 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ thioester formation for protein ligation using α-methylcysteine†
Fabienne
Burlina
abc,
George
Papageorgiou
d,
Caroline
Morris
d,
Peter D.
White
e and
John
Offer
*d
aUPMC Univ Paris 06, UMR 7203 Laboratoire des BioMolécules, 4 place Jussieu, 75005 Paris, France
bCNRS, UMR 7203 Laboratoire des BioMolécules, France
cENS, Département de Chimie, UMR 7203 Laboratoire des BioMolécules, Paris, France
dThe National Insitiute for Medical Research, The Ridgeway, London, UK. E-mail: joffer@nimr.mrc.ac.uk; Tel: +44 (0)20 8816 2082
eMerck Chemicals, Padge Road, Beeston, Notts, NG9 2JR, UK
First published on 20th November 2013
Abstract
The progress of total chemical protein synthesis has been hampered by difficulties in preparing peptide thioesters by standard Fmoc peptide synthesis. The amino acid, α-methylcysteine, sited at the C-terminus of a peptide can substitute for a thioester in peptide ligation reactions. C-terminal α-methylcysteine is fully compatible with Fmoc peptide synthesis and its use in ligation is very simple and robust. Its potential is demonstrated with the synthesis of model proteins.
Introduction
The peptide bond has a half-life in the order of 102 years,1 consequently, the peptide framework is generally considered to be very stable. There are celebrated exceptions, for example, the degradation of asparagine and aspartyl residues via an intramolecular mechanism.2 Intramolecularity is also responsible for the rapid hydrolysis of amide bonds observed for proximity assisted hydrolysis.3 Alternatively, torsional strain can raise the reactivity of amide bonds such as those in 2-quinuclidone,4 Kirby's amide,5 tetramethylpiperidine6 and others. These examples illustrate the dramatic increase in reactivity when the nitrogen lone pair is decoupled from resonance. Biologically relevant reactive amides include the core amide of penicillin as well as the splice junctions of inteins.7 The reactivity of amide bonds to thiols has also been extensively studied because of their biological relevance.8With the widespread adoption of native chemical ligation (NCL) many previously inaccessible protein targets have been synthesised.9,10 There is an emerging demand for proteins with post-translational modification (PTM) whose synthesis is not always compatible with the use of Boc protocols, although strategies are being developed to overcome this.11 The demand for deprotection conditions compatible with a full range of PTMs has driven the development of Fmoc/tBu compatible linkers for peptide thioester synthesis.12 The approaches used to prepare peptide thioesters with Fmoc protocols can be classified into two types, those that use a safety-catch approach, such as the sulfamylbutyryl approach of Kenner,13,14 the Dawson linker15 and hydrazide linker16 and those that exploit an intramolecular O,S or N,S acyl transfer step to transform a stable bond into a thioester.12b,17 Many of these linkers are converted to a thioester in a two-step reaction, rearrangement occurs irreversibly under strong acidic conditions followed by exchange to give a peptide thioester suitable for use in ligation.18 However, a subset of these linkers undergo acyl transfer under neutral conditions and can therefore be used directly in ligation. Botti and co-workers demonstrated O,S acyl transfer and ligation under neutral conditions.19N,S acyl transfer from tertiary amide bonds has recently been used for ligation.20,21 Furthermore, Macmillan and co-workers reported N,S acyl transfer and thioester exchange at cysteine sites in peptides using conditions of high temperature and acidic pH22 and recently demonstrated a peptide lactamization.23
We wanted a linker that would be fully compatible with conventional Fmoc peptide synthesis but would give a weakly activated C-terminus suitable for direct use in NCL after deprotection. Previous approaches have used N-substituted amides.20,21 However, we sought to avoid the difficulties involved in acylating sterically-hindered secondary amines and the propensity of N-substituted amino acids for diketopiperazine formation by investigating the effect on ligation of a Cα-substituted residue. The proposed, energetically unfavoured equilibrium at cysteine residues between amide and thioester at neutral pH, could perhaps be promoted by alkyl substitution of cysteine, providing a route for simultaneous N,S acyl transfer and ligation (Scheme 1a). The gem dimethyl effect, where alkyl substitution (not restricted to disubstitution) has been shown to be responsible for dramatic increases in rate observed in intramolecular reactions has been widely used in organic chemistry to improve cyclizations.24 Furthermore, the N,O homologue, isobucaine, possessed promising acyl transfer properties.25 We therefore investigated the acyl transfer properties of two Cα-substituted cysteine analogues; gem-dimethyl (gdm) and α-methylcysteine (Scheme 1a).
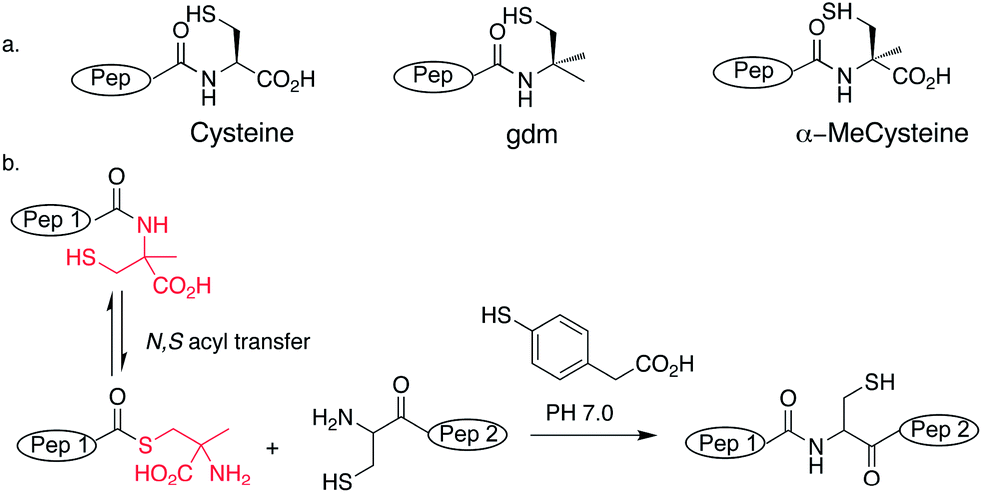 | ||
| Scheme 1 (a) Structures of cysteine analogues used. (b) Proposed mechanism of in situ thioester formation from α-methylcysteine and ligation. | ||
Results and discussion
The ‘gem-dimethyl’ (gdm) moiety (Scheme 1a) was synthesised and coupled onto the C-terminus of a short peptide sequence LAPAG (ESI†). LAPAG-gdm was added to neutral ligation buffer containing the peptide CFAPLV and the ligation product (LAPAGCFAPLV) was observed to form rapidly under NCL conditions and proceed to completion in 48 h (Fig. 1 and ESI, Fig. S1†). This demonstrated that introduction of gdm at the C-terminus of a peptide provided access to peptide thioesters via N,S acyl transfer. However, the strategy employed proved unwieldy as it was necessary to couple gdm to the protected peptide post-synthesis. Attempts to synthesize peptides using gdm modified trityl resin were unsuccessful possibly because of residual nucleophilicity of the trityl anchored sulphur atom; additionally there was extensive oxidation of the thioether to sulfoxide. We concluded that although the ligation properties of the gdm were encouraging its reactivity would have to be tempered for it to be stable as a linker during Fmoc solid phase synthesis. Additionally we noted that if TCEP was added to the ligation buffer, extensive desulfurisation occurred, as has been observed previously.26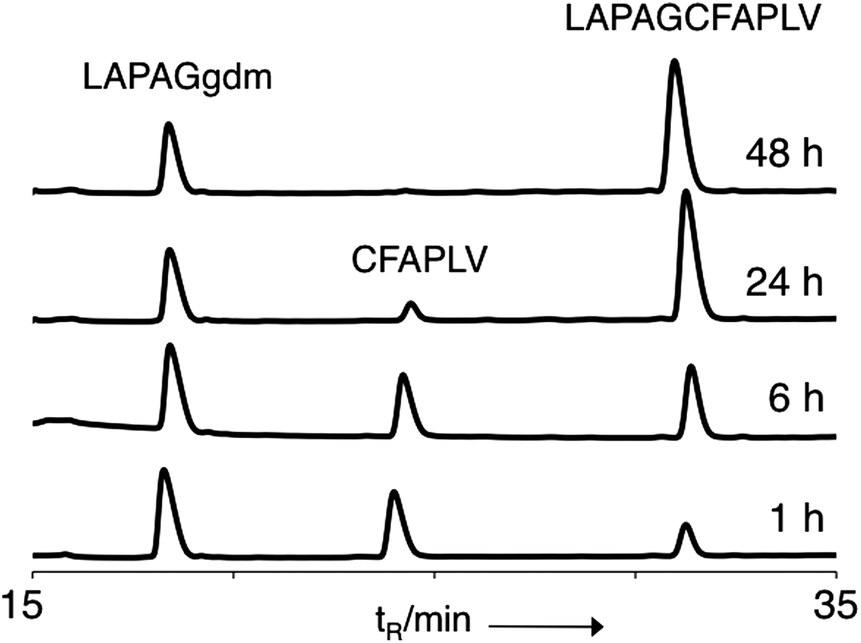 | ||
| Fig. 1 Ligation with gdm. Analytical HPLC time course of the reaction. Ligation conditions: [LAPAGgdm] = 10 mM, [CFAPLV] = 5 mM in phosphate buffer (200 mM; pH 7.5), 2 mM EDTA, 2% benzylmercaptan, 30 mM MPAA. | ||
When we considered alternatives to gdm, alpha amino acids appeared attractive for several reasons, chiefly, compatibility with standard SPPS conditions. We chose α-methylcysteine (Scheme 1), as it closely resembles gdm, with a methyl group replaced with a carboxyl group, both in the expectation that the reactivity of gdm could be tuned down to a more practical level and because the carboxylic group provided a more conventional site of attachment to the resin than sulfur. Trityl side-chain protected α-methylcysteine would be stable throughout a peptide synthesis, but potentially, after deprotection, N,S acyl transfer and ligation could occur. Substituting the acidic hydrogen of cysteine with a methyl group has additional benefits of both preventing β-elimination to dehydroalanine which causes low yields during Fmoc SPPS of C-terminal cysteine peptides.27 Furthermore, we were able to use standard coupling chemistry to subsequently acylate resin-attached α-methylcysteine. Although Cα-substituted amino acids can often be difficult to acylate because of increased steric hindrance,28 we coupled a range of amino acids to α-methylcysteine using standard automated peptide coupling procedures of DIC/HOBt and did not observe deletion sequences. The presence of an additional amino acid residue at the C-terminus wouldn't impair peptide solubility as is sometimes the case with thioesters. In the first instance Fmoc α-methylcysteine(Trt)-OH was loaded onto Wang resin using standard procedures (ESI†).29 2-Chlorotrityl resin was also loaded with α-methylcysteine. However, Wang resin gave better results for long peptides. Model peptides LAPAGαMeC-OH and LAPAAαMeC-OH were synthesised, both gave single species with correct mass on TFA cleavage. The peptides were added to ligation buffer with the test peptide CFAPRGKR-NH2 using a peptide concentration of 10 mM and neutral pH with 60 mM mercaptophenylacetic acid (MPAA) as the thiol additive and TCEP as reducing agent. In contrast to the gdm peptide, no desulfurisation was observed with the α-methylcysteine peptides in presence of TCEP. In both cases, the ligation product appeared rapidly as observed by analytical HPLC and the reactions proceeded cleanly to completion with no side reactions such as hydrolysis detected (ESI, Fig. S2 and S3†). Furthermore, no formation of peptide MPAA thioester was detected by HPLC suggesting that the rate-limiting step occurred during MPAA thioester formation. A control reaction performed in parallel under the same conditions with LAPAGC-OH gave only a minor amount of ligated product after 24 h, comparable to the amount of reaction product observed for LAPAGαMeC-OH after 10 min (ESI, Fig. S4†). This shows the importance of cysteine Cα-substitution for efficient N,S acyl transfer in ligation conditions.
These model reactions demonstrated the feasibility of performing ligation with C-terminal α-methylcysteine peptides. However, the peptide concentrations used were not representative of those generally used for protein synthesis. We therefore decided to examine the scope of this reaction with more challenging protein examples. Firstly we investigated the synthesis of bovine pancreatic trypsin inhibitor (BPTI), a benchmark for chemical protein synthesis14,30 and a target of interest for biophysical folding studies.31 BPTI has a full range of side-chain functional groups and a Gly–Cys ligation junction. In addition to employing α-methylcysteine we wanted to apply backbone protection in this synthesis, because it has been shown not only to assist peptide synthesis by preventing on-resin aggregation, but also to dramatically improve post-synthesis peptide solubility in aqueous solution.32 However, it has proven difficult to incorporate into N-terminal peptide thioesters using Boc SPPS.33 Hmb was incorporated into the N-terminal fragment at Gly28 using standard automated coupling. Acetylation of Hmb prevented its loss during acid deprotection and resin-cleavage, the acetyl group was in turn removed by the addition of aqueous hydrazine to the peptide in a neutral buffered solution, followed by HPLC purification. α-Methylcysteine, in contrast to a thioester, was stable to hydrazine treatment demonstrating its potential compatibility with hydrazine labile protecting groups. Ligation of the two pieces, HmbBPTI(1–37)αMeC and BPTI(38–58) proceeded cleanly to completion with MPAA as thiol additive at neutral pH (Fig. 2). This synthesis demonstrated the feasibility of protein synthesis with α-methylcysteine and was the first time backbone protection has been applied in a ligation.
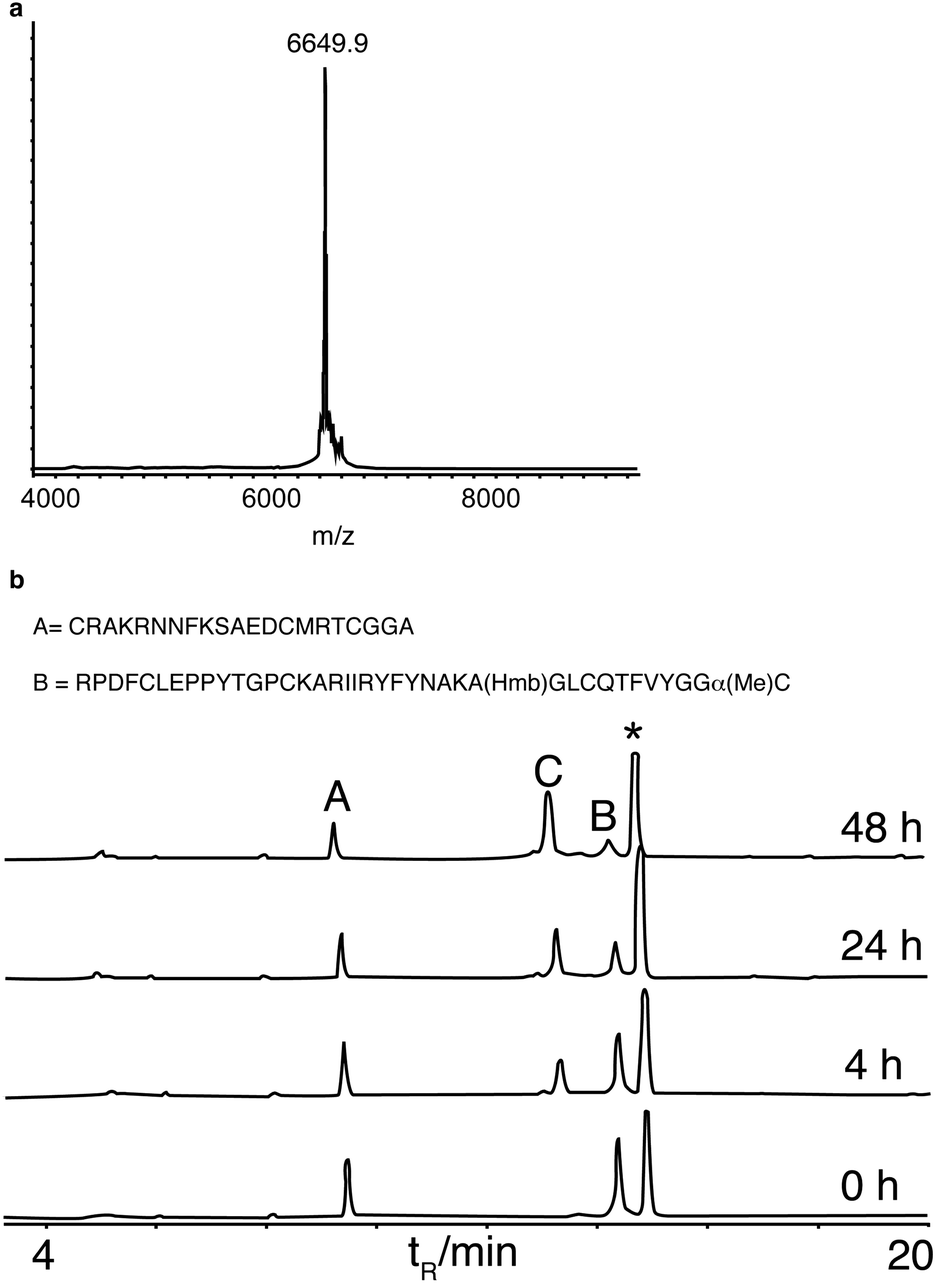 | ||
| Fig. 2 Synthesis of HmbBPTI. (a) MALDI-TOF mass spectrum of the ligation product (calculated average mass = 6650 Da). (b) Analytical HPLC time course of the ligation, A = BPTI(38–58), B = HmbBPTI(1–37)αMeC, C = ligation product,* MPAA. Ligation conditions: [A] = 3.25 mM, [B] = 2 mM in phosphate buffer (200 mM; pH 7.0), 2 mM EDTA, 6 M guanidine·HCl, 50 mM TCEP, 60 mM MPAA, 40 °C. HPLC conditions: RP-C18, 0–75% B (0.1% TFA, 10% H2O in CH3CN) in 23 min, 1 ml min−1. | ||
We next synthesised murine KC (mKC), the mouse functional homologue of interleukin 8, the first protein synthesised by NCL,34 to test the compatibility of α-methylcysteine peptides with more demanding ligation conditions. mKC has a central His–Cys site as a ligation junction. The N-terminal fragment aggregated above concentrations of 1 mM in ligation buffer containing 6 M guanidine. Therefore, ligation was attempted at a lower peptide concentration (0.4 mM). Ligation proceeded cleanly and went to completion as monitored by analytical HPLC (Fig. 3). Surprisingly, the ligation rate was similar to that of the model peptides at 10 mM and identical buffer conditions. This observation suggests that the reaction is dominated by a slow N,S unimolecular rearrangement with first order concentration dependence. This is in contrast to classical NCL conditions (using preformed peptide thioesters) that are very concentration dependent, presumably because of the rate limiting role of bimolecular thioester exchange.35 The ligation reactions using α-methylcysteine peptides were sluggish in comparison to NCL using preformed thioester peptides but no side reactions were detected. This reaction resembles the ‘low power’ peptide approach.36 In the low power philosophy, side reactions such as hydrolysis and epimerisation are minimised by using only weakly activated esters and a proximity assisted unimolecular step. Low reactivity is also an important consideration for long-term storage of protein fragments. Although many impressive protein targets have been achieved using the favoured ligation junctions of Gly and His, for example, diptericin,13c phospholipase A237 and cytochrome b562,38 the scope of the reaction would be very limited if it was only applicable to the two most favourable sites. Therefore we looked at leucine as it is one of a subset of amino acids that give a slower ligation rate when positioned at the ligation site.10 We chose a fragment (26–63) ColE1-Rop protein, used before as a model target for ligation.26b,39 The ligation reaction was saturated with MPAA to a concentration of 300 mM, considerably higher than we had used previously, but standard for other N,S transfer based ligations.20,21 The reaction was performed at pH 6 and proceeded smoothly to completion in a practical timescale (30 h, Fig. 4) comparable to the BPTI ligation with Gly at the ligation site at 60 mM MPAA. Again no side reactions such as thioester hydrolysis were observed. Leu is a much more demanding ligation residue than Gly and these results therefore suggested that the important factor in this reaction was the concentration of MPAA. In addition it indicates that this ligation is of promising scope and should be applicable to a wide range of ligation sites. A matrix of reactions was carried out to investigate the impact of ligation conditions (pH and MPAA concentration) (Table 1). This set of experiments indicates that MPAA concentration is the major determinant of reaction rate although lower pH (6 instead of 7) favours reaction also, possibly by trapping the acyl transfer in the thioester form with the amine protonated.12b The effect of pH was more apparent for Leu than for His or Gly. Under the optimized conditions (MPAA, 300 mM, pH 6), ligation with the control peptide LAPAGC-OH was still negligible after 24 h (ESI, Fig. S5†). It should be possible to increase reactivity by increasing the bulk of the substitution at the Cα position further. Simple modifications of this basic design can be envisaged to tailor the desired reactivity.40
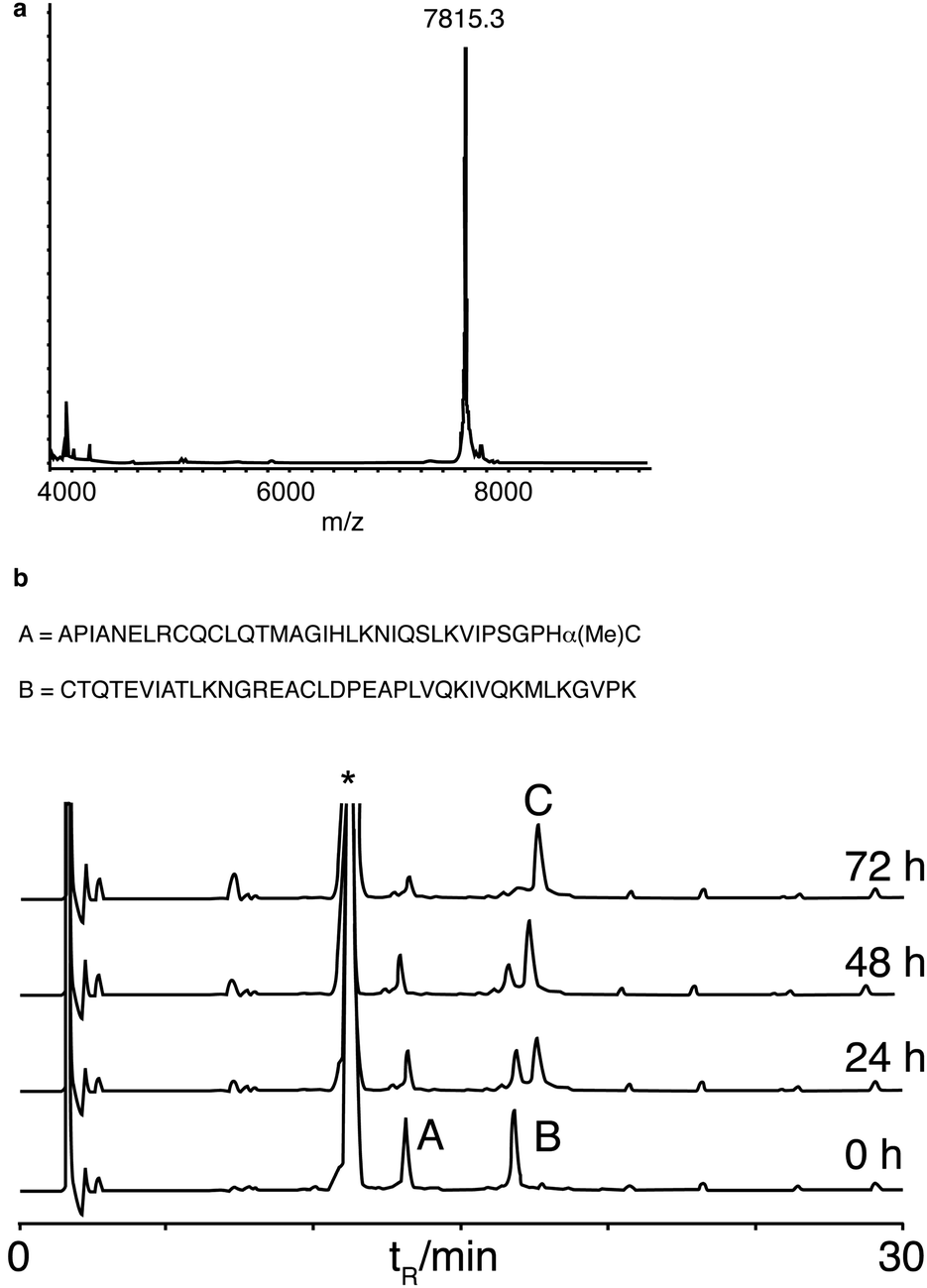 | ||
| Fig. 3 Synthesis of mKC. (a) MALDI-TOF mass spectrum of the ligation product (calculated average mass = 7816 Da). (b) Analytical HPLC time course of ligation. A = mKC(1–34)αMeC, B = mKC(35–72), C = product, * MPAA. Ligation conditions: [A] = 0.44 mM, [B] = 0.42 mM in phosphate buffer (200 mM; pH 7.0), 2 mM EDTA, 6 M guanidine·HCl, 50 mM TCEP, 60 mM MPAA, 40 °C. HPLC conditions: RP-C18, 20–50% B (0.1% TFA, 10% H2O in CH3CN) in 30 min, 1 ml min−1. | ||
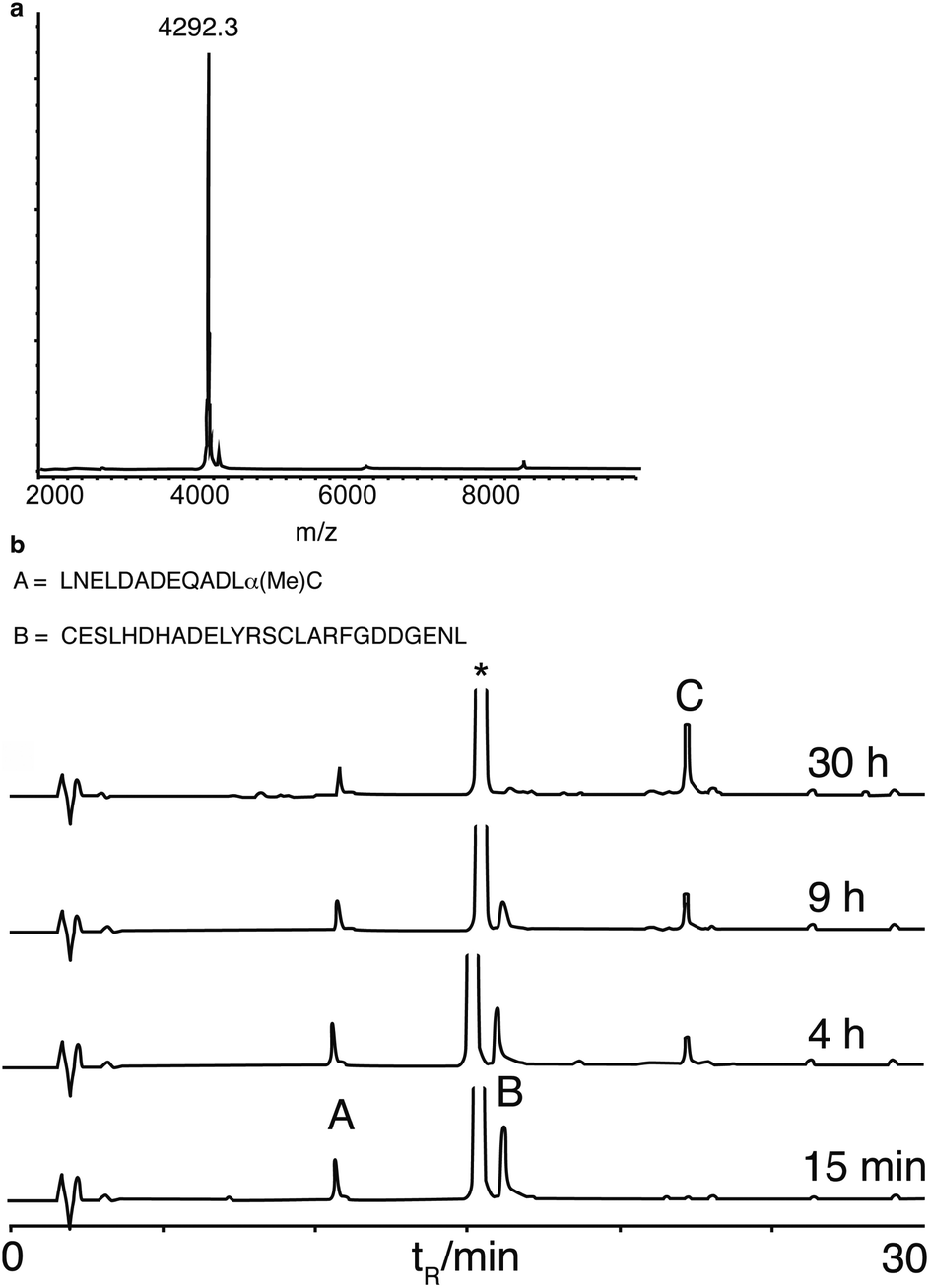 | ||
| Fig. 4 Synthesis of ROP. (a) MALDI-TOF mass spectrum of the ligation product (calculated average mass = 4294 Da). (b) Analytical HPLC time course of the ligation, A = ROP(26–37)-α(Me)C, B = ROP(38–63), C = ligation product,* MPAA. Ligation conditions: [A] = 3 mM [B] = 2.7 mM in phosphate buffer (200 mM; pH 6.0), 2 mM EDTA, 6 M guanidine·HCl, 50 mM TCEP, 300 mM MPAA, 40 °C. HPLC conditions: RP-C18, 15–45% B (0.1% TFA, 10% H2O in CH3CN) in 30 min, 1 ml min−1. | ||
| Protein | [pep] (mM) | pH | [MPAA] (mM) | Ligation site | Timea (h) |
|---|---|---|---|---|---|
| a Time of completion was evaluated from the analytical HPLC time course of the reaction. No secondary product was detected in any of the reactions. | |||||
| BPTI | 2 | 7 | 60 | Gly | 48 |
| BPTI | 2 | 7 | 300 | Gly | 32 |
| BPTI | 2 | 6 | 300 | Gly | 24 |
| mKc | 0.4 | 7 | 60 | His | 72 |
| mKc | 0.4 | 6 | 300 | His | 48 |
| Rop | 3 | 7 | 60 | Leu | 120 |
| Rop | 3 | 6 | 60 | Leu | 120 |
| Rop | 3 | 7 | 300 | Leu | 72 |
| Rop | 3 | 6 | 300 | Leu | 30 |
Conclusions
The use of C-terminal α-methylcysteine as a thioester precursor is a powerful new technology for chemical ligation because of its simplicity. α-Methylcysteine can be loaded onto the resin of choice like any standard amino acid, subsequent acylation is quantitative with standard couplings and after deprotection and characterization, the peptides can be used directly without further manipulation. In contrast to the sulfonamide safety catch linker the peptide can be acetylated and treated with hydrazine post-synthesis allowing the application of Hmb and other protecting group strategies.41 The deprotected α-methylcysteine functions as a weakly activated thioester equivalent. The ligation rate can be modulated by simply changing the MPAA concentration. We are currently exploring the scope and compatibility of this approach with the synthesis of PTM proteins.Acknowledgements
We thank Richard Raz for assistance with peptide synthesis. This work was supported by a grant-in-aid from the MRC number U117592730.Notes and references
- (a) D. Kahne and W. C. Still, J. Am. Chem. Soc., 1988, 110, 7529–7534 CrossRef CAS; (b) R. A. R. Bryant and D. E. Hansen, J. Am. Chem. Soc., 1996, 118, 5498–5499 CrossRef CAS.
- J. L. Radkiewicz, H. Zipse, S. Clarke and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 3499–3506 CrossRef CAS PubMed.
- (a) J. W. Suggs and R. M. Pires, Tetrahedron Lett., 1997, 38, 2227–2230 CrossRef CAS; (b) N. M. Fernandes, F. Fache, M. Rosen, P.-L. Nguyen and D. E. Hansen, J. Org. Chem., 2008, 73, 6413–6416 CrossRef CAS PubMed.
- K. Tani and B. M. Stoltz, Nature, 2006, 441, 731–734 CrossRef CAS PubMed.
- A. J. Kirby, I. V. Komarov, P. D. Wothers and N. Feeder, Angew. Chem., Int. Ed., 1998, 37, 785–786 CrossRef CAS.
- (a) J. Clayden, Y. J. Y. Foricher and H. K. Lam, Eur. J. Org. Chem., 2002, 3558–3565 CrossRef CAS; (b) M. Hutchby, C. E. Houlden, M. F. Haddow, S. N. Tyler, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2012, 51, 548–551 CrossRef CAS PubMed; (c) J. Clayden, Nature, 2012, 481, 274–275 CrossRef CAS PubMed.
- A. Romanelli, A. Shekhtman, D. Cowburn and T. W. Muir, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6397–6402 CrossRef CAS PubMed.
- A. Fersht, J. Am. Chem. Soc., 1971, 93, 3504–3514 CrossRef CAS.
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- (a) Y. Makimura, T. Kiuchi, M. Izumi, S. Dedola, Y. Ito and Y. Kajihara, Carbohydr. Res., 2012, 364, 41–48 CrossRef CAS PubMed; (b) Z. P. Gates, J. R. Stephan, D. J. Lee and S. B. Kent, Chem. Commun., 2013, 49, 786–788 RSC.
- (a) F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS PubMed; (b) D. Macmillan, A. Adams and B. Premdjee, Isr. J. Chem., 2011, 51, 885–899 CrossRef CAS PubMed.
- (a) R. Ingenito, E. Bianchi, D. Fattori and A. Pessi, J. Am. Chem. Soc., 1999, 121, 11369–11374 CrossRef CAS; (b) R. R. Flavell, M. Huse, M. Goger, M. Trester-Zedlitz, J. Kuriyan and T. W. Muir, Org. Lett., 2002, 4, 165–168 CrossRef CAS PubMed; (c) Y. Shin, K. A. Winans, B. J. Backes, S. B. H. Kent, J. A. Ellman and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121, 11684–11689 CrossRef CAS.
- F. Burlina, C. Morris, R. Behrendt, P. White and J. Offer, Chem. Commun., 2012, 48, 2579–2581 RSC.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- G. M. Fang, J. X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347–10350 CrossRef CAS PubMed.
- J. Kang and D. Macmillan, Org. Biomol. Chem., 2010, 8, 1993–2002 CAS.
- (a) H. Hojo, Y. Onuma, Y. Akimoto, Y. Nakahara and Y. Nakahara, Tetrahedron Lett., 2007, 48, 25–28 CrossRef CAS PubMed; (b) H. Hojo, Y. Murasawa, H. Kartayama, T. Ohira, Y. Nakahara and Y. Nakahara, Org. Biomol. Chem., 2008, 6, 1808–1813 RSC.
- P. Botti, M. Villain, S. Manganiello and H. Gaertner, Org. Lett., 2004, 6, 4861–4864 CrossRef CAS PubMed.
- (a) N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238–5241 CrossRef CAS PubMed; (b) N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2011, 51, 209–213 CrossRef PubMed; (c) W. Hou, X. Zhang, F. Li and C.-F. Liu, Org. Lett., 2011, 13, 386–389 CrossRef CAS PubMed.
- K. Sato, A. Shigenaga, K. Tsuji, S. Tsuda, Y. Sumikawa, K. Sakamoto and A. Otaka, ChemBioChem, 2011, 12, 1840–1844 CrossRef CAS PubMed.
- (a) J. Kang, J. P. Richardson and D. Macmillan, Chem. Commun., 2009, 407–409 RSC; (b) J. Masania, J. Li, S. J. Smerdon and D. Macmillan, Org. Biomol. Chem., 2010, 8, 5113–5119 RSC.
- (a) B. Cowper, D. J. Craik and D. Macmillan, ChemBioChem, 2013, 14, 809–812 CrossRef CAS PubMed; (b) D. Macmillan, M. De Cecco, N. L. Reynolds, L. F. Santos, P. E. Barran and J. R. Dorin, ChemBioChem, 2011, 12, 2133–2136 CrossRef CAS PubMed.
- A. J. Kirby, Adv. Phys. Org. Chem., 1980, 17, 183–278 CAS.
- J. R. Reasenberg and S. D. Goldberg, J. Am. Chem. Soc., 1945, 67, 933 CrossRef CAS.
- (a) N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed; (b) H. Rohde, J. Schmalisch, Z. Harpaz, F. Diezmann and O. Seitz, ChemBioChem, 2011, 12, 1396–1400 CrossRef CAS PubMed.
- J. Lukszo, D. Patterson, F. Albericio and S. A. Kates, Lett. Pept. Sci., 1996, 3, 157–166 CrossRef CAS.
- Y. Fu, L. G. J. Hammarström, T. J. Miller, F. R. Fronczek, M. L. McLaughlin and R. P. Hammer, J. Org. Chem., 2001, 66, 7118–7124 CrossRef CAS PubMed.
- R. J. Topping, I. Nuiry, J. Mastriona and J. A. Moss, Tetrahedron Lett., 2008, 49, 2907–2910 CrossRef CAS PubMed.
- (a) W. Lu, M. A. Starovasnik and S. B. Kent, FEBS Lett., 1998, 429, 31–35 CrossRef CAS; (b) N. Fotouhi, N. G. Galakatos and D. S. Kemp, J. Org. Chem., 1989, 54, 2803–2817 CrossRef CAS; (c) C. M. Gross, D. Lelievre, C. K. Woodward and G. Barany, J. Pept. Res., 2005, 65, 395–410 CrossRef CAS PubMed.
- N. Metanis and D. Hilvert, Angew. Chem., Int. Ed., 2012, 51, 5585–5588 CrossRef CAS PubMed.
- M. Quibell, W. G. Turnell and T. Johnson, J. Org. Chem., 1994, 59, 1745–1750 CrossRef CAS.
- E. C. Johnson and S. B. Kent, J. Am. Chem. Soc., 2006, 128, 7140–7141 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- P. E. Dawson, M. Churchill, M. R. Ghadiri and S. B. H. Kent, J. Am. Chem. Soc., 1997, 119, 4325–4329 CrossRef CAS.
- (a) M. Brenner, Pept., Proc. Eur. Pept. Symp., 8th, 1967, 1–7 CAS; (b) D. S. Kemp, Biopolymers, 1981, 20, 1793–1804 CrossRef CAS; (c) C.-F. Liu and J. P. Tam, J. Am. Chem. Soc., 1994, 116, 4149–4153 CrossRef CAS.
- T. M. Hackeng, C. M. Mounier, C. Bon, P. E. Dawson, J. H. Griffin and S. B. H. Kent, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7845–7850 CrossRef CAS.
- D. W. Low, M. G. Hill, M. R. Carrasco, S. B. Kent and P. Botti, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6554–6559 CrossRef CAS PubMed.
- D. S. Kemp and R. I. Carey, J. Org. Chem., 1993, 58, 2216–2222 CrossRef CAS.
- D. Fiset and A. B. Charette, RSC Adv., 2012, 2, 5502–5505 RSC.
- W. C. Chan, B. W. Bycroft, D. J. Evans and P. D. White, J. Chem. Soc., Chem. Commun., 1995, 2209–2210 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of peptides, experimental conditions and HPLC for ligations described herein. See DOI: 10.1039/c3sc52140k |
| This journal is © The Royal Society of Chemistry 2014 |