 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switchable π-coordination and C–H metallation in small-cavity macrocyclic uranium and thorium complexes†‡
Polly L.
Arnold
*a,
Joy H.
Farnaby
a,
Rebecca C.
White
a,
Nikolas
Kaltsoyannis
*b,
Michael G.
Gardiner
*c and
Jason B.
Love
*a
aEaStCHEM School of Chemistry, University of Edinburgh, West Mains Road, Edinburgh EH9 3JJ, UK. E-mail: Polly.Arnold@ed.ac.uk; Jason.Love@ed.ac.uk; Fax: +44 (0)131 6504743; Tel: +44 (0)131 6504762
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: N.Kaltsoyannis@ucl.ac.uk; Fax: +44 (0)207 6797463; Tel: +44 (0)207 6794670
cSchool of Chemistry, University of Tasmania, Private Bag 75, Hobart, Tasmania 7001, Australia. E-mail: Michael.Gardiner@utas.edu.au; Fax: +61 3 6226 2858; Tel: +61 3 6226 2404
First published on 2nd December 2013
Abstract
New, conformationally restricted ThIV and UIV complexes, [ThCl2(L)] and [UI2(L)], of the small-cavity, dipyrrolide, dianionic macrocycle trans-calix[2]benzene[2]pyrrolide (L)2− are reported and are shown to have unusual κ5:κ5 binding in a bent metallocene-type structure. Single-electron reduction of [UI2(L)] affords [UI(THF)(L)] and results in a switch in ligand binding from κ5-pyrrolide to η6-arene sandwich coordination, demonstrating the preference for arene binding by the electron-rich UIII ion. Facile loss of THF from [UI(THF)(L)] further increases the amount of U–arene back donation. [UI(L)] can incorporate a further UIII equivalent, UI3, to form the very unusual dinuclear complex [U2I4(L)] in which the single macrocycle adopts both κ5:κ5 and η6:κ1:η6:κ1 binding modes in the same complex. Hybrid density functional theory calculations carried out to compare the electronic structures and bonding of [UIIII(L)] and [UIII2I4(L)] indicate increased contributions to the covalent bonding in [U2I4(L)] than in [UI(L)], and similar U–arene interactions in both. MO analysis and QTAIM calculations find minimal U–U interaction in [U2I4(L)]. In contrast to the reducible U complex, treatment of [ThCl2(L)] with either a reductant or non-nucleophilic base results in metallation of the aryl rings of the macrocycle to form the (L−2H)4− tetraanion and two new and robust Th–C bonds in the –ate complexes [K(THF)2ThIV(μ-Cl)(L−2H)]2 and K[ThIV{N(SiMe3)2}(L−2H)].
Introduction
The organometallic chemistry of the early actinides has seen rich growth in recent times.1–5 However, compared to the d-block, the study of arene complexes of the f-block is still in its infancy. We recently reported the spontaneous reduction of arenes mediated by the disproportionation of two UIII centres to yield the uranium–arene complexes [{X2U}(μ-C6H5R)] (X = N(SiMe3)2 or OAr, R = H, Me, SiH3, Ph, BBN).6 In this type of arene compound, the actinide-to-ligand back donation from molecular orbitals with δ-symmetry is becoming recognised as a major component of the bonding.7–14 These ‘softer’ δ-interactions between the metal and ligand provide an important opportunity to probe covalency in actinide bonding, which has implications in the separation of the transuranics and lanthanides from the early actinides in nuclear waste.15–19 Organo-thorium complexes exhibiting arene interactions are even rarer, with only a few complexes reported, for example [Li(DME)3][η6-{1,3-[(2-C4H3N)(CH3)2C]2C6H4}ThCl3]20 and the cationic [(XA2)Th(CH2SiMe3)(η6-C6H6)][B(C6F5)4], (XA2 = 4,5-bis(2,6-diidopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene).21Pyrrolic macrocyclic ligands have been shown to support a diverse array of f-block chemistry,22 including dinitrogen cleavage,23,24 stable alkyl complexes,25 and the first f-block example of the reductive disproportionation of CO2 to carbonate and CO.26 Focusing on actinide chemistry, we (Love and Arnold) used an octadentate, tetraanionic Pacman-shaped Schiff-base pyrrole macrocycle to make UIII and NpIII iodides [(AnIIII)2(L)]n that showed magnetic coupling between the AnIII centres,27 and mononuclear [UIV(L)].28,29 Although pyrrolide ligands can coordinate to the metal centre in a κ1 mode through the N atom or in a κ5 mode, similar to classical cyclopentadienyl coordination to a metal, the κ1 mode is by far the more common of the two.15,30–37 The trans-calix[2]benzene[2]pyrrolide, (L)2−, combines two pyrrolide heterocycles with two aryl rings connected via dimethylmethane linkers.38 The lack of extended conjugation in this macrocycle, as compared to porphyrins, grants it a large degree of flexibility, allowing the possibility of either σ- or π-bonding of the ligand to the metal centre through either the pyrrolide or arene. Complexes of this ligand are limited to two examples of SmIII; [SmCl(L)] displays a η6-arene:κ1-pyrrolide binding mode to the SmIII cation (Scheme 1).39 Treatment of [SmCl(L)] with LiMe resulted in isolation and partial characterisation of a mixture of products, including [SmIII(L−H)(THF)] the product of single aryl-metalation of the ligand and [SmIII(L)(L′H)] which incorporates a partially protonated and N-confused macrocycle. In the reaction of [SmCl(L)] with NaH, [Sm(L)(L′H)] was the sole isolable reaction product.
 | ||
| Scheme 1 Sm(III) chemistry of (L)2−. | ||
With this precedent we anticipated that the conformational flexibility of this ligand would enable the study of An–arene interactions in a well-defined macrocyclic environment. Herein, we report the syntheses of new uranium(III), uranium(IV) and thorium(IV) complexes of L, that display unusual and new binding modes of the ligand in mono and dinuclear conformations. We demonstrate the suitability of (L)2− for the stabilisation of both mono and dinuclear complexes of UIII that display a preference for bis(arene) sandwich-type coordination. We also demonstrate facile double aryl metallation on thorium to form complexes with new and robust Th–C bonds.
Results and discussion
Synthesis of [AnIVX2(L)]
The trans-calix[2]benzene[2]pyrrole (H2L), was synthesised according to the literature procedure,38 and was deprotonated in situ with KH to yield the potassium salt K2L. The reaction between K2L and equimolar AnX4(solvent)2 (An = Th, X = Cl, solvent = DME; An = U, X = I, solvent = Et2O) in THF at 80 °C for up to 3 days, followed by work-up to remove KX yielded the new compounds [ThCl2(L)] 1 as a yellow solid in 66% yield and [UI2(L)] 2 as a red solid in 53% yield (Scheme 2). The synthesis of 2 proceeds cleanly irrespective of alkali metal salt or solvent (THF or arene) employed; for example, 2 is also readily accessible from Li2L in toluene (see ESI‡). The 1H NMR spectra of both 1 and 2 show the macrocyclic ligand to have C2v symmetry. In 2, the resonances are broadened and contact shifted (110 to −16 ppm) due to presence of the paramagnetic UIV f2 ion and the aryl protons closest to the metal centre are not observed. The methyl groups of 1 and 2 are observed as two singlets of equal intensity due to the magnetic inequivalence of the endo and exo faces of the ligand upon metallation. Single crystals suitable for X-ray diffraction were grown from a THF solution of 1 layered with n-hexane and allowed to stand at ambient temperature for 3 days. For 2, single crystals were grown at −30 °C from a saturated toluene solution. | ||
| Scheme 2 Syntheses of [ThCl2(L)], 1 and [UI2(L)], 2. | ||
The molecular structures of 1 and 2 (Fig. 1 and ESI‡ respectively), are isomorphous and show a new binding mode for (L)2− with a 1,3-alternating double cone macrocycle conformation with endo metallocene-type binding of the An(IV) cation κ5:κ5 between the two pyrrolide rings and an empty arene cavity. The average An–[C4N]centroid distances and the metallocene angles [κ5-C4N]cent–An–[κ5-C4N]cent are similar to those seen in the cyclopentadienyl AnIV complexes [Cp*2AnX2].40,41 The distances to the ipso-carbons of the rings (Th1–C9, 3.024(2) Å, Th1–C29, 3.017(2) Å; U1–C9, 3.045(5) Å and U1–C29, 3.022(5) Å) are too long to suggest an agostic interaction.42 The ligand binding in 1 and 2 contrasts to that seen in the SmIII complex [SmCl(L)] (Scheme 1) which displays a 1,3-alternating conformation but with substantial flattening of the pyrrolide rings in a shallow double cone conformation, with an endo bound η6:κ1:η6:κ1 SmIII cation in the arene cavity. The binding mode of the singly aryl metallated [Sm(THF)(L−H)] is nominally similar to 1 and 2 in that it has κ5:κ5 pyrrolide binding, but in the SmIII case it is a result of the cation being located deeper within the macrocyclic cavity; the contraction of the pyrrolide cone gives κ5:κ5 binding with a [κ5-C4N]cent–Sm–[κ5-C4N]cent angle of 136.25(5)° and concomitant splaying of the arene rings on metallation.39 This contrast in macrocycle binding between actinide(IV) and samarium(III) is notable given their similar ionic radii.43
 | ||
| Fig. 1 Solid state structure of 1 (displacement ellipsoids are drawn at 50% probability) (a) front view and (b) side view. For clarity, H atoms are omitted. Selected bond distances (Å) and angles (°) for 1: Th1–Cl1 2.6745(7), Th1–Cl2 2.6564(7), Th1⋯C9 3.024(2), Th1⋯C29 3.017(2), Th1–[κ5-C4N]cent(ave) 2.557, Cl1–Th1–Cl2 = 84.16(2), C9–Th1–C29 120.50(7), [κ5-C4N]cent–Th–[κ5-C4N]cent 163.60; 2: U1–I1 3.0708(4), U1–I2 3.0573(4), U1⋯C9 3.045(5), U1⋯C29 3.022(5), U1–[κ5-C4N]cent(ave) 2.480, I1–U1–I2 81.51(1), C9–U1–C29 118.39(1), [κ5-C4N]cent–U–[κ5-C4N]cent 163.26. | ||
Reduction of [UIVI2(L)] to [UIIII(THF)(L)]
The reduction of [UIVI2(L)], 2 by KC8 in THF at low temperature resulted in a colour change from red to dark green. Filtration to remove KI and washing to remove trace H2L allowed the isolation of the UIII complex [UI(THF)(L)] 3 as a dark brown solid in a moderate, 30% yield (Scheme 3). In coordinating solvents, 3 exists as the thermally stable solvate [UI(S)(L)] (S = THF, pyridine, dioxane). Single crystals suitable for X-ray diffraction were grown from a saturated THF solution at −30 °C and the solid state structure of 3 is shown in Fig. 2. In the absence of coordinated solvent, 3 shows very limited solubility in hydrocarbon solvents and decomposes in diethyl ether. Coordinated solvent is removed under vacuum to yield [UI(L)] 3a, as supported by the EI mass spectrum and combustion analysis. We were also able to obtain single crystals of the unsolvated complex 3a by layering a THF solution with n-hexane at ambient temperature (Fig. 2b). | ||
| Scheme 3 Reduction of [UIV(L)I2] 2 to [UIII(L)I(THF)] 3. | ||
 | ||
| Fig. 2 Solid state structures of 3 (a), front view and 3a (b), side view (displacement ellipsoids drawn at 50% probability). For clarity, H atoms and solvent of crystallisation are omitted. Selected bond distances (Å) and angles (°) for 3: U1–I1 3.2092(9), U1–O1 2.697(6), U1–N1 2.530(6), U1–N2 2.501(7), U1–[aryl]cent(ave) 2.669, U1–Cavg 3.001, C–C 1.360(11)–1.413(10), [aryl1]cent–U1–[aryl2]cent = 171.61, N1–U1–N2 115.2(2), N2–U1–I1 88.74, N1–U1–O1 76.02 and 3a: U1–I1 3.1112(15), U1–N1 2.468(10), U1–N2 2.438(10), U1–[aryl]cent(ave) 2.612, U1–Cavg 2.97, 2.95, C–C 1.382(19)–1.44(2), [aryl1]cent–U1–[aryl2]cent = 173.55, N1–U1–N2 118.2(4), N1–U1–I1 121.31, N2–U1–I1 119.45. | ||
As in 2, the 1H NMR spectrum of 3 shows resonances that are consistent with an approximately C2v symmetric macrocyclic environment, with the methyl groups observed as two magnetically inequivalent singlets of equal intensity; in contrast to 2, all of the aryl protons are observed. The ligand resonances for 3 are contact shifted and broadened, as expected for a UIII complex, but in a spectral window (19 to −35 ppm) narrower than for 2. In 3, resonances for coordinated solvent were not observed, indicative perhaps of a fluxional process at ambient temperature on the 1H NMR timescale, in keeping with the lability of the coordinated solvent.
In contrast to the SmIII chemistry,39 reduction of 2 to 3 proceeds cleanly and results in a dramatic change in macrocycle conformation. With the change in oxidation state from UIV to UIII, ligand binding switches from the unusual κ5:κ5 metallocene binding in 1 and 2 to the η6:κ1:η6:κ1 bis(arene) binding seen in [SmCl(L)], showing a clear preference for arene binding in uranium(III). This bis(arene) sandwiched structural motif is reminiscent of that seen in the bis(arene) complexes of the zero oxidation state lanthanides and early transition metals [M(η-Bz*)2] (Bz* = 1,3,5-(But)3C6H3), uniquely accessible by metal vapour synthesis.44–46 Complex 3 also represents are a rare example of a monomeric uranium(III) cation stabilised by a single dianionic ligand and with a single halide site for subsequent metathesis chemistry.
The U–N, U–O, and U–I distances in 3 are unremarkable. The [aryl]cent–U1–[aryl]cent angles, 172° in 3 and 174° in 3a are almost linear and similar to that found in [SmCl(L)] (176°). The U1–Cavg distances in both 3 (3.001 Å) and 3a (2.970 and 2.950 Å) are longer than those of the inverse μ-arene sandwich complexes of uranium6 and are comparable to the UIII aryloxide complex, [U(O-2,6-Pri2C6H3)3]2,47 U1–Cavg of 2.92(2) Å, which exists as a π-arene bridged dimer in the solid state, and to the two UIII adducts of hexamethylbenzene.48,49 The C–C(aryl) distances (range 1.360(11)–1.413(10) Å in 3) are unchanged from 2 or benzene (average 1.40 Å).50,51
Interestingly, the loss of coordinated THF from 3 to form 3a results in a significant shortening of U–L bonds, for example U1–N1 contracts from 2.530(6) to 2.468(10) Å and the lengthening of some of the arene C–C bonds in 3a (C–C 1.382(19)–1.44(2) Å) is now notable, and suggests a greater stabilising back donation from the UIII centre into the arene groups. The change of π-binding and incorporation of UIII into the arene cavity on reduction of 2 to 3 causes the interplanar angle of the arene cavity to decrease significantly from 21.88° to 18.05°. An even smaller value of 15.55° is observed for 3a and thus this metric can be used as a measure of the uranium–arene interaction.
Synthesis of [U2IIII4(L)]
Given the stability of 3 it was obvious to question whether it could be synthesised directly from a UIII precursor. While reactions between K2L or Li2L and UI3 in THF result in the formation of 3, a major by-product, identified as the very unusual dinuclear complex [UIII2I4(L)], 4 is observed. Moreover, the reaction of base-free UI3 (two equivalents) with Li2L in toluene afforded 4 as the sole ligand-containing product which was isolated as an analytically pure material in a 22% yield (Scheme 4). The 1H NMR spectrum of 4 displays resonances that are consistent with a single ligand environment of C2v symmetry. These resonances are contact shifted and broadened, with the width of the spectral window (60 to −35 ppm) intermediate between those of 2 and 3, and, as in 2, the aryl protons closest to the κ5:κ5 uranium cation are not observed. Single crystals of dinuclear UIII4 were grown from hot benzene (Fig. 3); these data show that the macrocycle bridges the two [UIIII2]+ units with both κ5:κ5 and η6:κ1:η6:κ1 binding modes. The stabilisation of not one but two uranium(III) centres by this single dianionic ligand was unexpected and is remarkable. | ||
| Scheme 4 Synthesis of [U2I4(L)] 4. | ||
 | ||
| Fig. 3 Solid state structure of 4 (displacement ellipsoids are drawn at 50% probability). For clarity, H atoms are omitted. Selected bond distances (Å) and angles (°) for 4: U1–I1 3.1096(6), U1–N1 2.669(5), U1–[aryl1]cent(ave) 2.799, U1–[aryl2]cent(ave) 2.748, U1–Cavg 3.105, C–C 1.383(9)–1.410(8), U2–Cavg 2.869, [aryl1]cent–U1–[aryl2]cent 164.99, U2–I2 3.0432(8), U2–I3 3.0256(8), U2–[κ5-C4N]cent(ave) 2.583, [κ5-C4N]cent–U2–[κ5-C4N]cent 139.03, U1⋯U2 3.8639(5). | ||
The κ5:κ5 pyrrolide binding of U2 in 4 is similar to that seen in 2, although the U2–[κ5-C4N]cent(ave) distance of 2.583 Å is longer (2.480 Å in 2) and the metallocene angle is much smaller, [κ5-C4N]cent–U2–[κ5-C4N]cent of 139° (cf. 163° in 2) resulting in a more classical, bent-sandwich geometry. Likewise, the η6:κ1:η6:κ1 bis-arene bonding of U1 in 4 is very similar to that seen in 3 and 3a; the interplanar arene angle is 16.41° in 4, intermediate between 3 (18.05°) and 3a (15.55°). The U1–[aryl]cent(ave) distances are also longer (2.799 and 2.748 Å) than in 3 (2.669 and 2.612 Å) and the [aryl1]cent–U1–[aryl2]cent angle narrower (164.99° cf. 172° in 3). The U1⋯U2 separation of 3.8639(5) Å in 4 is dictated by the macrocycle geometry and is significantly longer than the shortest reported example, the amido-bridged UIII–UIII complex, [U(C8H8)]2[μ-η4:η4-HN(CH2)3N(CH2)2N(CH2)3NH] (3.3057(9) Å).75
The use of toluene as a solvent for UI3 reaction chemistry can result in decreased product yields due to the formation of insoluble aggregates.52,53 However, it is necessary in our case to prevent a solvent-dependent equilibrium between 3 and UI3 and 4 (Scheme 5). Dissolution of crystals of 4 in d8-thf results in an immediate colour change from brown to purple and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05 ratio of 3:4 was determined by 1H NMR spectroscopy. Complex 3 is stable in hydrocarbons, even after loss of coordinated solvent, but the mixing of 3 with an equimolar amount of UI3 in d8-toluene results in the slow formation of 4 (3:4, 1:0.02 after 1 h, 1:0.13 after 15 h). We have not been able to determine the resting position of the equilibrium because of poor solubility and the solution behaviour of 3 and 4 necessarily limits the choice of reaction and work-up solvents for this chemistry.
:0.05 ratio of 3:4 was determined by 1H NMR spectroscopy. Complex 3 is stable in hydrocarbons, even after loss of coordinated solvent, but the mixing of 3 with an equimolar amount of UI3 in d8-toluene results in the slow formation of 4 (3:4, 1:0.02 after 1 h, 1:0.13 after 15 h). We have not been able to determine the resting position of the equilibrium because of poor solubility and the solution behaviour of 3 and 4 necessarily limits the choice of reaction and work-up solvents for this chemistry.
 | ||
| Scheme 5 Equilibrium behaviour of 3 and 4 in solution. | ||
Complexes 3 and 4 are thermally robust and isolable, albeit in moderate yields, free from metallation or oxidation products, and represent an important synthetic entry into UIII chemistry in a new and versatile macrocyclic ligand environment.
Electronic structure and bonding of [UIIII(L)] and [U2IIII4(L)]
In order to probe the electronic structure and bonding in 3a and 4 we turned to quantum chemistry in the form of hybrid density functional theory (PBE0). Geometry optimisations proceeded smoothly to yield structures in excellent agreement with those found by X-ray crystallography: e.g. for 3a (C1 symmetry) U–I = 3.150 Å, U–N(av) = 2.455 Å and U–C(av) = 2.926 Å; for 4 (C2v symmetry) U1–I = 3.119 Å, U2–I = 3.025 Å, U1–N = 2.644 Å, U2–N = 2.718 Å, U1–C(av) = 3.126 Å, U2–C(av) = 2.870 Å and U1–U2 = 3.768 Å (see ESI‡ for tabular comparison).The calculated interplanar arene angles in 3a and 4 are 13.61° and 14.68° respectively, very similar to the experimental values of 15.55° and 16.41°, respectively. The average C–C(arene) bond lengths are also very similar for 3a and 4 at 1.395 Å and 1.392 Å respectively.
A valence molecular orbital (MO) energy level diagram for 3a is presented in Fig. 4. The highest three orbitals (140–142) have predominantly uranium 5f character (84, 91 and 94% respectively), as anticipated for a UIII system. The highest occupied pyrrolide π-based levels separate into two pairs (138–139 and 136–137) with very little metal contribution. Below these orbitals come the iodide p-based levels; 134 and 135 (pπ) and 133 (pσ). Finally, a block of six orbitals (127–132) are seen, which feature U–N σ bonding (MOs 127 and 129) and four arene π-based levels, the most stable of which (MOs 128 and 130) clearly display uranium–arene δ bonding.
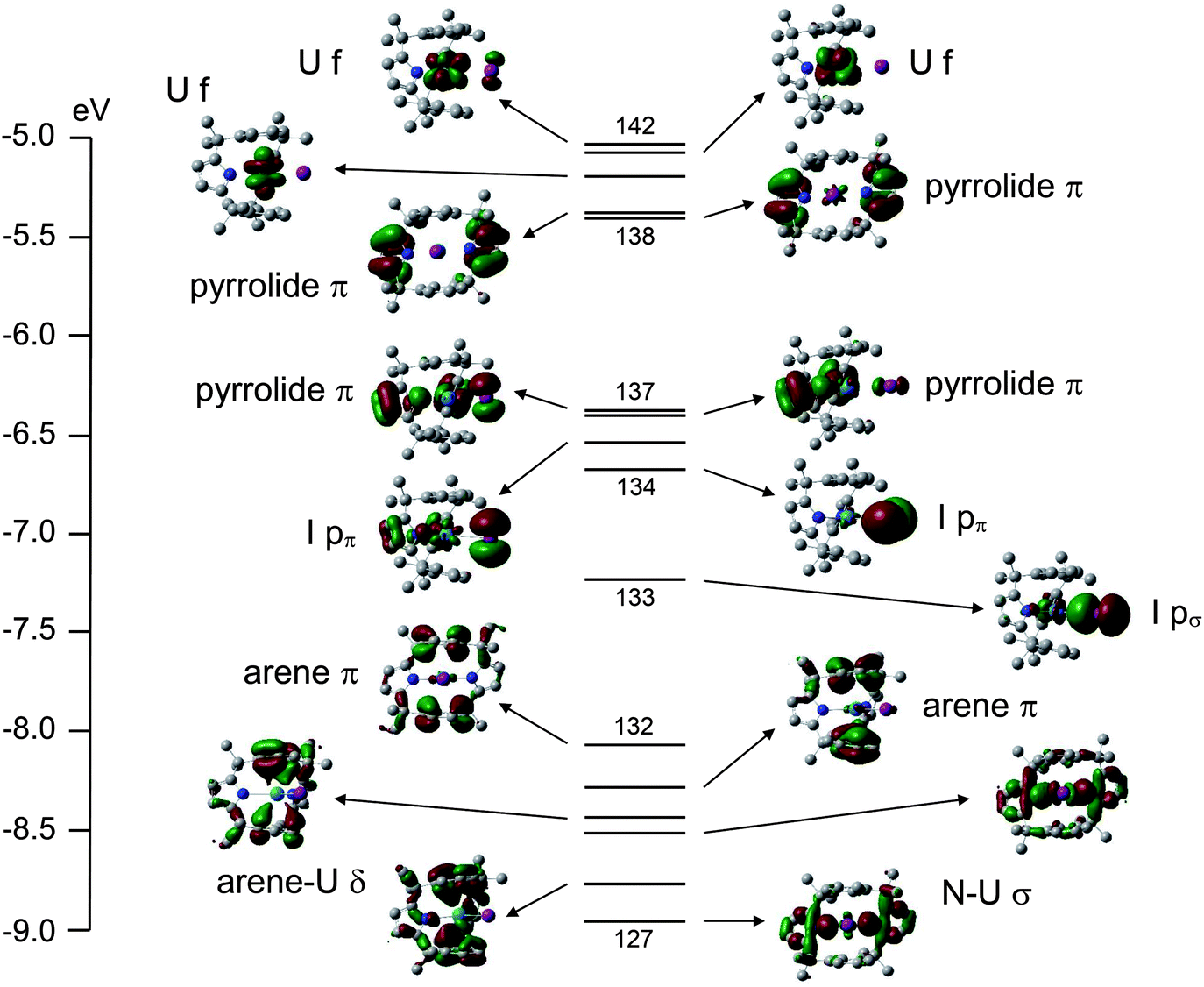 | ||
| Fig. 4 Valence MO energy level diagram for 3a. α spin MOs numbers 127–142 (highest occupied MO) are shown, with an isosurface cutoff of 0.035 au. The principal character of each orbital is also indicated. | ||
The valence molecular orbital structure of 4 is presented in Fig. 5. The increased number of metal and halogen atoms yields a more crowded valence region, but the character of the orbitals can once again be discerned. The principal difference between Fig. 4 and Fig. 5 is the energy of the pyrrolide π orbitals; in 3a they are less stable than the iodide p-based MOs whereas in 4 the opposite is true; the energies of the iodide p-based levels are approximately the same in the two systems, whereas the pyrrolide π orbitals are ca. 2 eV more stable in 4 than 3a.
 | ||
| Fig. 5 Valence MO energy level diagram for 4. α spin MOs numbers 145–170 (highest occupied MO) are shown, with an isosurface cutoff of 0.035 au. The principal character of each orbital is also indicated. | ||
Table 1 contains the average uranium atomic orbital contribution to the I p-based and U–arene δ bonding orbitals of 3a and 4. Although arguably a rather crude measure, these data indicate slightly larger metal contributions to the orbitals of 4vs. their 3a analogues, suggestive of enhanced covalency in the former.§
| MO type | 3a | 4 |
|---|---|---|
| I pσ | 12 (10d, 2s) | 14 (7d, 3s, 2f, 2p) |
| I pπ | 7 (6f, 1d) | 10 (8f, 2d) |
| U–arene δ | 7 (d) | 8.5 (d) |
Single point calculations on (L)2− in its geometry in 3a and 4 reveal that it is 58.8 kJ mol−1 more stable in the former (at the SCF level). In both cases the four highest occupied MOs are the pyrrolide π levels which contribute to Fig. 4 and 5, and these are slightly more stable in (L)2− in its 3a geometry than in that of 4 (e.g. the HOMO of (L)2−//3a is 35.2 kJ mol−1 more stable than the HOMO of (L)2−//4). Clearly, however, this situation is reversed when the macrocycle incorporates uranium – as noted above, the pyrrolide π-based orbitals are about 2 eV (ca. 200 kJ mol−1) more stable in 4 than 3a. The explanation almost certainly lies in the extent to which the uranium atomic orbitals are mixed into the pyrrolide levels. A good comparison is MO 139 of 3a (Fig. 4) with MO 151 of 4 (Fig. 5); the former has a very minor (4%) non-bonding 5f contribution whereas the latter features 15% uranium 6d character in what is clearly a metal–pyrrolide δ bonding interaction. We can therefore conclude that at least part of the driving force for the macrocycle to adopt the more constrained geometry in 4 than in 3a is the covalent bonding it gains with U2 in the former.
Tables containing the principal character and orbital contributions to the frontier MOs in 3a and 4 are included in the ESI,‡ alongside enlarged pictures of the MOs from Fig. 4 and 5.
The charges on the uranium atoms in 3a and 4 have been calculated in several different ways, and the results are collected in Table 2. It is typically the case that there are rather large differences between the absolute values of partial atomic charges calculated in different ways, though often the trends are similar and this is the case here. For all three charge analysis schemes, the uranium atom is most positive in 3a, while in 4, the arene-coordinated U1 is more positive than the π-pyrrolide coordinated U2. These data support the MO composition analysis described above; a higher partial charge is associated with less covalency, and hence we conclude that the bonding in 4 is more covalent than in 3a. Within 4 the lower charge on U2 suggests this atom is, overall, more covalently bonded than U1.
| Hirshfeld | Mulliken | QTAIM | |
|---|---|---|---|
| 3a | +0.50 | +0.78 | +2.04 |
| 4 U1 | +0.46 | +0.66 | +1.99 |
| 4 U2 | +0.43 | +0.48 | +1.86 |
A less traditional approach, certainly within f element systems, to assessing the relative extent of ionicity and covalency is the Quantum Theory of Atoms-in-Molecules (QTAIM) which we have recently applied to a variety of uranium compounds.54–60 We have found the properties of bond critical points (BCPs) to be valuable additions to discussions of f element–ligand bonding, in particular the electron and energy densities. These are collated for selected BCPs of 3a and 4 in Table 3.¶
| Complex | Parameter | ρ | H | δ(A,B) |
|---|---|---|---|---|
| 3a | U–I | 0.039 | −0.007 | 0.542 |
| U–N(av) | 0.071 | −0.016 | 0.486 | |
| 4 | U1–I | 0.040 | −0.007 | 0.548 |
| U2–I | 0.047 | −0.009 | 0.650 | |
| U1–N | 0.044 | −0.003 | 0.321 | |
| U2–N | 0.037 | −0.001 | 0.264 |
As with most BCP data for actinide compounds, the absolute values of both metrics are small, suggesting the uranium–ligand bonds are largely ionic. That said, we can use these data to assess relative covalency, and can relate them to the MO and partial charge arguments developed above. This is best done for the U–I bonds (which are obviously free of the complicating effects of macrocycle rearrangement) and comparison of the U–I data in 3a with 4 suggests that the U1–I bond in 4 is very similar to that in 3a. The slightly larger (absolute) values of ρ and H for U2–I indicate slightly greater covalency, in agreement with the charge data in Table 2. The κ1 U–N interaction is clearly significantly weaker in 4 (U1–N), as the bond distance is c. 0.2 Å longer than in 3a. This is reflected in ρ and H, which are both (absolutely) smaller in 4. At the QTAIM level the differences between the κ1 and κ5 U–N interactions in 4 are comparable with the differences between U1–I and U2–I, although now U2 has the smaller BCP metrics.
Also given in Table 3 are the delocalisation indices δ(A,B) for the selected bonds. δ(A,B) is the average number of electrons shared between atoms A and B, and is the bond order when atoms A and B are connected by a bond path, as is the case here. These data very much support the conclusions from the BCP analysis, as δ(A,B) for U–I in 3a and U1–I in 4 are very similar, whereas U2–I has a slightly larger δ(A,B), and the U–N bond order in 3a is larger than in 4.
It is noteworthy that QTAIM analysis does not locate a U–U bond path in 4 and hence, by the theory's rigorous definition of chemical bonding, 4 does not contain a U–U bond. This is certainly in keeping with analysis of the valence orbital structure, which finds little evidence of MOs with contributions from both uranium atoms. To a large extent, Fig. 5 suggests that the two uranium centres have independent electronic structures.
Reactivity of [ThCl2(L)]
The π/δ-acceptor capabilities and flexibility of this macrocycle led us to study the reduction chemistry of 1. Treatment of a THF solution of 1 with two equivalents of K/naphthalenide at room temperature over 16 h resulted in a colour change from yellow to red-brown and the precipitation of KCl (Scheme 6). Prompt filtration, followed by diffusion of n-hexane into the filtrate allowed analytically pure crystals of the new colourless complex [K(THF)2Th(μ-Cl)(L−2H)]2, 5 to be isolated in 31% yield. The insolubility of 5 in common solvents precluded analysis by 1H or 13C{1H} NMR spectroscopy, but single crystal X-ray diffraction revealed a dimeric ThIV complex of the tetraanionic form of the macrocycle formed as a result of double aryl metallation (L−2H = {CMe2(NC4H2)CMe2}2(C6H3-1,3)2) (Fig. 6). Complex 5 is also the product of the reaction of 1 with two equivalents of K metal in THF at 80 °C for 16 h. | ||
| Scheme 6 Syntheses of [K(THF)2Th(μ-Cl)(L−2H)]2, 5 and K[Th(N′′)(L−2H)], 6. | ||
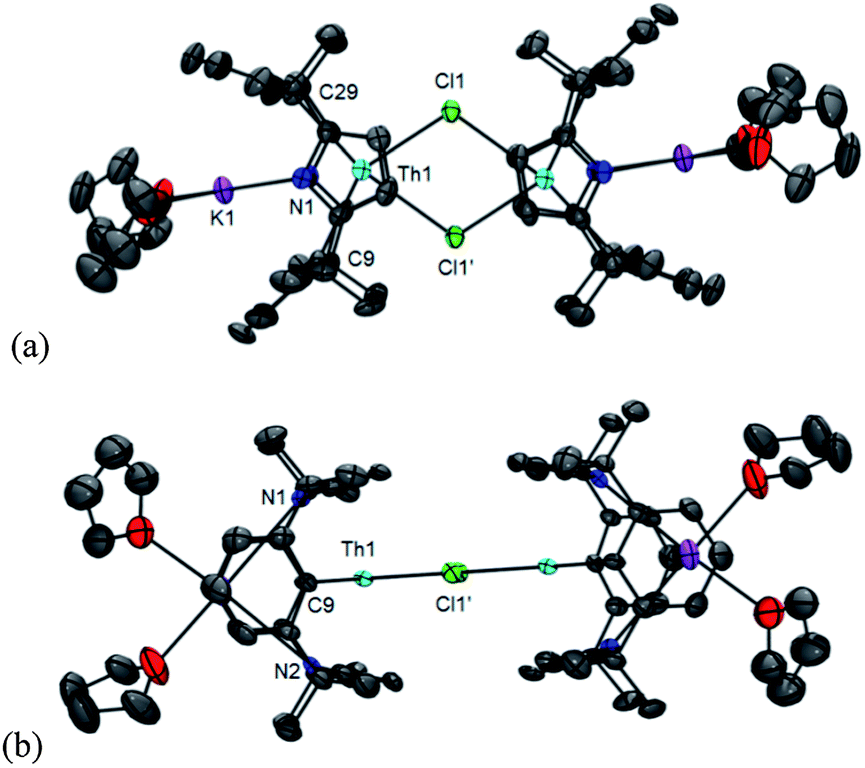 | ||
| Fig. 6 Solid state structure of 5 (displacement ellipsoids drawn at 50% probability): (a) top view, and (b) side view. For clarity, H atoms and solvent of crystallisation are omitted. Selected bond distances (Å) and angles (°): Th1–Cl1 2.840(2), Th1–Cl1′ 3.060(2), Th1–C9 2.659(8), Th1–C29 2.669(9), [κ5-C4N]cent(ave)–Th1 2.550, K–[aryl]cent(ave) 3.190, Cl1–Th1–Cl2 68.96(6), C9–Th1–C29 112.21(3), [κ5-C4N]cent–Th1–[κ5-C4N]cent 164.98. | ||
With the aim of making a more soluble analogue, 1 was treated with two equivalents of KN(SiMe3)2 (KN′′). This reaction proceeds cleanly to form K[Th(N′′)(L−2H)], 6 as identified by a single crystal X-ray diffraction experiment (Fig. 7). Like 5, 6 has undergone double aryl metallation of the macrocycle, loss of one chloride ligand, and salt incorporation, in this case KN′′; the reaction between 1 and 3 eq. KN′′ allows the isolation of 6 as a pale pink powder in a 29% yield. The 1H NMR spectrum of 6 is similar to 1, with a single set of symmetrical ligand resonances and an additional single resonance for the SiMe3 protons, but is devoid of the aryl-H resonances assigned to C9–H and C29–H, consistent with metallation (see Fig. 1, 6 and 7). The 13C{1H} NMR spectrum shows a resonance at 211.7 ppm, which is shifted to higher frequency from 121.6 ppm in 1, and is assigned as the Th-bound ipso-aryl carbon. This resonance is comparable to those reported for [Cp*2Th(o-MeC6H4)Cl] at 224.8 ppm,61 and [Th({2-CH2NMe2}C6H4)4] at 230.8 ppm (ref. 62) but at a higher frequency to other metalated- or alkyl-ThIV complexes.63,64
 | ||
| Fig. 7 Solid state structure of 6, (a) asymmetric unit and (b) K-bridged extended structure (displacement ellipsoids drawn at 50%). For clarity, H atoms are omitted. Selected bond distances (Å) and angles (°): Th1–C9 2.691(16), Th1–C29 2.693(12), Th1–N3 2.375(12), Th1–[κ5-C4N]cent(ave) 2.547, K1–C9 3.299(1), K1–C29 3.240(1), K1′–C25 3.299(1), K1′–C26 3.080(1), K1′–C27 3.187(1), [κ5-C4N]cent–Th–[κ5-C4N]cent 164.39, C9–Th1–C29 119.58(4). | ||
The molecular structures of 5 and 6 show a κ5:η1:κ5:η1 ligand bonding mode, but with two new Th–C bonds to C9 and C29, from the double metallation of the macrocycle. The potassium counter-ion occupies the cavity subtended by the two aryl rings in both cases. Complex 5 is a chloride-bridged dimer in the solid state, whereas complex 6 exists as a K-bridged linear polymer (Fig. 7). There is no significant change to pyrrolide binding on metallation, but the arene cavity has expanded in 5 and 6 compared to 1; the interplanar angle between the arene rings is 20.02° in 1 but 63.66° in 5 and 56.48° in 6. The constrained geometry of the macrocycle is reflected in the long Th–C distances in 5 (Th1–C9 2.659(8) Å, Th1–C29 2.669(9) Å) and 6 (Th1–C9 2.691(16) Å, Th1–C29 2.693(12) Å), longer than those found in the similarly metallated complex [Li(DME)3][ThCl3(η6-C6H4{1,3-CMe2[(2-C4H3N)CMe2})]] (2.612(8) Å).20
In the reactions between 1 and either K or KN′′, potassium cation incorporation occurs readily in the arene cavity of the macrocycle, and allows clean substitution chemistry of the halide ligands, although it also facilitates double C–H metallation of the ligand aryl groups by the ThIV centre. There is no evidence to suggest that these reactions proceed through a ThIII oxidation state.65 The inaccessibility of the ThIII oxidation state (−3.0 eV vs. SHE)66,67 is doubtless a factor in the different reactivity of complexes 1 and 2 with reductants.
Conclusions
We have shown that the small-cavity macrocycle (L)2− is capable of binding an actinide cation in either a κ5:κ5 pyrrolide–metallocene or η6:κ1:η6:κ1 bis(arene) manner. This is unusual as the normal binding mode of a pyrrolide ligand is κ1 through the N atom, a feature that has been exploited by us and others in κ1-pyrrole-based macrocycles (e.g. Pacman and expanded porphyrins) for supporting new actinide chemistry. As such, the switchable coordination of (L)2− provides a unique and well-defined molecular environment in which to interrogate the π and δ bonding interactions between these ligands and actinide metal cations. This feature is best exemplified in the preference for π-arene bonding in the lower oxidation state UIII complexes [UI(THF)(L)] 3 and desolvated [UI(L)] 3a which clearly display covalent uranium–arene δ-bonding.The use of base-free conditions allows the isolation of the very unusual dinuclear complex [U2I4(L)] 4 in which the macrocycle bridges two [UIIII2]+ units and adopts both κ5:κ5 and η6:κ1:η6:κ1 binding modes.
Hybrid DFT studies of 3a and 4 suggest very similar U–arene interactions in these complexes. These computational data indicate slightly larger covalency contributions to the bonding in the UIII2(L) complex 4 than in the UIII(L) complex 3a with the pyrrolide π-orbitals significantly stabilised (ca. 200 kJ mol−1 lower in energy) by forming a metal–pyrrolide δ bonding interaction that contains ca. 15% uranium 6d character. Therefore, at least part of the driving force for the macrocycle to adopt the more constrained geometry in 4 than in 3a is the gain in covalent bonding in the former. Analysis of the calculated charges on the uranium atoms by a number of methods support the greater covalency in the π-pyrrolide-bound U atom, which has a lower positive charge than the π-arene bound U atom.
Further computational analysis by the QTAIM method suggests that in 4, the two UIII centres have approximately independent electronic structures, and the degree of covalency in the U–L bonds is greater in the pyrrolide-bound U than the arene-bound U.
In contrast, the Th chemistry of (L)2− is dominated by ligand metalation processes due to the inaccessibility of Th(III) and the proximity of the aryl C–H bonds to the Th metal centre. Rare new Th–aryl bonds are formed in [K(THF)2Th(μ-Cl)(L−2H)]25 and K[Th(N′′)(L−2H)] 6 in which the macrocycle has retained the κ5:κ5 binding mode to ThIV whilst a potassium cation resides in the arene cavity occupied by UIII in complexes 3 and 4. It is likely that alkali metal cation complexation into the π-framework of the macrocycle facilitates this new reaction chemistry.
This small-cavity macrocycle has enabled access to two different metal – 6π-electron binding modes in homobimetallic (UIII) and heterobimetallic (KIThIV) actinide complexes for the first time. The ability of the macrocycle to adopt different binding modes and switch between them, should facilitate new low oxidation state U chemistry. Further work is in progress to explore the cooperative bimetallic reactivity of 4 and the new hydrocarbyl reaction chemistry that (L−2H)4− can support.
Acknowledgements
The authors thank the University of Edinburgh, the EPSRC for financial support and the University of Tasmania for study leave support for MGG. NK thanks the UCL High Performance Computing Facility (Legion@UCL) and associated support services.Notes and references
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341 CrossRef CAS PubMed.
- P. L. Arnold, Chem. Commun., 2011, 47, 9005 RSC.
- M. Ephritikhine, Organometallics, 2013, 32, 2464 CrossRef CAS.
- M. Ephritikhine, C. R. Chim., 2013, 16, 391 CrossRef CAS PubMed.
- K. R. D. Johnson and P. G. Hayes, Chem. Soc. Rev., 2013, 42, 1947 RSC.
- P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668 CrossRef CAS PubMed.
- P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108 CrossRef CAS.
- P. L. Diaconescu and C. C. Cummins, Inorg. Chem., 2012, 51, 2902 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2004, 126, 14533 CrossRef CAS PubMed.
- W. J. Evans, C. A. Traina and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 17473 CrossRef CAS PubMed.
- M. J. Monreal, S. I. Khan, J. L. Kiplinger and P. L. Diaconescu, Chem. Commun., 2011, 47, 9119 RSC.
- D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454 CAS.
- D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 10388 CrossRef CAS PubMed.
- D. L. Clark, S. K. Grumbine, B. L. Scott and J. G. Watkin, J. Am. Chem. Soc., 1995, 117, 9089 CrossRef CAS.
- E. Kohler, W. Bruser and K. H. Thiele, J. Organomet. Chem., 1974, 76, 235 CrossRef.
- I. Infante, J. Raab, J. T. Lyon, B. Liang, L. Andrews and L. Gagliardi, J. Phys. Chem. A, 2007, 111, 11996 CrossRef CAS PubMed.
- G. Hong, F. Schautz and M. Dolg, J. Am. Chem. Soc., 1999, 121, 1502 CrossRef CAS.
- E. Glueckauf and H. A. C. McKay, Nature, 1950, 165, 594 CrossRef CAS.
- I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353 RSC.
- I. Korobkov, B. Vidjayacoumar, S. I. Gorelsky, P. Billone and S. Gambarotta, Organometallics, 2010, 29, 692 CrossRef CAS.
- C. A. Cruz, D. J. H. Emslie, C. M. Robertson, L. E. Harrington, H. A. Jenkins and J. F. Britten, Organometallics, 2009, 28, 1891 CrossRef CAS.
- B. M. Rambo and J. L. Sessler, Chem.–Eur. J., 2011, 17, 4946 CrossRef CAS PubMed.
- E. Campazzi, E. Solari, C. Floriani and R. Scopelliti, Chem. Commun., 1998, 2603 RSC.
- M. G. Gardiner and D. N. Stringer, Materials, 2010, 3, 841 CrossRef CAS.
- J. Wang, M. G. Gardiner, B. W. Skelton and A. H. White, Organometallics, 2005, 24, 815 CrossRef CAS.
- N. W. Davies, A. S. P. Frey, M. G. Gardiner and J. Wang, Chem. Commun., 2006, 4853 RSC.
- P. L. Arnold, N. A. Potter, N. Magnani, C. Apostolidis, J.-C. Griveau, E. Colineau, A. Morgenstern, R. Caciuffo and J. B. Love, Inorg. Chem., 2010, 49, 5341 CrossRef CAS PubMed.
- J. B. Love, Chem. Commun., 2009, 3154 RSC.
- P. L. Arnold, N. A. Potter, C. D. Carmichael, A. M. Z. Slawin, P. Roussel and J. B. Love, Chem. Commun., 2010, 46, 1833 RSC.
- D. L. Swartz II, L. P. Spencer, B. L. Scott, A. L. Odom and J. M. Boncella, Dalton Trans., 2010, 39, 6841 RSC.
- J. B. Love, A. J. Blake, C. Wilson, S. D. Reid, A. Novak and P. B. Hitchcock, Chem. Commun., 2003, 1682 RSC.
- E. Campazzi, E. Solari, R. Scopelliti and C. Floriani, Inorg. Chem., 1999, 38, 6240 CrossRef CAS PubMed.
- M. Ganesan, C. D. Bérubé, S. Gambarotta and G. P. A. Yap, Organometallics, 2002, 21, 1707 CrossRef CAS.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433 CrossRef CAS.
- T. Dubé, J. Guan, S. Gambarotta and G. P. A. Yap, Chem.–Eur. J., 2001, 7, 374 CrossRef.
- C. Liu, S. Zhou, S. Wang, L. Zhang and G. Yang, Dalton Trans., 2010, 39, 8994 RSC.
- M. Nishiura, T. Mashiko and Z. Hou, Chem. Commun., 2008, 2019 RSC.
- J. L. Sessler, W.-S. Cho, V. Lynch and V. Král, Chem.–Eur. J., 2002, 8, 1134 CrossRef CAS.
- S. Ilango, B. Vidjayacoumar and S. Gambarotta, Dalton Trans., 2010, 39, 6853 RSC.
- M. R. Spirlet, J. Rebizant, C. Apostolidis and B. Kanellakopulos, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 2135 CrossRef.
- C. R. Graves, E. J. Schelter, T. Cantat, B. L. Scott and J. L. Kiplinger, Organometallics, 2008, 27, 5371 CrossRef CAS.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- F. G. N. Cloke, Chem. Soc. Rev., 1993, 22, 17 RSC.
- F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 1997, 119, 7899 CrossRef.
- M. N. Bochkarev, Chem. Rev., 2002, 102, 2089 CrossRef CAS PubMed.
- W. G. Van der Sluys, C. J. Burns, J. C. Huffman and A. P. Sattelberger, J. Am. Chem. Soc., 1988, 110, 5924 CrossRef CAS.
- M. Cesari, U. Pedretti, Z. Zazzetta, g. Lugli and W. Marconi, Inorg. Chim. Acta, 1971, 5, 439 CrossRef CAS.
- P. C. Blake, M. F. Lappert, R. G. Taylor, J. L. Atwood and H. Zhang, Inorg. Chim. Acta, 1987, 139, 13 CrossRef CAS.
- J. Plíva, J. W. C. Johns and L. Goodman, J. Mol. Spectrosc., 1990, 140, 214 CrossRef.
- S. G. Kukolich, J. Am. Chem. Soc., 1995, 117, 5512 CrossRef CAS.
- C. D. Carmichael, N. A. Jones and P. L. Arnold, Inorg. Chem., 2008, 47, 8577 CrossRef CAS PubMed.
- D. L. Clark, A. P. Sattelberger, S. G. Bott and R. N. Vrtis, Inorg. Chem., 1989, 28, 1771 CrossRef CAS.
- P. L. Arnold, Z. R. Turner, N. Kaltsoyannis, P. Pelekanaki, R. M. Bellabarba and R. P. Tooze, Chem.–Eur. J., 2010, 16, 9623 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352 CrossRef CAS PubMed.
- M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Chem. Sci., 2013, 4, 1189 RSC.
- I. Kirker and N. Kaltsoyannis, Dalton Trans., 2011, 40, 124 RSC.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036 CrossRef CAS PubMed.
- A. R. E. Mountain and N. Kaltsoyannis, Dalton Trans., 2013, 42, 13477 RSC.
- M. J. Tassell and N. Kaltsoyannis, Dalton Trans., 2010, 39, 6719 RSC.
- A. F. England, C. J. Burns and S. L. Buchwald, Organometallics, 1994, 13, 3491 CrossRef CAS.
- L. A. Seaman, E. A. Pedrick, T. Tsuchiya, G. Wu, E. Jakubikova and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 10589 CrossRef CAS PubMed.
- I. Korobkov, A. Arunachalampillai and S. Gambarotta, Organometallics, 2004, 23, 6248 CrossRef CAS.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, Chem.–Eur. J., 2011, 17, 12669 CrossRef CAS PubMed.
- D. E. Bergbreiter and J. M. Killough, J. Am. Chem. Soc., 1978, 100 CrossRef CAS.
- S. G. Bratsch and J. J. Lagowski, J. Phys. Chem., 1986, 90, 307 CrossRef CAS.
- J. W. Bruno, D. G. Kalina, E. A. Mintz and T. J. Marks, J. Am. Chem. Soc., 1982, 104, 1860 CrossRef CAS.
- T. Le Borgne, M. Lance, M. Nierlich and M. Ephritikhine, J. Organomet. Chem., 2000, 598, 313 CrossRef CAS.
Footnotes |
| † Celebrating 300 years of Chemistry at Edinburgh. |
| ‡ Electronic supplementary information (ESI) available: Full synthetic and crystallographic details; computational methodology and converged cartesian atomic coordinates enlarged MO images, tabular comparison of theoretical and experimental geometries, and MO composition tables for 3a and 4. CCDC 951384–951390 and 964833. For ESI and crystallographic data in CIF or other electronic format see See DOI: 10.1039/c3sc52072b |
| § Although one of us (NK) has recently argued that orbital mixing and covalency (in the generally accepted chemical sense at least, if not the Heitler-London definition) should be separated in compounds of elements toward the middle of the actinide series, it is more likely that they are synonymous in uranium chemistry. |
| ¶ The satisfactory location of all the metal–carbon BCPs in carbocyclic sandwich molecules is notoriously difficult, and so it proved with 3a and 4. Hence these data are considered unreliable and are not included here. |
| This journal is © The Royal Society of Chemistry 2014 |