 Open Access Article
Open Access ArticleRuthenium-catalyzed ortho-C–H bond alkylation of aromatic amides with α,β-unsaturated ketones via bidentate-chelation assistance†‡
Guy
Rouquet
and
Naoto
Chatani
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: chatani@chem.eng.osaka-u.ac.jp; Fax: +81-(0)6-6879-7396; Tel: +81-(0)6-6879-7397
First published on 19th March 2013
Abstract
A new chelation assisted reaction using a removable 8-aminoquinoline bidentate directing group that permits the ruthenium-catalyzed ortho-C–H bond alkylation of aromatic amides with various α,β-unsaturated ketones under straightforward conditions has been developed. This methodology represents the first efficient utilization of enones in the ortho directed ruthenium-catalyzed addition of C–H bonds to C–C double bonds. The reaction offers a broad scope and a high functional group tolerance.
Introduction
The successful development of new transition metal-catalyzed reactions for the formation of C–C bonds represents a crucial and long-standing challenge in synthetic organic chemistry.1 Among the many strategies available, catalytic C–H bond activation has recently emerged as one of the most promising and powerful methods for the construction of C–C bond frameworks.2 This statement was beautifully illustrated in 1993, when Murai published a report on the ruthenium-catalyzed ortho-C–H bond alkylation of aromatic ketones with olefins via a chelation assisted strategy. The reaction permits the direct and selective transformation of a C–H bond into a C–C bond through the addition to an olefin.3 This reaction can be considered to constitute an ideal pathway in terms of step and atom economy for the formation of C–C bonds and was rapidly expanded to other aromatic substrates.4 Advances were subsequently reported by Darses and Genet et al.5 through the development of an in situ-generated active catalyst prepared from [RuCl2(p-cymene)]2, which can be regarded as more flexible and practical than the ruthenium species (Ru3(CO)12, RuH2(PPh3)4, RuH2(CO)(PPh3)3…) traditionally used in these types of reactions. However, most olefin-bearing functional groups still remain unusable in these processes,6 and this severely limits the scope of this fundamental transformation in synthesis by virtue of the fact that some of the more important families of acceptors, such as α,β-unsaturated acceptors, cannot be used (eqn (1), Scheme 1). A few alternatives to address this issue have begun to appear in the literature,7 but, although promising, these methods are rather rare, limited in scope and require the use of expensive rhodium7a,c,d or rhenium7b catalysts. The search for new ruthenium-based catalytic systems to fulfil this ambition remains challenging and no alternatives have been reported so far, despite the strong appeal of this metal.8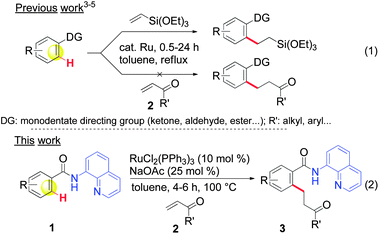 | ||
| Scheme 1 ortho-C–H bond alkylation of aromatic substrates with α,β-unsaturated ketones: monodentate vs. bidentate directing groups. | ||
In a first approach, we hypothesized that the lack of reactivity of a large range of olefins in the ruthenium-catalyzed reactions reported by Murai et al.4,6 may be due to the inappropriate nature of the monodentate directing groups used. Bidentate-type directing groups have recently emerged as a new tool in exploring reactions that have not been achieved with conventional monodentate directing groups through the publications of Daugulis et al.,9 which were followed by others,10 including some from our laboratory.11 We have, thus, anticipated that bidentate directing groups could open new horizons for expanding the scope of the ruthenium-catalyzed ortho-C–H bond alkylation of aromatic substrates.12 Herein, in support of our hypothesis, we report on a new chelation assisted strategy in which a removable 8-aminoquinoline bidentate directing group is used9b (as in 1, Scheme 1) that allows the first ruthenium-catalyzed ortho-C–H bond alkylation of aromatic amides via 1,4-addition to various α,β-unsaturated ketones under straightforward reaction conditions by using attractive RuCl2(PPh3)3 or [RuCl2(p-cymene)]2 catalysts (eqn (2), Scheme 1).13
Results and discussion
Preliminary studies
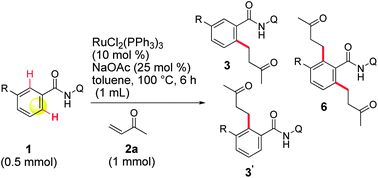
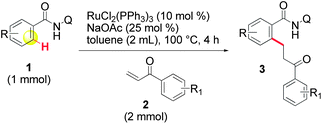
We initiated our studies by examining the feasibility of the ruthenium-catalyzed ortho-C–H bond alkylation of amide 1a with methyl vinyl ketone (MVK 2a) (Table 1). Our efforts were initially rewarded, when it was found that, when Ru3(CO)12 was used, the expected product 3aa was produced in 6% yield, but the cyclic 4aa was also produced in 6% yield (entry 3). The formation of 4aa could be attributed to a competitive β-hydride elimination that produces the alkenylated amide 5aa,14 which immediately undergoes an intramolecular Michael reaction. Our attempts to improve the yields in this reaction were fruitless and we finally abandoned further experiments using this catalyst.|
|
|||
|---|---|---|---|
| Entry | Catalysta | Additive | 3aa (%) |
| a 5 mol% of [RuCl2(p-cymene)]2 was used. b Numbers in parentheses are recovered 1a. c 6% of 4aa was isolated. d 30 mol% of PPh3. | |||
| 1 | RuH2(CO)(PPh3)3 | — | 0 (98) |
| 2 | RuH2(PPh3)4 | — | 0 (95) |
| 3 | Ru3(CO)12 | — | 6 (68)c |
| 4 | [RuCl2(p-cymene)]2 | — | 10 (84) |
| 5 | [RuCl2(p-cymene)]2 | NaHCO2/PPh3d | 50 (43) |
| 6 | [RuCl2(p-cymene)]2 | NaOAc/PPh3d | 90 (0) |
| 7 | [RuCl2(p-cymene)]2 | NaOAc | 91 (0) |
| 8 | RuCl2(PPh3)3 | NaOAc | 94 (0) |
| 9 | RuCl2(PPh3)3 | Et3N | 54 (32) |
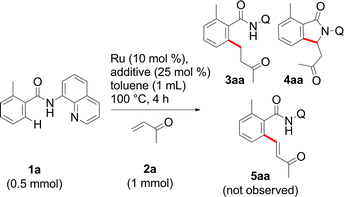
[RuCl2(p-cymene)]2 then, attracted our attention when we noticed its ability to cleanly produce 3aa as the exclusive product but in a poor yield (entry 4). To our delight, the yield of 3aa was improved when the system of Darses and Genet et al. (entry 5) was applied.5a In part inspired by the success of carboxylate-assisted metal-catalyzed C–H functionalization reactions,15 we discovered that optimum efficiency could be achieved by replacing sodium formate with sodium acetate (entry 6). Interestingly, neither 5aa nor 4aa were obtained under these conditions, while Ackermann, Dixneuf and others, reported that Ru(II) catalysts are powerful catalysts in Fujiwara–Moritani type reactions with α,β-unsaturated acceptors.2r,16 Furthermore, it was not necessary to use triphenylphosphine for a successful reaction in the case of o-substituted amides (entry 7).17a After further investigations of the scope, the combination RuCl2(PPh3)3/NaOAc (entry 8) provided more satisfactory results and, thus, was chosen to develop our methodology.17b Other metalated carboxylate bases were effective but they failed to provide better results17b and, contrary to all expectations, some organic bases, such as Et3N (entry 9), were found to be reactive. The crucial role of the bidentate structure was then highlighted, thanks to a survey of different directing groups (Fig. 1) and the reaction appears to be specific to the 8-aminoquinoline motif.
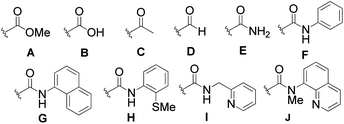 | ||
| Fig. 1 Ineffective directing groups. | ||
Scope and limitation
With optimized conditions in hand, the scope of the reaction was expanded to some representative amides. o-Substituted amides were initially examined (Table 2, entries 1–5) and high yields were obtained with o-phenyl (1b) or o-CF3 groups (1c). Difficulties were encountered in the cases of o-methoxy (1d) and o-fluorine (1e) groups, which afforded lower yields. Longer reaction times or higher temperatures failed to improve these results. It has been observed in the literature that, in the same configuration, cleavage of the C–O bond may occur5c,18 and that o-fluorine can affect reactivity.5c However, these groups are more easily tolerated in our system and no reduced compound 3fa or 1f corresponding to the cleavage of the C–O bond were detected. In the case of p-substituted amides (entries 6–9), the formation of di-alkylated amides 6 was preferred. We were unable to avoid the formation of compound 6, even when the amount of 2a used (entry 6a) or the reaction times were reduced and finally decided to optimize the formation of compound 6 by using 3 equivalents of 2a. In this manner, 6 was consistently obtained in quantitative yields, while the formation of the di-alkylated product was not absolutely predominant in previous studies.5,6,18|
|
||||
|---|---|---|---|---|
| Entry | t (h) | 1, R | 3 (%) | 6 (%) |
| a Numbers in parentheses are recovered 1. b [RuCl2(p-cymene)]2 (5 mol%) was used. c MVK 2a: 1 equiv. d MVK 2a: 3 equiv. e 1 mmol scale. | ||||
| 1 | 4 | 1a, 2-Me | 3aa, 94 | — |
| 2 | 4 | 1b, 2-Ph | 3ba, 91 (<5) | — |
| 3 | 4 | 1c, 2-CF3 | 3ca, 98 | — |
| 4b | 4 | 1d, 2-OMe | 3da, 63 (23) | — |
| 5 | 4 | 1e, 2-F | 3ea, 60 (25) | — |
| 6ac | 4 | 1f, 4-H | 3fa, 43 (31) | 6fa, 21 |
| 6bd | 4 | 1f, 4-H | 3fa, 0 | 6fa, 96 |
| 7d | 6 | 1g, 4-Me | 3ga, 0 | 6ga, 98 |
| 8d | 6 | 1h, 4-CO2Me | 3ha, 0 | 6ha, 97 |
| 9d,e | 6 | 1i, 4-OMe | 3ia, 0 | 6ia, 97 |
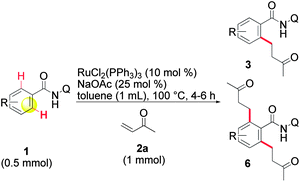
A series of more delicate m-substituted amides were investigated next (Table 3). In this arrangement, a second C–H activation leading to 6 is, also, likely to occur. Indeed, the m-methyl amide 1j gave the desired 3ja as the major product, but 6ja was also isolated (entry 1a). The formation of 6ja could be slightly minimized by using the bulky base NaOPiv (entry 1b). In the case of the m-phenyl (1k) and m-trifluoromethyl (1l), the alkylation took place exclusively at the less sterically demanding position, affording 3ka and 3la in high yields (entries 2 and 3). In contrast, in the case of an m-methoxy group (entry 4a) the formation of 6ma was strongly facilitated. Moreover, the mono-alkylation product was obtained as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers 3ma and 3′ma. However, the formation of 6ma may cloud the real value of this ratio and it was not possible to reverse this by using NaOPiv (entry 4b). This effect can be overcome by protecting the oxygen with a trifluoromethyl group (entry 5a),11d,18 affording 3na in a good yield without a trace of the regioisomer 3′na. In addition, only a very small amount of 6na was isolated when the reaction was carried out in the presence of NaOPiv (entry 5b). This reverse of the regioselectivity was not observed with an m-dimethylamino group (entry 6),18 therefore, only 3oa was isolated. The reaction offers a high tolerance to halogen atoms (entries 7–10), even the delicate iodide 1p survived in our reaction conditions. In general terms, without regard to the fluorine-containing 1s, the yield of 6 increases as the size of the halogen substituent decreases but, once again, it can be lowered by using NaOPiv. In the special case of the fluorine-containing 1s (entries 10a and b), the regioselectivity of the alkylation was completely reversed, as evidenced by the exclusive formation of 3′sa, irrespective of the base used. The effects that control this reactivity and the ratio of 3, 3′ and 6, are not well defined, as of this writing. Nonetheless, the results for m-substituted amides appear to be in line with those reported in the literature for the same type of reactions.5c,18 It is entirely possible that a second complexation of the metal by the lone pair of electrons on the m-substituent could direct the activation to the most substituted side. A greater stabilization of the oxidative-addition intermediate, thanks to an additional coordination to the metal, can also explain this reactivity.
:1 mixture of regioisomers 3ma and 3′ma. However, the formation of 6ma may cloud the real value of this ratio and it was not possible to reverse this by using NaOPiv (entry 4b). This effect can be overcome by protecting the oxygen with a trifluoromethyl group (entry 5a),11d,18 affording 3na in a good yield without a trace of the regioisomer 3′na. In addition, only a very small amount of 6na was isolated when the reaction was carried out in the presence of NaOPiv (entry 5b). This reverse of the regioselectivity was not observed with an m-dimethylamino group (entry 6),18 therefore, only 3oa was isolated. The reaction offers a high tolerance to halogen atoms (entries 7–10), even the delicate iodide 1p survived in our reaction conditions. In general terms, without regard to the fluorine-containing 1s, the yield of 6 increases as the size of the halogen substituent decreases but, once again, it can be lowered by using NaOPiv. In the special case of the fluorine-containing 1s (entries 10a and b), the regioselectivity of the alkylation was completely reversed, as evidenced by the exclusive formation of 3′sa, irrespective of the base used. The effects that control this reactivity and the ratio of 3, 3′ and 6, are not well defined, as of this writing. Nonetheless, the results for m-substituted amides appear to be in line with those reported in the literature for the same type of reactions.5c,18 It is entirely possible that a second complexation of the metal by the lone pair of electrons on the m-substituent could direct the activation to the most substituted side. A greater stabilization of the oxidative-addition intermediate, thanks to an additional coordination to the metal, can also explain this reactivity.
|
|
|||
|---|---|---|---|
| Entry | 1, R | 3 (%) | 6 (%) |
|
a Numbers in parentheses are recovered 1.
b NaOPiv (25 mol%) as base.
c Isolated as a mixture of 3ma and 3′ma (NMR ratio 3ma:3′ma = 3:1).
|
|||
| 1a | 1j, Me | 3ja, 77 | 6ja, 17 |
| 1bb | 1j, Me | 3ja, 84 (6) | 6ja, 8 |
| 2 | 1k, Ph | 3ka, 91 | 6ka, 0 |
| 3 | 1l, CF3 | 3la, 98 | 6la, 0 |
| 4a | 1m, OMe | 30c | 6ma, 35 |
| 4bb | 1m, OMe | 42c | 6ma, 43 |
| 5a | 1n, OCF3 | 3na, 63 (14) | 6na, 18 |
| 5bb | 1n, OCF3 | 3na, 70 (10) | 6na, 8 |
| 6 | 1o, NMe2 | 3oa, 61(22) | 6oa, 0 |
| 7a | 1p, I | 3pa, 52 | 6pa, 21 |
| 7bb | 1p, I | 3pa, 58 | 6pa, 13 |
| 8a | 1q, Br | 3qa, 35 | 6qa, 54 |
| 8bb | 1q, Br | 3qa, 65 | 6qa, 24 |
| 9a | 1r, Cl | 3ra, 22 | 6ra, 70 |
| 9bb | 1r, Cl | 3ra, 43 | 6ra, 38 |
| 10a | 1s, F | 3′sa, 24 | 6sa, 44 |
| 10bb | 1s, F | 3′sa, 29 | 6sa, 61 |
This concise overview was then expanded by testing a series of polysubstituted amides (Scheme 2). A wide range of original molecular frameworks were amenable for use. We also noted an ease of access to bulky C–H bonds (8aa and 8ba), as well as the possibility of using heteroaromatic substrates. Interestingly the indole core furnished a di-alkylated by-product 8′ia.
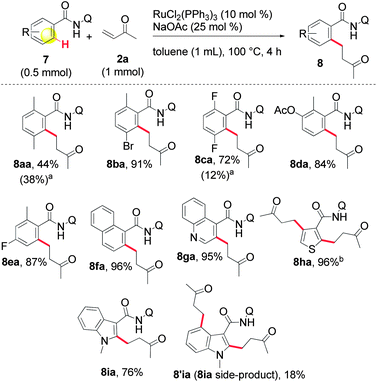 | ||
| Scheme 2 Alkylation of various amides with MVK 2a. a Recovered 7 in parentheses. b 3 equiv. of MVK 2a were used. | ||
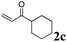
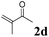
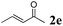
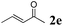
The second part of our work consisted in varying the nature of the acceptor molecules (Table 4). The reactivities of aliphatic enones 2b and 2c were very close to that of MVK 2a when examined in a reaction with amide 1a and the final products 3ab and 3ac were produced in very good yields. However, more sterically hindered double bonds proved to be more reluctant. For example, high temperatures were necessary (140 °C, entry 3a vs. 3b) to totally consume 1a if 2d was used. As expected, bulkier internal double bonds 2e and 2f are more resilient and the alkylation proceeded with moderate efficacy at a temperature of 140 °C. The more electron poor aromatic 1c provided similar results when it was reacted with 2e. Under the same conditions, olefins that were non-conjugated with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (styrene, n-hexene) failed to react with 1a. This suggests that the CO bond plays an important role in the reaction mechanism.
O bond (styrene, n-hexene) failed to react with 1a. This suggests that the CO bond plays an important role in the reaction mechanism.
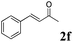
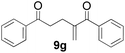
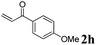
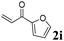
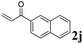
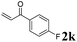
Phenyl vinyl ketone (PVK, 2g) and some derivatives of PVK were then considered (Table 5). From a general point of view, as the acceptors become more electrophilic, the yields of product 3 decrease. In this respect, the more electron-rich 2h and 2i afforded 3ah and 3ai in good yields, whereas enones bearing an electron-withdrawing group (2j–m) yielded 3aj and 3ak in moderate yields and 3al and 3am in low yields. We also noted that when reacted with PVK, the more electron deficient amide 1c (entry 2) provided better results than 1a. PVK derivatives offered a singular reactivity, in that we were able to isolate a side-product (as 9g derived from 2g) in a yield of around 20%. The side-product 9g, arising from the self-condensation of 2g, may be formed via a Rauhut–Currier reaction (Morita–Baylis–Hillman analogous), which is known to afford these types of adducts from α,β-unsaturated ketones.19 Nucleophilic species that are present in the reaction medium can react with 2g and thus initiate this background process.
|
|
|||
|---|---|---|---|
| Entry | 1, R | 2 | 3 , , (%) |
| a Numbers in parentheses are recovered 1. b 3ah–m were isolated by HPLC. c Longer reaction times or higher temperatures did not improve the yields. | |||
| 1 | 1a, 2-Me |
|
3ag, 71 (22) |
| 2 | 1c, 3-CF3 |
|
3cg, 88 |
| 3 | 1a, 2Me |
|
3ah, 78 (16) |
| 4 | 1a, 2-Me |
|
3ai, 81 (12) |
| 5 | 1a, 2-Me |
|
3aj, 61 (35) |
| 6 | 1a, 2-Me |
|
3ak, 51 (32) |
| 7 | 1a, 2-Me |
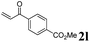
|
3al, 31 (60) |
| 8 | 1a, 2-Me |
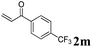
|
3am, 14 (60) |
The potential of the present methodology was expanded through the facile conversion of the amide moiety into a variety of different useful functional groups.9–11 For example, to illustrate this statement, the use of Schwartz's reagent allowed the formation of aldehydes 10 from amides 3 under mild conditions (Scheme 3), even with the bulky amide 3aa. Under these conditions, the prior protection of the ketone was required to achieve good yields. The carboxylic acid can also be obtained via basic hydrolysis of the N-methylated amide (Scheme 3).9e
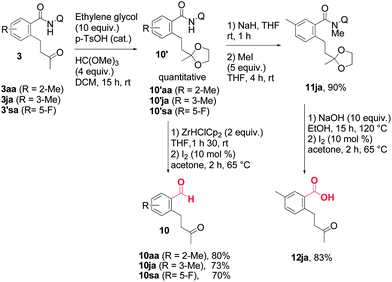 | ||
| Scheme 3 Cleavage of the 8-aminoquinoline directing group. | ||
Mechanistic investigation
In the last stage of our study, some aspects of the mechanism of the reaction were investigated. When the reaction was run with isotopically labeled substrates 13, a fast H/D scrambling was detected, indicating that C–H bond cleavage is not likely the rate determining step and is thus reversible in nature (eqn (3) and (4)). Contrary to our expectations, no clear evidence for the incorporation of deuterium into product 13a was found. This left some doubts about the origin of the incorporated hydrogen.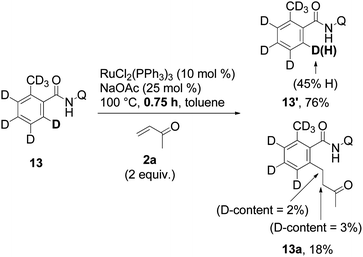 | (3) |
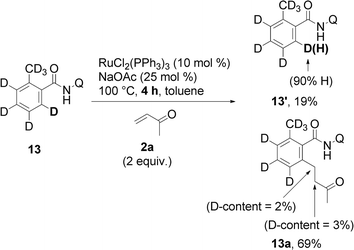 | (4) |
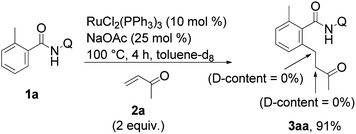
No trace of deuterium incorporation was detected when the reaction was performed in toluene-d8 with amide 1a (eqn (5)).
 | (5) |
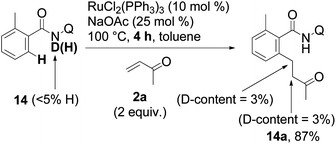
We then investigated whether the hydrogen of the N–H bond may act as a source of hydrogen. When substrate 14 was employed, a poor deuterium incorporation was observed (eqn (6)).
 | (6) |
Then the reaction was performed with deuterium-containing substrate 15 (eqn (7)). A larger incorporation of deuterium into the final product was finally observed, compared with eqn (4). Incorporation of hydrogen into the recovered starting material (15′) was also observed as in eqn (4), but not to the same degree.20
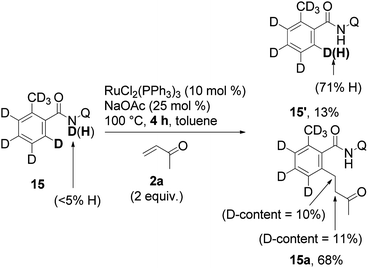 | (7) |
When the reaction was carried out in absence of acceptor 2a (eqn (8)), H/D exchange was detected. It therefore appears that the cleavage of the C–H bond can take place, even in absence of the acceptor.
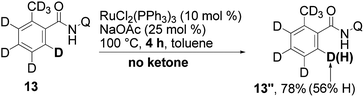 | (8) |
Interestingly, when the same reaction was carried out with substrate 14, 30% of deuterium incorporation at the ortho position of the amide was found (eqn (9)). It appears that cleavage of the N–H bond likely occurs in the mechanism.
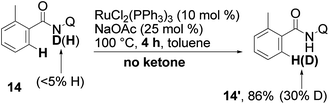 | (9) |
The solvent did not have any impact on these processes, as evidenced by the total absence of deuterium incorporation when the reaction was run in toluene-d8 (eqn (10)).
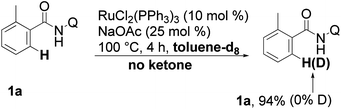 | (10) |
Almost no hydrogen incorporation took place when the reaction was carried out with 15 (eqn (11)), confirming that hydrogen incorporation in eqn (8) was the result of an exchange with the hydrogen of the N–H bond.
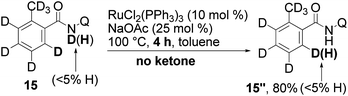 | (11) |
Competition experiments intended to shed light on electronic effects (Table 6) tend to support the view that the reaction is facilitated by an electron-deficient ring. However, our attempts to rationalize electronic effects through a Hammett plot were unsuccessful and a very bad correlation was obtained. This suggests that substituent effects are not governed by the Hammett equation and, thus, the reactivity is likely ruled by a subtle combination of electronic, steric and coordination effects.
|
|
||||
|---|---|---|---|---|
| Entry | 1AA, R1 | 1BB, R2 | 3AA (%) | 3BB (%) |
| a Yields were estimated from 1H NMR spectra. | ||||
| 1 | 1l, CF3 | 1j, Me | 3la, 80 | 3ja, <6 |
| 2 | 1k, Ph | 1j, Me | 3ka, 59 | 3ja, 14 |
| 3 | 1l, CF3 | 1m, OMe | 3la, 81 | 3ma, <6 |
| 4 | 1l, CF3 | 1o, NMe2 | 3la, 71 | 3oa, <6 |
| 5 | 1o, NMe2 | 1j, Me | 3oa, 24 | 3ja, 34 |
The original mechanism of the chelation-assisted ruthenium-catalyzed ortho alkylation of aromatic substrates with olefins, suggested by Murai and co-workers,4,6 involves the formation of a key ruthenium-hydride intermediate via activation of the C–H bond. Reversible insertion of the olefin into the Ru–H bond (hydrometalation type mechanism), followed by a reductive elimination of the alkyl group,21 leads to the formation of the expected C–C bond. As things stand, we cannot assert definitively that such a mechanism occurs in the present reaction, despite that some similarities are observed. In particular, the involvement of a ruthenium-hydride species still has to be clarified. It appears too soon to draw any conclusions concerning the nature of the mechanism and more investigations are currently being pursued.
Conclusion
In summary, the ability of an 8-aminoquinoline bidentate directing group in promoting a new ruthenium-catalyzed C–H bond ortho alkylation of aromatic amides with various α,β-unsaturated ketones under straightforward reaction conditions is reported. This methodology highlights the fact that bidentate directing groups can be used as an efficient tool for achieving transformations that proceed with difficulty when conventional methods are used and represents the first utilization of a family of α,β-unsaturated acceptors in the “ortho directed ruthenium-catalyzed addition of C–H bonds to C–C double bonds”. Thus, a new way to expand the scope of the ruthenium-catalyzed transformations initiated by Murai et al. 20 years ago has been achieved.3,22 It is also noteworthy that no ruthenium-catalyzed C–H bond alkylation of secondary benzamides with olefins via chelation assistance by using an amide directing group has been reported to date.23 Furthermore, the attractiveness and flexibility of the catalyst systems used, open numerous perspectives for the further development of this strategy. The present method offers a high functional group tolerance, provides fast and economical access to very interesting and functionalizable molecular backbones, and is enhanced by the potential transformation of the amide moiety to various functional groups. The mechanism of the reaction remains unclear and more investigations into this aspect of the reaction are currently in progress. Extensions of the methodology are also currently being pursued in our laboratory and will be reported in due course.Acknowledgements
This work was supported, in part, by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis” from the Ministry of Education, Culture, Sports, Science and Technology.Notes and references
- (a) J. Tsuji, Transition Metal Reagents and Catalysts: Innovations in Organic Synthesis, Wiley-VCH, Chichester, 2000 Search PubMed; (b) Transition Metals for Organic Chemistry, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- Some selected recent reviews: (a) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826 CrossRef CAS; (b) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS; (c) Y. J. Park and C. H. Jun, Bull. Korean Chem. Soc., 2005, 26, 871 CrossRef CAS; (d) Top. Organomet. Chem.: Directed Metallation, ed. N. Chatani, Springer, Heidelberg/New York, 2007, vol. 24 Search PubMed; (e) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013 CrossRef CAS; (f) L. Ackermann, R. Vincente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (g) P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15, 5874 CrossRef CAS; (h) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (i) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS; (j) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS; (k) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (l) L. Ackermann, Chem. Commun., 2010, 46, 4866 RSC; (m) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS; (n) K. Hirano and M. Miura, Synlett, 2011, 294 CAS; (o) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (p) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (q) J. Yamaguchi, A. D. Yamaguchi and H. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS; (r) P. Beatrice, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef; (s) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (t) D. Y. K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452 CrossRef CAS.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- For an overview: F. Kakiuchi, in Top. Organomet. Chem.: Directed Metallation, ed. N. Chatani, Springer, Heidelberg/New York, 2007, vol. 24, 1 Search PubMed.
- (a) R. Martinez, R. Chevalier, S. Darses and J. P. Genet, Angew. Chem., Int. Ed., 2006, 45, 8232 CrossRef CAS; (b) R. Martinez, J. P. Genet and S. Darses, Chem. Commun., 2008, 3855 RSC; (c) R. Martinez, M. O. Simon, R. Chevalier, C. Pautigny, J. P. Genet and S. Darses, J. Am. Chem. Soc., 2009, 131, 7887 CrossRef CAS; (d) M. O. Simon, R. Martinez, J. P. Genet and S. Darses, J. Org. Chem., 2010, 75, 208 CrossRef CAS. For other systems, see also: (e) Y. Guari, S. Sabo-Etienne and B. Chaudret, J. Am. Chem. Soc., 1998, 120, 4228 CrossRef CAS; (f) S. Bush and W. Leitner, Chem. Commun., 1999, 2305 RSC.
- F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda, N. Chatani and S. Murai, Bull. Chem. Soc. Jpn., 1995, 68, 62 CrossRef CAS.
- (a) S. G. Lim, J. A. Ahn and C. H. Jun, Org. Lett., 2004, 6, 4687 CrossRef CAS; (b) Y. Kuninobu, Y. Nishina, K. Okaguchi, M. Shouho and K. Takai, Bull. Chem. Soc. Jpn., 2008, 81, 1393 CrossRef CAS; (c) L. Yang, C. Correia and C. Li, Org. Biomol. Chem., 2011, 9, 7176 RSC; (d) L. Yang, B. Qian and H. Huang, Chem.–Eur. J., 2012, 18, 9511 CrossRef CAS.
- (a) T. Naota, H. Takaya and S.-I. Murahashi, Chem. Rev., 1998, 98, 2599 CrossRef CAS; (b) B. M. Trost, F. D. Toste and A. B. Pinkerton, Chem. Rev., 2001, 101, 2067 CrossRef CAS; (c) B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630 CrossRef CAS.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS; (c) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7 CrossRef CAS; (d) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS; (e) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS.
- (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS; (b) R. Giri, N. Maugel, B. M. Foxman and J.-Q. Yu, Organometallics, 2008, 27, 1667 CrossRef CAS; (c) F.-R. Gou, X.-C. Wang, P.-F. Huo, H.-P. Bi, Z.-H. Guan and Y.-M. Liang, Org. Lett., 2009, 11, 5726 CrossRef CAS; (d) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958 CrossRef CAS; (e) Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414 CrossRef CAS; (f) G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192 CrossRef CAS; (g) Y. Zhao and G. Chen, Org. Lett., 2011, 13, 4850 CrossRef CAS; (h) W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076 CrossRef CAS; (i) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS; (j) Y. Xie, Y. Yang, L. Huang, X. Zhang and Y. Zhang, Org. Lett., 2012, 14, 1238 CrossRef CAS; (k) M. R. Yadav, R. K. Rit and A. K. Sahoo, Chem.–Eur. J., 2012, 18, 5541 CrossRef CAS; (l) S.-Y. Zhang, G. He, Y. Zhao, K. Wright, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2012, 134, 7313 CrossRef CAS; (m) G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944 CrossRef CAS; (n) Y. Zhao, G. He, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2948 CrossRef CAS; (o) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507 CrossRef CAS; (p) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2012, 14, 3724 CrossRef CAS; (q) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS; (r) N. Rodríguez, J. Á. Romero-Revilla, M. A. Fernández-Ibáñez and J. C. Carretero, Chem. Sci., 2013, 4, 175 RSC.
- (a) S. Inoue, H. Shiota, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2009, 131, 6898 CrossRef CAS; (b) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS; (c) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070 CrossRef CAS; (d) H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 14952 CrossRef CAS; (e) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 14, 354 CrossRef CAS; (f) K. Shibata, N. Hasegawa, Y. Fukumoto and N. Chatani, ChemCatChem, 2012, 4, 1733 CrossRef CAS; (g) Y. Aihara and N. Chatani, Chem. Sci., 2013, 4, 664 RSC.
- Bidentate directing groups are notably known for their ability to retard the β-hydride elimination of the metal and to stabilize high oxidation state metalated species (ref. 9b). Therefore, in a first approach, we thought that they could help allow the formation and exploitation of new types of metalated intermediates.
- During the preparation of our manuscript, a relatively similar procedure for the alkylation of phenylpyridine derivatives with unactivated alkenes was reported: M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013 DOI:10.1002/anie.201208446.
- Formation of dehydrogenative products in Ru3(CO)12 catalyzed C–H activation was already reported: F. Kakiuchi, T. Sato, M. Yamauchi, N. Chatani and S. Murai, Chem. Lett., 1999, 19 CrossRef CAS.
- For a review, see: L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS.
- Some recent representative papers: (a) P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 3075 RSC; (b) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728 CrossRef CAS; (c) B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Org. Lett., 2012, 14, 736 CrossRef CAS; (d) K. Padala and M. Jeganmohan, Org. Lett., 2012, 14, 1134 CrossRef CAS; (e) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC.
- (a) However, if m- or p-substituted amides are used, a catalytic amount of PPh3 is necessary for the reaction to proceed (see optimization tables in the ESI‡); (b) see ESI‡ for detailed optimization investigations.
- M. Sonoda, F. Kakiuchi, N. Chatani and S. Murai, Bull. Chem. Soc. Jpn., 1997, 70, 3117 CrossRef CAS.
- (a) A review dealing with the Rauhut–Currier reaction: C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069 CrossRef CAS; (b) the formation of this side-product was not observed with aliphatic enones investigated in our work; (c) side-products 9 derived from other PVK derivatives are described in the ESI‡.
- H/D exchange with the hydrogens of the olefin may also occur, as was already reported by Murai and co-workers for similar alkylation reactions with triethoxyvinylsilane (see ref. 6 and the publication mentioned in ref. 21).
- In the case of aromatic ester substrates, it was suggested that the formation of the C–C bond proceeds through a nucleophilic migration of the alkyl group, which is facilitated by electron-withdrawing groups on the aromatic ring, see: F. Kakiuchi, H. Ohtaki, M. Sonoda, N. Chatani and S. Murai, Chem. Lett., 2001, 918 CrossRef CAS.
- For a pioneering study, see: L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728 CrossRef CAS.
- For selected recent reports with other metals, see: (a) R. Aufdenblatten, S. Diezi and A. Togni, Monatsh. Chem., 2000, 131, 1345 CrossRef CAS; (b) L. Ilies, Q. Chen, X. Zeng and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 5221 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Irina Petrovna Beletskaya for her contribution to metal-catalyzed reactions. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and spectroscopic data. See DOI: 10.1039/c3sc50310k |
| This journal is © The Royal Society of Chemistry 2013 |