740 | 0.76 | 5.6, 8.5, 12.6 | 4.3 | 8.2 | 0 |
| CS-3-A | 2120 | 0.94 | 5.4, 8.2, 11.9 | 3.9 | 6.9 | 0 |
| CS-6-A | 600 | 0.27 | 5.7, 12.4 | 2.8 | — | 0.72 |
2.2 Characterization
TEM images were obtained using an FEI Tecnai F20ST/STEM instrument operated at 200 keV. The preparation of samples for TEM analysis involved their sonication in ethanol for 2 to 5 min and deposition on a 400 mesh lacy carbon coated copper grid. SEM images were obtained using a Hitachi Table top Microscope TM-1000. Nitrogen adsorption isotherms were measured at −196 °C on ASAP 2010 volumetric adsorption analyzers manufactured by Micromeritics (Norcross, GA, USA) using nitrogen of 99.998% purity. CO2 adsorption isotherms were obtained at both 0 and 23 °C on an ASAP 2020 volumetric adsorption analyzer manufactured by Micromeritics. Before adsorption measurements, each sample was degassed under vacuum for at least 2 h at 200 °C. The specific surface area of the samples was calculated using the Brunauer–Emmett–Teller (BET) method within the relative pressure range of 0.05–0.20. Pore size distributions were calculated from nitrogen adsorption data by the DFT method provided by Micromeritics. The thermogravimetric (TG) measurements were performed on a TA Instruments TGA Q500 thermogravimetric analyzer using a high-resolution mode. The TG curves were recorded in flowing air with a heating rate of 10 °C min−1 from 30 to 800 °C. The X-ray diffraction (XRD) measurements were carried out for the carbonized samples using a PANanalytical, Inc. X'Pert Pro (MPD) Multi-Purpose Diffractometer with Cu Kα radiation (1.5406 Å) at an operating voltage of 45 kV.3. Results and discussion
3.1 Surface area and pore size distributions
Recent reports have shown that microporous carbon materials with pores less than 1 nm are suitable for CO2 capture at ambient temperature and pressure.20,22 Interestingly, the recently extended Stöber method to the synthesis of carbons21 afforded spheres with micropores below 1 nm. This finding motivated us to explore the CO2 adsorption properties of these carbon spheres.Two batches of phenolic resin spheres (PS) were synthesized at room temperature (RT, 23 °C) and 30 °C using a slightly modified recipe reported by Liu et al.21 These polymeric spheres were subjected to thermal treatment under nitrogen to obtain carbon spheres (CS). Activated carbon materials (ACMs) with extremely high surface area were prepared from PS and CS using KOH activation. Nitrogen adsorption isotherms measured on these carbon materials at −196 °C are presented in Fig. 1; panels A and B show isotherms for carbons prepared at RT and 30 °C, respectively. All isotherms are type I, which is reflected by an adsorption plateau at low relative pressure, high amount of adsorbed nitrogen, and the absence of hysteresis, indicating that these materials are microporous. The specific surface area and pore structure parameters for these carbons are listed in Table 1. The surface area for CS-6-A is 600 m2 g−1. A relatively small increase in the surface area after KOH activation indicates that the activation process of CS-6 was not effective, which was caused by poor infiltration of relatively hydrophobic CS with aqueous solution of KOH; in this case, activation occurred mainly at the surface of CS. On the other hand, the KOH activation of PS and CS-3 produced highly microporous carbons (CS-P-A and CS-3-A) with surface areas of 2400 and 1960 m2 g−1, respectively. PS and CS-3 possess hydrophilic domains, which facilitate the process of infiltration of microporous spheres with KOH solution and consequently enhance their activation.
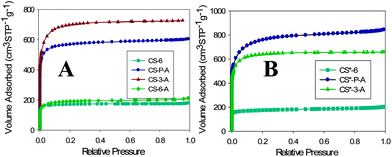 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms for carbons synthesized at (A) room temperature and (B) 30 °C. | ||
Pore size distribution (PSD) curves obtained from nitrogen adsorption by density functional theory (DFT) assuming slit-like pores are presented in the ESI, Fig. S1.† As can be seen from Fig. S1 (ESI†), CS-6 exhibits PSD centered at 0.5 nm, whereas the PSD curves for CS-3-A and CS-P-A show multiple peaks centered mainly at 0.5, 0.9 and 1.2 nm. This type of PSD curves is also observed for the CS* series. Interestingly, all carbon materials (CS and CS*) possess a significant fraction of micropores below 1 nm. The cumulative surface area and cumulative pore volume distributions are shown in Fig. 2A and B for the CS series, respectively. As can be seen from Fig. 2, the highest surface area for CS-P-A is observed for pores below 0.8 nm. However, in the case of CS-3-A the cumulative surface area is the highest in the range of larger micropores (above 0.8 nm) because of larger volume of these pores (Fig. 2). A similar behavior can be observed for the CS* carbons (ESI, Fig. S2†), although in this case CS*-P-A possessed the highest surface area in the whole range of micropores. The CO2 adsorption uptake follows the cumulative surface area and pore volume behavior up to 0.8 nm. This observation will be further discussed in the section devoted to CO2 adsorption.
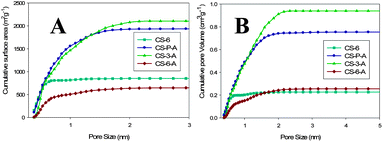 | ||
| Fig. 2 Cumulative surface area (A) and pore volume (B) plotted against the pore width for the CS series of carbons calculated from N2 adsorption by the DFT method. | ||
3.2 Morphology and phase structures
Transmission electron microscopy (TEM) was used to examine the particle morphology of the carbons studied. As can be seen in Fig. 3, the as-synthesized PS and CS-6 showed spherical morphology with the average diameter of 600 and 550 nm, respectively. The smaller diameter of CS-6 is due to the material shrinkage during carbonization. However, the KOH activated samples display flake-like morphology except for CS-6-A, indicating that the thermal treatment of polymer spheres in the presence of KOH resulted in structural disintegration. In contrast, the CS-6 sample is more stable and preserved the spherical morphology during KOH activation (see SEM images in Fig. S3, ESI†). The wide angle X-ray diffraction (XRD) patterns for the carbon samples obtained for the CS series are shown in Fig. S4 (see ESI†). Two broad and wide peaks at a 2θ of around 25 and 44 degrees are observed. Those broad diffraction peaks suggest that no pronounced graphitization occurred during the carbonization process. | ||
| Fig. 3 TEM images of (A) as-synthesized PS, (B) CS-600, (C) CS-3-A and (D) CS-P-A. | ||
3.3 CO2 adsorption
The CO2 adsorption capacities of microporous carbons were investigated at 0 and 23 °C under atmospheric pressure (1 bar). The CO2 adsorption isotherms measured under ambient conditions for both CS and CS* series are shown in Fig. 4. The CO2 adsorption capacities of the CS-6 and CS-3-A 23 °C are moderately high, 3.8 and 3.9 mmol g−1, respectively; whereas, the corresponding capacities of CS*-6 and CS*-3-A are 3.4 and 4.0 mmol g−1. As can be seen from Fig. 4 the CO2 capacities of the carbons obtained by direct activation of polymer spheres (CS-P-A and CS*-P-A) showed highest values, 4.3 and 4.6 mmol g−1, respectively. These CO2 adsorption capacities are very high in comparison to previously reported data for carbon materials under ambient conditions; the highest reported capacity under ambient conditions is 4.8 mmol g−1.22 Interestingly, the CS-P-A and CS*-P-A samples show ultra-high CO2 capacities at 0 °C and 1 bar. As can be seen from Fig. 4 the CO2 uptake capacities for the aforementioned carbons are 8.2 and 8.9 mmol g−1, respectively. It is noteworthy that these CO2 adsorption capacities are among the highest ever reported values for nanoporous carbons at 0 °C and 1 bar. To the best of our knowledge, the value of 8.9 mmol g−1 was not reported yet for carbons at 0 °C and 1 bar. It is also important to note that this value is higher than the CO2 capacities obtained for many MOFs at ambient pressure and 0 °C.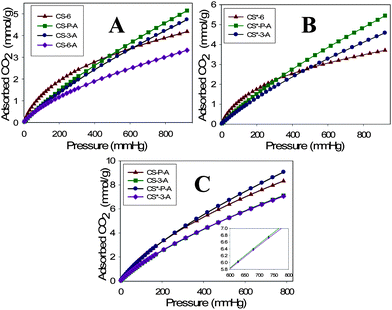 | ||
| Fig. 4 CO2 adsorption isotherms obtained at 23 °C for carbons synthesized at (A) room temperature and (B) 30 °C; and (C) CO2 adsorption isotherms obtained at 0 °C for selected carbon samples. | ||
The CO2 adsorption uptake at lower pressures purely depends on the fraction of fine micropores (∼0.5 nm). The PSD curves for CS-6 and CS*-6 show maxima at ∼0.46 and 0.5 nm, respectively. These samples showed very high adsorption up to 200 mmHg pressure, which indicates the importance of fine micropores in CO2 adsorption at low pressures. Also, it is noteworthy that CO2 adsorption capacities under ambient conditions are closely related to the cumulative micropore volume (PV0.8) calculated for pores up to 0.8 nm (see Table S1†). As can be seen from this table, the CO2 adsorption capacities are larger for the carbons with high PV0.8 volumes.
It is essential to show that CO2 is physically (reversibly) adsorbed on the carbon studied. The possible chemical adsorption of CO2 on these carbons could take place on nitrogen species and KOH residue. To check this issue elemental analysis (EA) and thermogravimetric analysis (TGA) were performed to determine N% and the amount of inorganic residue, respectively. The results of elemental analysis are shown in Table 1. As can be seen from this table, only CS-6 and CS-6-A samples contain N of ∼1.5 and 0.72%, respectively. The origin of this nitrogen is NH3 used as a catalyst for polymerization of resorcinol and formaldehyde. However, none of the activated samples possessed N except CS-6-A. It seems that the KOH activation of polymer and carbon spheres (CS-3) caused almost complete removal of nitrogen species. Since hydrophobic CS-6 spheres were poorly infiltrated with KOH, nitrogen species were not completely removed during the activation process. The excess KOH after activation was removed by acidic treatment. Thermo-gravimetric analysis (see Fig. S5, ESI†) of activated samples after acid treatment gave very low inorganic residues indicating residual KOH was effectively washed out (<1.0 weight%). Thus, the CO2 adsorption capacities of the activated carbons studied are mainly caused by the well-developed microporosity in the range below 0.8 nm.
4. Conclusions
This study shows that direct KOH activation of phenolic resin spheres obtained by a slightly modified Stöber method produces carbons with large fraction of micropores below 0.8 nm and high surface area, which exhibit superior CO2 adsorption capacities under ambient conditions. Namely, the CO2 uptake by these carbons can reach ∼8.9 mmol g−1 at 0 °C and 1 bar. This value is higher than the CO2 adsorption capacities reported so far for the most MOF samples and the highest ever reported value for carbonaceous materials. This superior CO2 capacity is mainly due to the presence of small micropores (<0.8 nm).Acknowledgements
The TEM images were obtained at the (cryo) TEM facility of the Liquid Crystal Institute, Kent State University, supported by the Ohio Research Scholars Program “Research Cluster on Surfaces in Advanced Materials”. The authors thank Dr Min Gao for technical support with the TEM experiments. N.W. wishes to acknowledge Dr Daniel Sherman at Saint-Gobain NorPro (Ohio) for permission to use a Table SEM for recording images of the samples studied during his CPT program.References
- R. D. Piacentini and A. S. Mujumdar, Drying Technol., 2009, 27, 629 CrossRef CAS.
- D. D'Alessandro, B. Smit and J. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef CAS.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS.
- S. E. Jee and D. S. Sholl, J. Am. Chem. Soc., 2009, 131, 7896 CrossRef CAS.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2009, 130, 2902 CrossRef.
- S. Choi, J. H. Drese, P. M. Eisenberger and C. W. Jones, Environ. Sci. Technol., 2011, 45, 2420 CrossRef CAS.
- K. S. Walton, A. R. Millward, D. Dubbeldam, H. Frost, J. J. Low, O. M. Yaghi and R. Q. Snurr, J. Am. Chem. Soc., 2008, 130, 406 CrossRef CAS.
- J. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H. Jeong, P. B. Balbuena and H. Zhou, Coord. Chem. Rev., 2011, 255, 1791 CrossRef CAS.
- G. Hao, W. Li, D. Qian and A. Lu, Adv. Mater., 2010, 22, 853 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS.
- A. O. Yazaydin, R. Q. Snurr, T. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 18198 CrossRef CAS.
- P. Küsgens, M. Rose, I. Senkovska, H. Fröde, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325 CrossRef.
- H. Yang, Y. Yuan and S. C. E. Tsang, Chem. Eng. J., 2012, 185–186, 374 CrossRef CAS.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781 CrossRef CAS; W. Xing, C. Liu, Z. Zhou, L. Zhang, J. Zhou, S. Zhuo, Z. Yan, H. Gao, G. Wang and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 7323 Search PubMed.
- L. Wang and R. T. Yang, J. Phys. Chem. C, 2012, 116, 1099 CAS.
- J. Silvestre-Albero, A. Wahby, A. Sepulveda-Escribano, M. Martinez-Escandell, K. Kaneko and F. Rodriguez-Reinoso, Chem. Commun., 2011, 47, 6840 RSC; A. Wahby, M. Ramos-Fernandez, M. Martinez-Escandell, A. Sepulveda-Escribano, J. Silvestre-Albero and F. Rodriguez-Reinoso, ChemSusChem, 2010, 3, 974 CrossRef CAS.
- V. Presser, J. McDonough, S. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059 CAS.
- X. Hu, M. Radosz, K. A. Cychosz and M. Thommes, Environ. Sci. Technol., 2011, 45, 7068 CrossRef CAS.
- Y. Liu and J. Wilcox, Environ. Sci. Technol., 2012, 46, 1940 CrossRef CAS.
- J. Liu, S. Z. Qiao, H. Liu, J. Chen, A. Orpe, D. Zhao and G. Q. Lu, Angew. Chem., Int. Ed., 2011, 50, 5947 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ta00388k |
| This journal is © The Royal Society of Chemistry 2013 |