 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anhydrides as α,β-unsaturated acyl ammonium precursors: isothiourea-promoted catalytic asymmetric annulation processes†
Emily R. T.
Robinson
,
Charlene
Fallan
,
Carmen
Simal
,
Alexandra M. Z.
Slawin
and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
First published on 1st March 2013
Abstract
The asymmetric annulation of a range of α,β-unsaturated acyl ammonium intermediates, formed from isothiourea HBTM 2.1 and anhydrides with either 1,3-dicarbonyls, β-ketoesters or azaaryl ketones gives either functionalised esters (upon ring opening), dihydropyranones or dihydropyridones in good yields (up to 93%) and high enantioselectivity (up to 97% ee).
Introduction and background
Asymmetric organocatalysis has developed tremendously as a synthetic strategy within the last decade and a range of methodologies and catalysts have emerged that provide functionalised products with high levels of stereocontrol.1 Ideally a given organocatalyst architecture should be able to participate in a range of reaction processes and display diverse modes of reactivity, while showing good catalytic efficiency and delivering products with high levels of enantioselectivity. Within this area, isothioureas,2 initially employed by Birman and Okamoto as efficient O-acyl transfer reagents,3 have been utilised in a range of kinetic resolution,4 asymmetric desymmetrisation,5C-acylation and C-carboxylation processes,6 as well as O-silylation reactions.7 Recent advances have showcased the utility of isothioureas to generate ammonium enolates8 from carboxylic acids and their applications in aldol-9 and Michael-lactonisation processes (Fig. 1A).10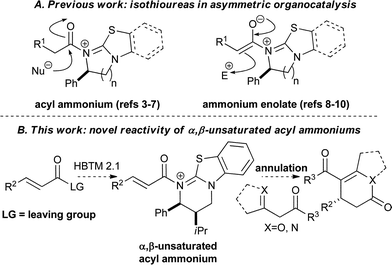 | ||
| Fig. 1 Proposed access to enantioenriched annulation products via an unexplored α,β-unsaturated acyl ammonium species. | ||
Building upon these precedents, this work demonstrates the previously unexplored ability of isothioureas to generate asymmetry by promoting the addition of a range of nucleophiles to a stereodefined α,β-unsaturated acyl ammonium species (Fig. 1B). While Peters and Ye have invoked α,β-unsaturated acyl ammoniums as precursors to dienolate formation,11 to the best of our knowledge there are currently no processes that form C–C bonds directly via such intermediates. Related work in the literature has shown that NHCs12 can catalytically generate α,β-unsaturated acyl azolium intermediates through an internal redox process from alkynals,13 from enals using a stoichiometric oxidant,14 directly from α,β-unsaturated acyl fluorides or enol esters,15 or alternatively from α-bromoenones.16 The oxidative approach from enals has been applied to a range of asymmetric C–C bond-forming reactions including aza-Claisen,17 Coates-Claisen,18 cyclopropanation19 and Michael addition processes.20 Given the recognised difficulties in accessing a wide variety of ynals and enals for such NHC-catalysed approaches and limitations associated with the scope and generality of such processes,21 we envisaged a direct strategy to generate an α,β-unsaturated acyl ammonium species from readily available α,β-unsaturated carboxylic acids or their anhydrides. Described herein are our results concerning isothiourea-promoted asymmetric addition of 1,3-diketones, β-ketoesters and azaaryl ketones to α,β-unsaturated acyl ammonium intermediates for the preparation of a range of functionalised esters, stereodefined dihydropyranones and dihydropyridones in highly enantioenriched form (up to 97% ee).
Proof of concept and reaction optimisation: annulations using diketone nucleophiles
Initial investigations focused upon generation of an α,β-unsaturated acyl ammonium directly from cinnamic acid 1 (Scheme 1A), via in situ activation using 4-methoxybenzoic anhydride (PMBA) and isothiourea HBTM 2.1 5 (20 mol%).22 Employing diketone 4 as the nucleophile provided dihydropyranone 2 in modest 25% isolated yield, albeit with an encouraging 95% ee. Cinnamic anhydride 3 was next evaluated as an alternative acyl ammonium precursor,23 giving dihydropyranone 2 in improved 49% yield and 95% ee (Scheme 1B). These initial findings served as a benchmark for further optimisation with the aim to lower catalyst loadings and improve isolated yields.24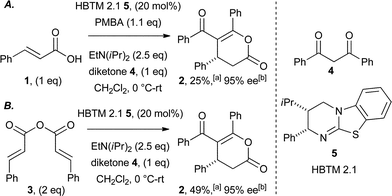 | ||
| Scheme 1 Initial proof of concept studies. aIsolated yield of 2; bdetermined by HPLC analysis. | ||
Further studies showed that in situ ring opening of dihydropyranone 2 with MeOH led to consistently higher isolated yields of the functionalised ester product 6 (Table 1). Variation of the base showed that EtN(iPr)2, DBU or PS-BEMP (polymer-supported 2-t-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine) could be used in this process, although DBU gave 6 with reduced enantioselectivity (entry 5).25 The use of PS-BEMP proved optimal, allowing the catalyst loading of HBTM 2.1 5 to be reduced to 1 mol% without compromising product enantioselectivity, albeit with reduced product yields (entry 11). Performing the reaction in THF led to a reduced yield (entries 4 and 9). While catalytic asymmetric Michael additions to nitro-olefins and enones are well documented,26 this strategy formally allows the asymmetric Michael addition of diketones to α,β-unsaturated carboxylic acid derivatives for which there is only limited precedent.27
|
|
||||
|---|---|---|---|---|
| Entry | HBTM 2.1 (mol%) | Base | Yielda | eeb (%) |
| a Isolated yield of 6. b Determined by HPLC analysis. c Conversion determined by 1H NMR spectroscopic analysis of the unpurified reaction mixture. d Reaction conducted in THF. | ||||
| 1 | 20 | EtN(iPr)2 | 77 | 96 |
| 2 | 10 | EtN(iPr)2 | 70 | 95 |
| 3 | 5 | EtN(iPr)2 | 62c | — |
| 4d | 5 | EtN(iPr)2 | 45 | 99 |
| 5 | 20 | DBU | 86 | 73 |
| 6 | 20 | PS-BEMP | 85 | 95 |
| 7 | 10 | PS-BEMP | 82 | 95 |
| 8 | 5 | PS-BEMP | 83 | 96 |
| 9d | 5 | PS-BEMP | 50 | 83 |
| 10 | 2.5 | PS-BEMP | 69 | 93 |
| 11 | 1 | PS-BEMP | 62 | 93 |
Reaction scope and generality
The generality of this process was next probed, initially through variation of the α,β-unsaturated anhydride component (Table 2). Using HBTM 2.1 5 (5 mol%) and a number of symmetrical diketones, this protocol tolerates a range of 2-, 3-, and 4-substituted β-aryl groups containing either electron-withdrawing or electron-donating groups, as well as heteroaryl substituents, giving the corresponding functionalised esters in good yield (up to 86%) and high enantioselectivity (90–97% ee, 6–14). Notable reactivity trends within this series indicate that anhydrides containing electron deficient β-aryl units give higher product conversion and isolated yields than their electron rich β-aryl counterparts (product 9). β-Alkyl substituents within the anhydride are also tolerated (a significant advantage over the NHC-catalysed systems that typically exhibit low enantioselectivity in similar transformations),20b although low temperatures are necessary to achieve optimal enantioselectivity, resulting in only moderate reaction efficiency and reduced product yields (15 and 16). Variation of the diketone functionality was next investigated (Table 2, 17–21). A range of substituted aryl and heteroaryl diketones participate in this reaction process, giving functionalised esters 17–21 in moderate to good yield and high enantioselectivity.28
Further investigations probed the stereospecificity of this asymmetric annulation protocol (Scheme 2). While (E,E)-cinnamic anhydride 3 gave functionalised ester (S)-6 in 83% yield and 96% ee (Scheme 2A), (Z,Z)-cinnamic anhydride 22 gave (R)-6 in reduced 41% yield and only 30% ee (Scheme 2B),29 indicating the necessity of the (E)-configuration for maximum enantiocontrol.
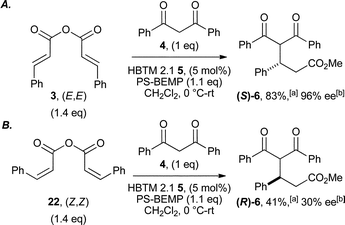 | ||
| Scheme 2 Stereospecificity of asymmetric annulation process. aIsolated yield of 6; bdetermined by HPLC analysis. | ||
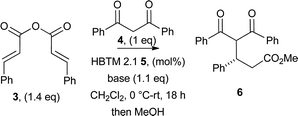
Subsequent studies probed the ability of non-symmetric dicarbonyls to participate in this protocol. Using cinnamic anhydride 3, 1-phenyl-1,3-dibutanone 23 generated a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of regioisomeric dihydropyranones 24 and 25 in 88% overall yield and 70% and 61% ee, respectively (Scheme 3A),30 while ethyl benzoylacetate 26 gave dihydropyranone 27 in 60% isolated yield as a single regioisomer and in 94% ee (Scheme 3B).
:30 mixture of regioisomeric dihydropyranones 24 and 25 in 88% overall yield and 70% and 61% ee, respectively (Scheme 3A),30 while ethyl benzoylacetate 26 gave dihydropyranone 27 in 60% isolated yield as a single regioisomer and in 94% ee (Scheme 3B).
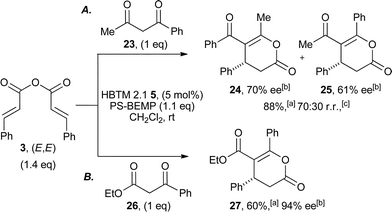 | ||
| Scheme 3 Regioselectivity of annulation using unsymmetrical nucleophiles. aIsolated yield; bdetermined by HPLC analysis. | ||
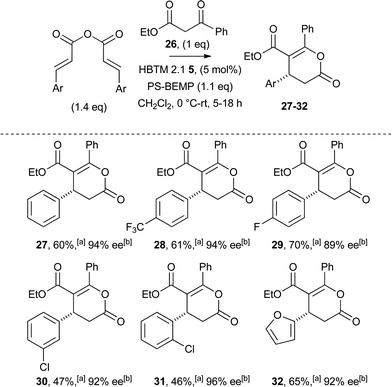
The generality of this process was next examined (Table 3), with β-aryl and β-heteroaryl substituents within the anhydride tolerated, in all cases giving the corresponding dihydropyranones 27–32 in acceptable yield (46–70%) and high ee (89–96%).31
Having probed the viability of this process, our attention turned to 1,3-dicarbonyl systems that are not tolerated in related NHC-catalysed processes. For example, aliphatic cyclic Michael donors such as 1,3-cyclohexanedione 33 have been reported by Bode to be ineffective in NHC-catalysis using α,β-unsaturated acyl azoliums.21 However, in this organocatalysed process, diketone 33 demonstrated favourable reactivity and provided 34 in high yield and good enantioselectivity (Scheme 4).
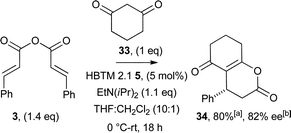 | ||
| Scheme 4 1,3-Cyclohexanedione as a Michael donor. aIsolated yield; bdetermined by HPLC analysis. | ||
Beyond 1,3-dicarbonyls: azaaryl ketones as nucleophiles
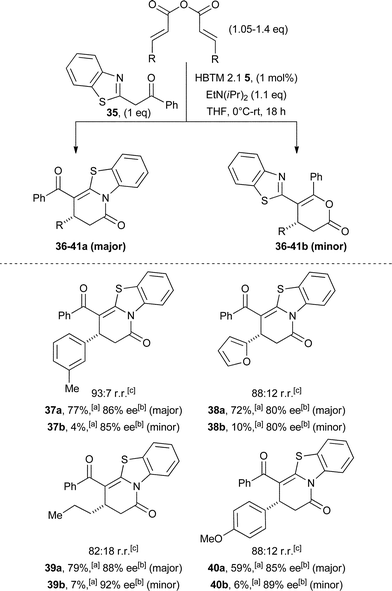
Encouraged by the novel and complementary reactivity of the isothiourea catalysis, the scope of this asymmetric annulation was extended beyond the use of 1,3-dicarbonyl nucleophiles. Gratifyingly, azaaryl ketone 35 proved a competent nucleophile and displayed improved reactivity compared with simple 1,3-dicarbonyls. This increased reactivity allows bench grade solvents in an open flask atmosphere to be employed, PS-BEMP can be replaced with more cost efficient EtN(iPr)2 and a lower catalyst loading of 1 mol% could be routinely employed. The use of this nucleophile led to intriguing regioselectivity. Cyclisation occurs preferentially through the benzothiazole nitrogen, generating dihydropyridone 36a as the major product, in addition to dihydropyranone 36b as the minor product (88:12 regioisomeric ratio). These heterocycles were readily separable by chromatography, with the major product 36a obtained in excellent yield and high enantioselectivity (97% ee after a single recystallisation, Scheme 5).
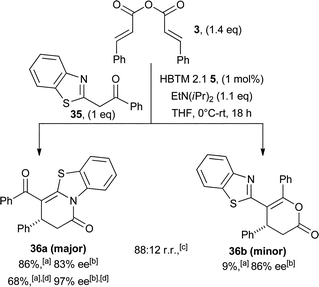 | ||
| Scheme 5 Asymmetric addition of azaaryl ketone 35. aIsolated yield; bdetermined by HPLC analysis; cdetermined from 1H NMR of unpurified reaction mixture; dfollowing a single recrystallisation. | ||
Next, the substrate scope with this nucleophile was examined with respect to the anhydride component (Table 4). The increased reactivity of the azaaryl ketone 35 provided a wide range of enantioenriched heterocycles in excellent yields and enantioselectivities. For example, while anhydrides bearing electron-rich and aliphatic substituents were modestly tolerated using diketone 4 (Table 2) in terms of both reactivity and enantioselectivity, the use of azaaryl ketone 35 with the same anhydrides leads to enantioenriched products in high yields under our reaction conditions (Table 4).
Mechanistic investigations
In related NHC-catalysed processes involving α,β-unsaturated acyl azolium intermediates two potential mechanistic pathways have been proposed; a Michael addition–lactonisation process with dicarbonyls, ketene acetals and enamines (favoured by Studer and Mayr)32 or alternatively an initial 1,2-addition followed by a [3,3]-Claisen rearrangement with Kojic acid and enamine derivatives (favoured by Bode)21 to facilitate the formation of enantioenriched products. Similarly, in our isothiourea-promoted annulation, related catalytic cycles depicted in Fig. 2(a) and (b), could potentially be responsible for the generation of the annulation products with high enantiocontrol. Both cycles involve an initial N-acylation of HBTM 2.1 5 with an anhydride to generate the corresponding α,β-unsaturated acyl ammonium 41. The s-cis conformation of ammonium 41 is presumably favoured, with the carbonyl oxygen hypothesised to adopt a syn-conformation with respect to the isothiourea S atom due to a stabilising non-bonding O–S interaction (no to σ*C–S).33 In pathway (a), Michael addition31 of diketone enolate 42 to the Re face of the α,β-unsaturated acyl ammonium 41 gives intermediate 43,34 subsequent proton transfer followed by lactonisation generates the desired dihydropyranone 44, which can be either isolated or subsequently ring opened with MeOH to generate the ester products 45 in high ee. Alternatively, pathway (b) demonstrates that 1,2-addition to the α,β-unsaturated acyl ammonium 42 to generate intermediate 46 followed by a [3,3]-rearrangement and subsequent proton transfer/lactonisation process would lead to the same enantioenriched products.![Proposed mechanisms of asymmetric dihydropyranone formation via (a) Michael addition–lactonisation or (b) 1,2-addition, [3,3]-rearrangement.](/image/article/2013/SC/c3sc50199j/c3sc50199j-f2.gif) | ||
| Fig. 2 Proposed mechanisms of asymmetric dihydropyranone formation via (a) Michael addition–lactonisation or (b) 1,2-addition, [3,3]-rearrangement. | ||
To gain insight into the favoured mechanistic pathway, potential intermediates were synthesised and subjected to the reaction conditions. Interestingly, Lupton has previously shown that 1,2-addition of NHCs to enol esters such as 47 facilitates formation of dihydropyranone 49 in the presence of NHC catalyst 48, albeit with moderate enantioselectivity (Scheme 6A). In this regard, 50 was prepared from cinnamoyl chloride and dicarbonyl 4. This potential [3,3]-rearrangement precursor was examined under the reaction conditions, resulting in no conversion into dihydropyranone 2 (Scheme 6B)15a–c after 24 h at room temperature. While this result does not rule out either mechanistic pathway, it indicates that 50 is not a likely intermediate in this process. Additionally the absence of any 1,2-addition products such as 50 in the 1H NMR of all unpurified reaction mixtures provides further evidence to support this view.
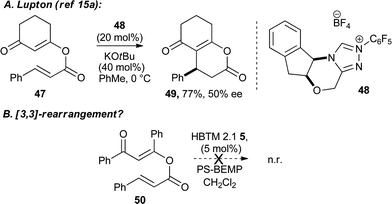 | ||
| Scheme 6 Probing the mechanism. | ||
Evidence supporting the intermediacy of an acyl ammonium species was next obtained. Treatment of trans-cinnamoyl chloride 51 with HBTM 2.1 5 in CH2Cl2 gave the α,β-unsaturated acyl ammonium salt 52 that was isolated in high yield (Scheme 7).35 X-ray crystallography confirmed the structure of 52 and provided further support for the syn geometry of the carbonyl oxygen and the isothiourea S atom, presumably due to the previously hypothesised stabilising non-bonding O–S interaction (no to σ*C–S).33
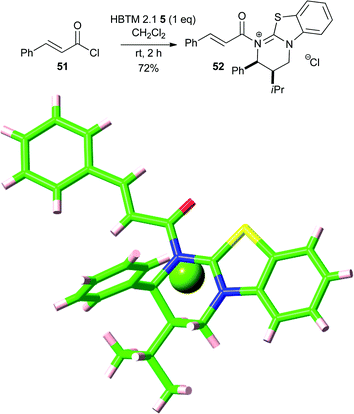 | ||
| Scheme 7 Isolation and representation of the X-ray crystal structure of α,β-unsaturated acyl ammonium salt 52. | ||
The isolated acyl ammonium salt 52 was next examined as a precatalyst, employing cinammic anhydride 3 and diketone 4 under our optimised reaction conditions (Scheme 8). The functionalised ester 6 was isolated in similar yield and enantioselectivity (82% yield, 91% ee) compared with the use of organocatalyst HBTM 2.1 5 (83% yield, 95% ee) in this process, consistent with acyl ammonium salt 52 being an intermediate in this asymmetric annulation.
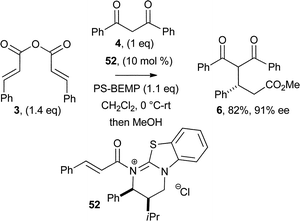 | ||
| Scheme 8 α,β-Unsaturated acyl ammonium salt 52 as organocatalyst. | ||
At this juncture we cannot rule out any plausible mechanistic pathway that involves an α,β-unsaturated acyl ammonium ion, although we currently favour a catalytic cycle involving a Michael addition–lactonisation sequence, described in Fig. 2(a). Further mechanistic investigations and DFT calculations to provide insight into the pathway in operation are underway and will be reported in due course.
Conclusions
To conclude, HBTM 2.1 5 promotes the asymmetric annulation of a range of nucleophiles including 1,3-diketones, β-ketoesters and azaaryl ketones to (E,E)-α,β-unsaturated anhydrides, giving either functionalised esters (upon ring opening), dihydropyranones, or dihydropyridones in good yields (up to 86%) and high enantioselectivity (up to 97% ee) via a postulated α,β-unsaturated acyl ammonium intermediate. Current research from this laboratory is directed toward developing alternative uses of isothioureas and other Lewis bases in asymmetric catalysis, and exploiting α,β-unsaturated acyl ammonium intermediates for a range of synthetic procedures.Acknowledgements
We thank the Royal Society for a University Research Fellowships (ADS), the EPSRC (ER) and the EU (CF and CS) for funding, as well as European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013) ERC grant agreement n° 279850. We also thank the EPSRC National Mass Spectrometry Service Centre (Swansea).Notes and references
- For selected reviews on organocatalysis see: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138–5175 CrossRef CAS; (b) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS; (c) A. Erkkilä, I. Majaner and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef; (d) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS; (e) M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS; (f) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS; (g) C. Gronal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS; (h) A. Moyana and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS; (i) F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406–2447 RSC.
- For a comprehensive review of the catalytic uses of guanidines, amidines and isothioureas see: J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 Search PubMed.
- (a) V. B. Birman, H. Jiang, X. Li, L. Guo and E. W. Uffman, J. Am. Chem. Soc., 2006, 128, 6536–6537 CrossRef CAS; (b) M. Kobayashi and S. Okamoto, Tetrahedron Lett., 2006, 47, 4347–4350 CrossRef CAS; (c) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS.
- For kinetic resolutions using anhydrides as acylating agents see: (a) V. B. Birman and L. Guo, Org. Lett., 2006, 8, 4859–4861 CrossRef CAS; (b) V. B. Birman and X. Li, Org. Lett., 2008, 10, 1115–1118 CrossRef CAS; (c) D. Belmessieri, C. Joannesse, P. A. Woods, C. MacGregor, C. Jones, C. D. Campbell, C. P. Johnston, N. Duguet, C. Concellón, R. A. Bragg and A. D. Smith, Org. Biomol. Chem., 2011, 9, 559–570 RSC; (d) X. Li, H. Jiang, E. W. Uffman, L. Guo, Y. Zhang, X. Yang and V. B. Birman, J. Org. Chem., 2012, 77, 1722–1737 CrossRef CAS. For kinetic resolutions using carboxylic acids as acylating agents utilising in situ formation of a reactive mixed anhydride see: (e) I. Shiina and K. Nakata, Tetrahedron Lett., 2007, 48, 8314–8317 CrossRef CAS; (f) I. Shiina, K. Nakata, M. Sugimoto, Y. Onda, T. Iizumi and K. Ono, Heterocycles, 2009, 77, 801–810 CrossRef CAS; (g) X. Yang and V. B. Birman, Adv. Synth. Catal., 2009, 351, 2301–2304 CrossRef CAS; (h) I. Shiina and K. Nakata, Heterocycles, 2010, 80, 169–175 CrossRef CAS; (i) I. Shiina, K. Nakata and Y. Onda, Eur. J. Org. Chem., 2008, 5887–5890 CrossRef CAS; (j) I. Shiina, K. Nakata, K. Ono, M. Sugimoto and A. Sekiguchi, Chem.–Eur. J., 2010, 16, 167–172 CrossRef CAS; (k) K. Nakata, Y. Onda, K. Ono and I. Shiina, Tetrahedron Lett., 2010, 51, 5666–5669 CrossRef CAS; (l) I. Shiina, K. Ono and K. Nakata, Chem. Lett., 2011, 40, 147–149 CrossRef CAS. For the kinetic resolution of β-lactams by asymmetric alcoholysis see: (m) V. D. Bumbu and V. B. Birman, J. Am. Chem. Soc., 2011, 133, 13902–13905 CrossRef CAS; by asymmetric acylation see: (n) X. Yang, V. D. Bumbu and V. B. Birman, Org. Lett., 2012, 13, 4755–4757 Search PubMed.
- (a) X. Yang, G. Lu and V. B. Birman, Org. Lett., 2010, 12, 892–895 CrossRef CAS; (b) X. Yang and V. B. Birman, Angew. Chem., Int. Ed., 2011, 50, 5553–5555 Search PubMed.
- For C-acylation and C-carboxylation reaction processes see: (a) F. R. Dietz and H. Gröeger, Synthesis, 2009, 4208–4218 CAS; (b) C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS; (c) P. A. Woods, L. C. Morrill, T. Lebl, A. M. Z. Slawin, R. A. Bragg and A. D. Smith, Org. Lett., 2010, 12, 2660–2663 CrossRef CAS; (d) C. Joannesse, L. C. Morrill, C. D. Campbell, A. M. Z. Slawin and A. D. Smith, Synthesis, 2011, 1865–1879 CAS; (e) B. Viswambharan, T. Okimura, S. Suzuki and S. Okamoto, J. Org. Chem., 2011, 76, 6678–6685 CrossRef CAS; (f) P. A. Woods, L. C. Morrill, R. A. Bragg and A. D. Smith, Chem.–Eur. J., 2011, 17, 11060–11067 CrossRef CAS.
- C. I. Sheppard, J. L. Taylor and S. L. Wiskur, Org. Lett., 2011, 13, 3794–3797 CrossRef CAS.
- (a) G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945–7946 CrossRef CAS; (b) G. S. Cortez, S. H. Oh and D. Romo, Synthesis, 2001, 1731–1736 CrossRef CAS; (c) S. H. Oh, G. S. Cortez and D. Romo, J. Org. Chem., 2005, 70, 2835–2838 CrossRef CAS; (d) H. Henry-Riyad, C. Lee, V. C. Purohit and D. Romo, Org. Lett., 2006, 8, 4363–4366 CrossRef CAS; (e) G. Ma, H. Nguyen and D. Romo, Org. Lett., 2007, 9, 2143–2146 CrossRef CAS; (f) H. Nguyen, G. Ma and D. Romo, Chem. Commun., 2010, 46, 4803–4805 RSC; (g) K. A. Morris, K. M. Arendt, S. H. Oh and D. Romo, Org. Lett., 2010, 12, 3764–3767 CrossRef CAS; (h) H. Nguyen, G. Ma, T. Gladysheva, T. Fremgen and D. Romo, J. Org. Chem., 2011, 76, 2–12 CrossRef CAS.
- (a) V. C. Purohit, A. S. Matla and D. Romo, J. Am. Chem. Soc., 2008, 130, 10478–10479 CrossRef CAS; (b) C. A. Leverett, V. C. Purohit and D. Romo, Angew. Chem., Int. Ed., 2010, 49, 9479–9483 CrossRef CAS.
- (a) D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS; (b) C. Simal, T. Lebl, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2012, 51, 3653–3657 Search PubMed; (c) L. C. Morrill, T. Lebl, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2012, 3, 2088–2093 RSC.
- There is only limited precedent for the potential intermediacy of α,β-unsaturated acyl ammonium ions in asymmetric catalysis. For dienolate formation that may proceed via an α,β-unsaturated acyl ammonium ion see: (a) P. S. Tiseni and R. Peters, Angew. Chem., Int. Ed., 2007, 46, 5325–5328 CrossRef CAS; (b) P. S. Tiseni and R. Peters, Chem.–Eur. J., 2010, 16, 2503–2517 CrossRef CAS; (c) L. T. Shen, L. H. Sun and S. Ye, J. Am. Chem. Soc., 2011, 133, 15894–15897 CrossRef CAS. Recently, α,β-unsaturated acyl ammonium intermediates have been accessed from α,β-unsaturated acid fluorides and employed in 1,3-dipolar cycloaddition reactions, albeit with low enantioselectivity, see: (d) S. Pandiancherri, S. J. Ryan and D. W. Lupton, Org. Biomol. Chem., 2012, 10, 7903–7911 RSC.
- For reviews see: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS; (b) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS; (c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC; (d) J. L. Moore and T. Rovis, in Topics in Current Chemistry, ed. B. List, Springer, Berlin, vol. 291, 2009, pp. 77–144 Search PubMed; (e) C. D. Campbell, K. B. Ling and A. D. Smith, in N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Catalysis by Metal Complexes 32, ed. C. S. J. Cazin, Springer, Berlin, 2010, pp. 263–297 Search PubMed; (f) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013 10.1039/c3cs35522e.
- (a) K. Zeitler, Org. Lett., 2006, 8, 637–640 CrossRef CAS. For a mechanistic investigation of the formation of α,β-unsaturated acyl azoliums from alkynals see: (b) J. Mahatthananchai, P. Zheng and J. W. Bode, Angew. Chem., Int. Ed., 2010, 49, 9479–9483 CrossRef CAS.
- (a) S. De Sarkar and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 9266–9269 CrossRef; (b) R. S. Reddy, J. N. Rosa, L. F. Veiros, S. Caddick and P. M. P. Gois, Org. Biomol. Chem., 2011, 9, 3126–3129 RSC; (c) S. D. Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef CAS.
- (a) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176–14177 CrossRef CAS; (b) L. Candish and D. W. Lupton, Org. Lett., 2010, 12, 4836–4839 CrossRef CAS; (c) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2011, 133, 4694–4697 CrossRef CAS; (d) C. Yao, D. Wang, J. Lu, T. Li, W. Jiao and C. Yu, Chem.–Eur. J., 2012, 18, 1914–1917 CrossRef CAS; (e) L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2013, 135, 58–61 Search PubMed.
- (a) C. Yao, D. Wang, J. Lu, T. Li, W. Jiao and C. Yu, Chem.–Eur. J., 2012, 18, 1914–1917 CrossRef CAS; (b) F.-G. Sun, L.-H. Sun and S. Ye, Adv. Synth. Catal., 2012, 353, 3134–3138 Search PubMed; (c) B. Zhang, P. Feng, Y. Cui and N. Jiao, Chem. Commun., 2012, 48, 7280–7282 RSC.
- B. Wanner, J. Mahatthananchai and J. W. Bode, Org. Lett., 2011, 13, 5378–5381 CrossRef CAS.
- J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810–8812 CrossRef CAS.
- A. Biswas, S. De Sarkar, L. Tebben and A. Studer, Chem. Commun., 2012, 48, 5190–5192 RSC.
- (a) Z. Q. Zhu and J. C. Xiao, Adv. Synth. Catal., 2010, 352, 2455–2458 CrossRef CAS; (b) Z.-Q. Rong, M.-Q. Jia and S.-L. You, Org. Lett., 2011, 13, 4080–4083 CrossRef CAS; (c) Z.-Q. Zhu, X.-L. Zheng, N.-F. Jiang, X. Wang and J.-C. Xiao, Chem. Commun., 2011, 47, 8670–8672 RSC. For a Michael-initiated cascade process see: (d) A. Biswas, S. De Sarkar, R. Frohlich and A. Studer, Org. Lett., 2011, 13, 4966–4969 CrossRef CAS.
- J. Mahatthananchai, J. Kaeobamrung and J. W. Bode, ACS Catal., 2012, 2, 494–503 Search PubMed.
- Screening of a range of isothioureas for catalytic activity and asymmetry in these processes showed that HBTM 2.1 was optimal.
- All α,β-unsaturated anhydrides in this manuscript were readily prepared from the corresponding α,β-unsaturated acid by EDCI-mediated coupling. See ESI† for further information.
- The absolute configuration within 2 was proven by comparison of its specific rotation [α]22D −7.6 (c 0.5 in CHCl3) with the literature {lit.20b [α]22D −6.5 (c 1.0 in CHCl3)}.
- Control studies showed that DBU alone can act as a catalyst for this transformation, as 0.6 equivalents of DBU in the absence of isothiourea catalyst afforded 75% consumption of diketone starting material.
- For selective reviews on asymmetric Michael additions see: (a) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 8033–8061 Search PubMed; (b) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 2, 171–196 Search PubMed; (c) J. Christoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688–1690 CrossRef CAS; (d) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701–1716 CrossRef CAS.
- The use of α,β-unsaturated acyl phosphonates as ester equivalents has been demonstrated see: (a) H. Jiang, M. W. Paixão, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 2775–2783 CrossRef CAS. For Michael additions of ethyl benzoylacetates to N-alkylmaleimides using chiral guanidines see: (b) Z. Jiang, W. Ye, Y. Yang and C.-H. Tana, Adv. Synth. Catal., 2008, 350, 2345–2351 CrossRef CAS.
- Aliphatic diketones such as 2,4-pentanedione led to products with low levels of enantiocontrol. With cinnamic anhydride, 2,4-pentanedione in the presence of HBTM 2.1 5 gave the corresponding ester in 64% yield and 37% ee. See ESI† for details.
- (Z,Z)-Cinnamic anhydride 22 was prepared by Lindlar reduction of ethyl 3-phenylpropiolate, ester hydrolysis to give (Z)-cinnamic acid and subsequent anhydride formation. For full experimental procedures see ESI.†.
- In situ ring opening by addition of methanol to dihydropyranones derived from unsymmetrical Michael donors led to a statistical mixture of diastereoisomers and so this strategy was not pursued further.
- Addition of ethyl acetoacetate to cinnamic anhydride 3 afforded the corresponding dihydropyranone in 72% yield, 70% ee; addition of ethyl benzoylacetate to crotonic anhydride afforded the corresponding hydropyranone in 45% yield, 65% ee. See ESI† for further information.
- For a mechanistic investigation regarding the preferred 1,4-addition of diketone enolates and alternative nucleophiles to α,β-unsaturated acyl azoliums see: R. C. Samantha, B. Maji, S. De Sarkar, K. Bergander, R. Fröhlich, C. Mück-Lichtenfeld, H. Mayr and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 5234–5238 Search PubMed . Additionally, a recent computational study highlights the similarity of the two potential mechanistic pathways: E. Lyngvi, J. W. Bode and F. Schoenebeck, Chem. Sci., 2012, 3, 2346–2350 Search PubMed.
- Such non-bonded S–O interactions have been widely recognised; for overviews see: (a) V. I. Minkin and R. M. Minyaev, Chem. Rev., 2001, 101, 1247–1266 CrossRef CAS; (b) K. A. Brameld, B. Kuhn, D. C. Reuter and M. Stahl, J. Chem. Inf. Model, 2008, 48, 1–24 CrossRef CAS. For nO to σ*C–S interactions discussed with respect to (acylimino)thiadiazoline derivatives see: (c) Y. Nagao, T. Hirata, S. Goto, S. Sano, A. Kakehi, K. Iizuka and M. Shiro, J. Am. Chem. Soc., 1998, 120, 3104–3310 CrossRef CAS. In the context of isothiourea-mediated catalysis the importance of these interactions have been recognised see: (d) V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS; (e) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 Search PubMed . See also ref. 9b.
- Re face addition to the α,β-unsaturated acyl ammonium is favoured for β-aryl substituents, with Si face addition favoured to β-alkyl substituents due to CIP priority changes.
- The acyl ammonium chloride salt 52 was characterised by single-crystal X-ray diffraction.†.
Footnote |
| † Electronic supplementary information (ESI) available: All spectroscopic data and procedures for novel compounds. CCDC 916302. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc50199j |
| This journal is © The Royal Society of Chemistry 2013 |