 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Depleted uranium catalysts for chlorine production†
Amol P.
Amrute
,
Frank
Krumeich
,
Cecilia
Mondelli
and
Javier
Pérez-Ramírez
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Wolfgang-Pauli-Strasse 10, CH-8093 Zurich, Switzerland. E-mail: jpr@chem.ethz.ch; Fax: +41 44 6331405; Tel: +41 44 6337120
First published on 4th February 2013
Abstract
This study demonstrates depleted uranium as a remarkable heterogeneous catalyst for the oxidation of HCl to Cl2. This reaction comprises a sustainable approach to valorise byproduct HCl streams in the chemical industry. Bulk α-U3O8 showed an outstanding stability against chlorination, which is crucial for its durability in catalytic tests. UO2 and γ-UO3 transformed into α-U3O8 under reaction conditions. Uranium deposition on different carriers by dry impregnation concluded the superiority of zirconia as support. HAADF-STEM investigations revealed that the uranium oxide on the surface of this carrier is present in the form of a film-like nanostructure with a thickness ranging from a monolayer to 1 nm as well as atomic dispersion. The effect of variables (temperature, feed O2/HCl ratio, metal loading, and Cl2 co-feeding) on the performance of U3O8/ZrO2 has been studied. The HCl conversion over this catalyst increased with reaction time as a likely consequence of in situ re-dispersion of the original uranium phase into atomically dispersed UOx. As demonstrated by H2-TPR, the uranium in the generated UOx phase is more oxidised than in the original U3O8. Such a highly dispersed active phase is produced faster in the uncalcined sample. The extraordinary stable Cl2 production over U3O8/ZrO2 at 773 K for 100 h on stream indicates its potential for application in high-temperature HCl oxidation. Under these conditions, other known catalytic materials suffer from significant deactivation.
Introduction
Uranium compounds have been used as heterogeneous and homogeneous catalysts.1,2 Their suitability for redox reactions is related to the wide range of oxidation states that uranium can assume (from II to VI), which in turn derives from the ability of its 5f-electrons to hybridise.3 Specifically for the heterogeneous catalysis field, uranium oxides (mostly U3O8) have been recognised since the 1920s for reactions of industrial relevance such as the oxidation of hydrocarbons and the partial oxidation of ethanol.4–6 Later efforts extended the scope of uranium-catalysed transformations to comprise the oxidative destruction of volatile chlorinated hydrocarbons,7,8 the oxidative coupling of ethylene, acetylene, and acetaldehyde,3 the esterification of formaldehyde,3 and NOx reduction.9 Relevantly, uranium-based materials were once used in industry for the hydrocracking of shale oil (UO3/Al2O3, UO3/CoMoO4)10 and in the ammoxidation of propylene to acrylonitrile (USbxOy).11–13Natural uranium consists of three isotopes, 238U, 235U, and 234U, in the relative abundance of 99.275, 0.720, and 0.005%, respectively.14238U and 234U are α-ray emitters, while 235U emits both α- and low-energy γ-rays. Alpha particles are much less penetrating than other forms of radiation, thus rendering uranium only a little hazardous (mainly from the γ-rays). Depleted uranium (DU), which is produced as a waste in the uranium enrichment process, is even considerably less radioactive (ca. 0.2–0.4% 235U) and, thus, less harmful. To generate the carbon-neutral energy source, the demand of enriched uranium as a fissile nuclear fuel can be expected to increase,2 which represents a strong incentive for the development of novel applications of DU.
The heterogeneously catalysed oxidation of HCl to Cl2 (Deacon reaction)15 is an attractive route to recycle chlorine from byproduct HCl streams in the chemical industry, namely in the production of polyurethanes and polycarbonates.16–18 Two industrial catalysts based on RuO2, featuring high activity at a relatively low temperature and remarkable stability, have been recently introduced: RuO2/SiO2/TiO2-rutile (by Sumitomo) and RuO2/SnO2–Al2O3 (by Bayer).19–27 The wide use of ruthenium catalysts for HCl oxidation is hindered by its high and fluctuating market price.16 This drawback triggered research efforts to develop alternative cost-effective systems. CeO2-based catalysts represent tangible steps along this direction.28,29
Uranium oxide-based catalysts for HCl oxidation have recently been patented.30,31 High single-pass HCl conversion at high temperature and practically negligible active phase loss have been claimed as the key characteristics of these systems. To assess the real potential of uranium-based catalysts for industrial application, further knowledge needs to be gathered. The optimal combination of active phase and support will be derived only based on a deeper understanding of activity and stability descriptors. The catalyst performance should be then put into perspective with respect to other known catalytic systems and evaluated in an industrially relevant time frame. Herein, we systematically investigated uranium oxides in bulk and supported forms for HCl oxidation. Catalytic tests at ambient pressure in a continuous flow fixed-bed reactor combined with detailed characterisation of the catalysts prior to and after reaction have been applied to gather a solid knowledge of the Deacon chemistry of these materials.
Results and discussion
Bulk uranium oxides
As starting point, the main binary oxides of uranium were considered in this study, namely, UO2, U3O8, and UO3. The X-ray diffractograms of the solids as well as their corresponding structures are displayed in Fig. 1a and b. According to the XRD phase analysis, the catalysts were identified as uranium dioxide (JCPDS 05-0550), α-triuranium octoxide (JCPDS 31-1424), and γ-uranium trioxide (JCPDS 31-1422) with small amounts of β- and α-forms. The crystal structure of UO2 is of fluorite type with face-centred cubic atomic arrangement. Uranium and oxygen atoms are octa- and tetrahedrally coordinated, respectively.14 α-U3O8, one of the two forms (α, β) of this oxide which are stable at ambient temperature,3 crystallises in an orthorhombic structure. All of the uranium atoms are coordinated with oxygen atoms forming pentagonal pyramids.1,14 γ-UO3, the most stable of the seven crystalline phases (α, β, δ, ε, γ, ζ, and η) of this oxide,3 belongs to the tetragonal crystal system and is characterised by octa- and dodecahedral coordination of uranium to oxygen. All of the three oxides possess a very low total surface area (SBET, Table 1).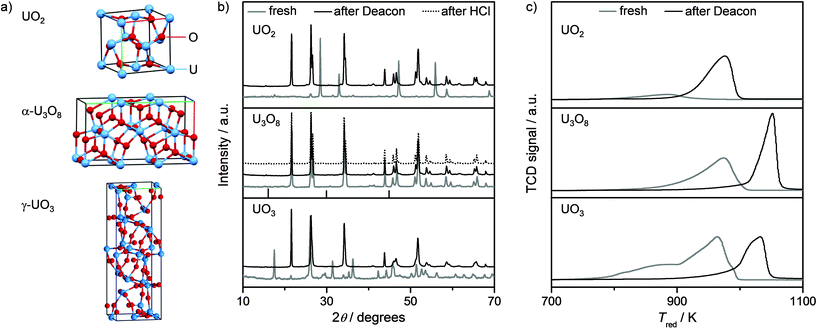 | ||
| Fig. 1 Structure of the uranium oxides (a) and characterisation results from powder XRD (b) and H2-TPR (c) of the samples in fresh form (blue lines), after HCl oxidation at 773 K (black lines), and after HCl treatment at 823 K (dotted lines). Vertical lines at the bottom of the U3O8 pattern show the positions of most intense reflections of UCl4. | ||
| Sample | Ua (wt%) | S BET (m2 g−1) | r (mol Cl2 h−1 mol U−1) | E appa (kJ mol−1) |
|---|---|---|---|---|
| a Determined by ICP-OES. b Conditions: W = 0.5 g (bulk oxides) or 0.25 g (supported catalysts), Tbed = 773 K, O2/HCl = 2, FT = 166 cm3 STP min−1, and t = 3 h (bulk oxides) or 1 h (supported catalysts). c Surface area of the supports in brackets. | ||||
| UO2 | 88.1 | 1 | 2.2 | 52 |
| γ-UO3 | 83.1 | 3 | 2.9 | 40 |
| α-U3O8 | 84.8 | 1 | 2.0 | 54 |
| U3O8/ZrO2 | 9.8 | 35 (47)c | 63.5 | 50 |
| U3O8/SiO2 | 9.5 | 136 (193) | 45.4 | 46 |
| U3O8/TiO2 | 9.4 | 30 (52) | 28.6 | 54 |
| U3O8/Al2O3 | 9.6 | 131 (191) | 27.3 | 57 |
The reducibility of these materials was studied under a diluted H2 flow up to 1100 K (Fig. 1c). The reduction profile of UO2 shows a little H2 consumption at ca. 880 K. As the XRD pattern of the reduced sample (Fig. S1 in the ESI†) was unaltered with respect to that of the fresh solid, this feature was attributed to the removal of oxygen species that are known to accommodate in the lattice of the fluorite structure of UO2 upon exposure to air.1 For α-U3O8, a single and broad peak centred at ca. 975 K was evidenced, which is assigned to the reduction of U3O8 to UO2.8 The reduction profile of γ-UO3 displays a broad signal composed by two main contributions at ca. 880 and 963 K, due to the transitions UO3 → U3O8 and U3O8 → UO2, respectively (Fig. 1c).32 The formation of UO2 from both α-U3O8 and γ-UO3 was confirmed by XRD (Fig. S1 in the ESI†).
These bulk uranium oxides were tested in the gas-phase oxidation of HCl at Tbed = 773 K and O2/HCl = 2 for 3 h. The rates of Cl2 production were stable at ca. 2 mol Cl2 h−1 mol U−1 for UO2 and α-U3O8 and ca. 3 mol Cl2 h−1 mol U−1 for γ-UO3. Normalisation of the rates by SBET of the fresh samples gives the values as 7 × 10−3, 8 × 10−3, and 3.4 × 10−3 mol Cl2 h−1 m−2 for α-U3O8, UO2, and γ-UO3, respectively. However, due to transformation of the latter two oxides into the former during reaction (vide infra), rates normalised by the SBET of the used sample are more relevant and lead to a value of 4 × 10−3 mol Cl2 h−1 m−2 in all cases. The dependence of the activity of these oxides on temperature was investigated between 673 and 823 K at O2/HCl = 2. The reaction rate scaled linearly with the temperature in the whole range. The apparent activation energy (Eappa) was estimated from the Arrhenius plots at 52, 54, and 40 kJ mol−1 for UO2, α-U3O8, and γ-UO3, respectively.
The used catalysts were characterised by the same techniques applied to the fresh samples in order to assess possible structural changes upon exposure to reaction conditions. Remarkably, XRD analysis indicated the absence of chlorinated phases in any of the used catalysts. However, we observed the complete conversion of UO2 and γ-UO3 into α-U3O8 (Fig. 1b). It is suggested that such transformation is due to oxidation by the excess gas-phase O2 for the former oxide and reduction by feed HCl for the latter. Indeed, treatment of UO2 and γ-UO3 in 20 vol% O2/N2 at 773 K for 3 h caused the complete transformation of UO2 into α-U3O8, while it did not affect the state of γ-UO3 (confirmed by XRD, Fig. S2 in the ESI†). All of the H2-TPR profiles of the uranium oxides after reaction feature a single reduction peak, attributed to the transformation of U3O8 into UO2 (Fig. 1c), in line with the identical bulk composition of the samples after HCl oxidation. The appearance of the peak at higher reduction temperature for used γ-UO3 and α-U3O8 is likely related to certain degree of surface chlorination and/or sintering. With regard to the former, the bulk α-U3O8 catalyst after Deacon reaction was calcined in static air at 773 K for 5 h (aimed at removing surface chlorine species) and then measured by H2-TPR. A reduction profile equivalent to that of the fresh α-U3O8 sample was obtained (Fig. S3a†), which confirmed that the change in reducibility is mainly due to surface chlorination. Further, TEM of α-U3O8 in fresh form and after Deacon indicated a slight increase in overall particle size for the latter (Fig. S3b and c†). Since calcination of U3O8 after Deacon reproduced the reduction profile of the fresh sample, the effect of sintering on reducibility of U3O8 seems to be negligible.
α-U3O8 was further assessed under harsher conditions, i.e. at O2/HCl = 0.5 and 0 (without gas-phase O2) at 823 K for 2 h on stream to evaluate its resistance to bulk chlorination and metal loss. The weight of the reactor before and after the tests remained practically unchanged, suggesting no loss of uranium. Furthermore, the diffractograms of the samples after these treatments indicated the preservation of a pure oxidic phase (Fig. 1b). The endothermic nature of the penetration of Cl atoms to deeper layers (ca. 2 eV) has been already found as a key reason for the robustness of RuO2 against bulk chlorination.22,28 Similar property could be responsible for the stability of α-U3O8 against bulk chlorination. In this line, chlorination of UO2 (which also revealed the absence of any chloride phase upon testing in O2/HCl = 0 at 823 K for 2 h), by Cl2 to form UCl4 has been reported highly endothermic (ΔG = 148.9 kJ mol−1).33 Thus, bulk uranium oxide represents an exceptionally stable high-temperature catalyst for HCl oxidation. This finding is particularly striking since CuO, Cr2O3, CeO2, and RuO2 undergo structural changes at high temperatures. In particular, after testing at 823 K and O2/HCl = 0.5 for 2 h, strong chlorination was detected (XRD analysis) for the first three oxides, while RuO2 underwent partial transformation into volatile RuO4 (ca. 20 wt% RuO2 loss).16 It is worth noting, though, that RuO2 is an outstanding low-temperature (473–673 K) catalyst and is extremely stable under its optimised operating conditions.20,34
Supported U3O8 catalysts
Based on the very promising performance of bulk α-U3O8, the next step of the work consisted of finding a suitable support for this uranium-based active phase. Monoclinic ZrO2, γ-Al2O3, SiO2, and TiO2-anatase were considered as carriers. The synthesis protocol comprised dry impregnation of these oxides with a uranium precursor (in an amount corresponding to a nominal loading of 10 wt% U), followed by calcination under the same conditions applied for the preparation of bulk α-U3O8 (see Experimental section).The supported U3O8 catalysts were screened in HCl oxidation at O2/HCl = 2 in the temperature range of 673–823 K (Fig. 2a). Blank experiments confirmed that the Deacon activity of the pure carriers was negligible under the conditions applied. The HCl conversion displayed a steady increase with the temperature for all supported catalysts, reaching values comprised between 21 and 47% at 823 K. U3O8/ZrO2 was the most active catalyst, followed by U3O8/SiO2 and, finally, U3O8/TiO2 and U3O8/Al2O3, which were comparably active. With respect to the bulk oxide, only the zirconia- and silica-supported materials offered improved performances (Fig. 2a). Still, as α-U3O8 was tested using twice the catalyst amount, a better comparison was drawn on the basis of the reaction rates per mol of U at 773 K. Accordingly, it appeared evident that any of the supports employed determined an activity enhancement, overall leading to 14–30 times higher rates (Table 1). As shown in the same table, the Eappa values (at 723–823 K and O2/HCl = 2) determined from the Arrhenius plots were in the range of 46–57 kJ mol−1 for the supported catalysts, thus being similar to α-U3O8. The dependence of the activity on the relative O2 content in the feed was studied over the two most promising catalysts, U3O8/ZrO2 and U3O8/SiO2 (Fig. 2b). In both cases, the HCl conversion increased upon raising the feed O2/HCl ratio and the formal reaction order of O2 was calculated as ca. 0.3. This behaviour is common to the vast majority of Deacon catalysts24,28 and indicates that catalyst re-oxidation is the limiting step.27 It is worth noting that the HCl conversion over U3O8/ZrO2 remained higher than that of U3O8/SiO2 at all O2/HCl ratios. Overall, the catalytic results indicate that zirconia is the most suitable carrier for uranium oxide.
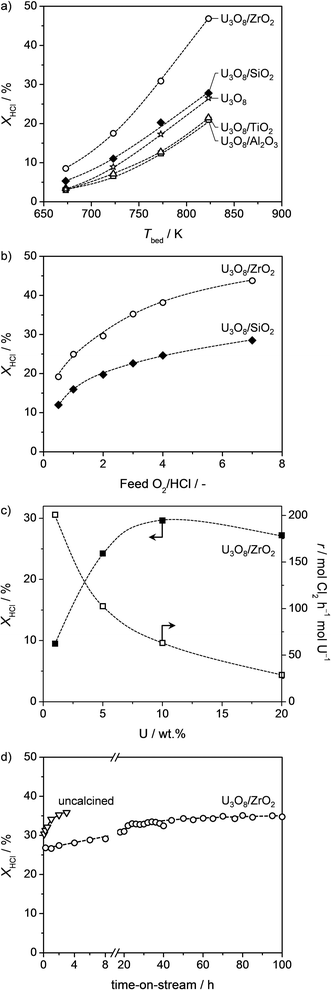 | ||
| Fig. 2 HCl conversion over U3O8-based catalysts versus (a) bed temperature at O2/HCl = 2 and (b) O2/HCl ratio at 773 K. HCl conversion and U-specific rate over U3O8/ZrO2versus the uranium loading (c) and HCl conversion over U3O8/ZrO2 and an as-impregnated zirconia-supported catalyst (uncalcined) versus time-on-stream (d) at 773 K and O2/HCl = 2. Data were acquired after 1 h under each condition for a–c. Other conditions are detailed in the Experimental section. | ||
In order to rationalise the activity differences, the supported U3O8 catalysts were characterised in fresh form and after use in the Deacon reaction. The uranium content, as determined by ICP-OES, was close to the nominal value of 10 wt% for all of the catalysts and remained unchanged in the used samples, indicating negligible uranium loss during HCl oxidation. The fresh alumina- and silica-based catalysts featured ca. 4 times larger SBET than the zirconia- and titania-based materials (Table 1). This deviation reflects the difference in surface area of the pure carriers, which was depleted to a similar extent upon uranium incorporation in all cases, likely due to pore blockage. The SBET of the catalysts was also unaltered upon use. Accordingly, the activity trend cannot be explained by differences and/or changes in the active phase content or textural characteristics.
XRD analysis of the fresh materials evidenced the formation of α-U3O8 over all supports with exception of titania (Fig. 3). In this latter case, a mixed UTiO5 phase was detected (JCPDS 49-1397).35 Furthermore, reflections specific to both the anatase and rutile forms of titania were observed, indicating that partial transformation of the carrier structure occurred during the high-temperature thermal activation of the as-impregnated solid. Thus, the loss in the support's surface area during catalyst preparation could be additionally ascribed to phase changes and structural reconstructions for U3O8/TiO2. Based on the much lower intensity of its diffraction lines, the uranium phase is supposed to be present in form of smaller nanostructures on ZrO2 compared to the other carriers, especially titania. The diffractograms of the samples after reaction revealed the absence of bulk chlorides (Fig. 3), extending the stability of α-U3O8 against chlorination also to the supported form. No changes were detected in the patterns of the TiO2 and Al2O3-supported catalysts upon use, while the reflections specific to α-U3O8 became less intense for U3O8/SiO2 and disappeared for U3O8/ZrO2. Since uranium was not lost upon reaction, these alterations might be substantiated by fragmentation of the α-U3O8 phase in tinier structures.
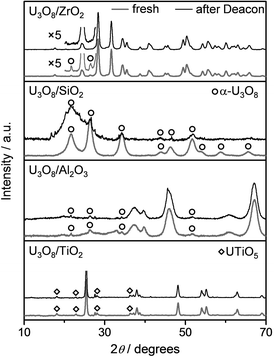 | ||
| Fig. 3 XRD patterns of supported U3O8 samples in fresh form (blue lines) and after Deacon reaction (black lines). Unmarked reflections belong to the corresponding carriers. | ||
In order to further tackle this point and as the XRD analysis hints to differences in the dispersion of the supported active phase as a possible main parameter for determining the activity levels, the two most active catalysts (U3O8/ZrO2 and U3O8/SiO2) were further investigated by electron microscopy (EM, Fig. 4 and 5). For fresh U3O8/ZrO2, aggregates of 20–30 nm sized support grains are visualised by HRTEM (Fig. 4a). However, inspection of surface regions even at higher magnification does not reveal a distinct uranium phase. Thus, based on the significant difference in the atomic numbers of U and Zr (ZU = 92 versus ZZr = 40), HAADF-STEM with Z-contrast was applied as a suitable tool to get information about the distribution of uranium-based phases (Fig. 5). Indeed, the uranium oxide species in the fresh U3O8/ZrO2 are clearly visualised as bright rim or spots (Fig. 5a and b). The presence of uranium in these rims was confirmed by EDXS analysis. Investigation of the surface structure at the edges and on the surface revealed that two types of uranium oxide dispersions are present in the fresh U3O8/ZrO2, namely, (i) a film-like nanostructure with a thickness ranging from a monolayer to 1 nm (Fig. 5a) and (ii) atomically dispersed uranium oxide as identified by bright spots (encircled) on the ZrO2 support (Fig. 5b). Moreover, analysis of the complete structure of these spots is not possible on the basis of HAADF-STEM and would require more specific methods such as STEM coupled with electron energy loss spectrometer (EELS).36 Nonetheless, based on the studies on identification of single atoms,36 the bright spots seem to be composed of a single uranium atom (likely with some O atoms bound to it) and therefore, in this study they are referred to as atomically dispersed UOx. Upon exposure to reaction conditions (for 5 h), the catalyst morphology seems to be altered. A film-like nanostructure is less visible and a concentration of bright spots of UOx appears to be increased (Fig. 5c), suggesting the transformation into tinier, better dispersed uranium oxide. Thus, uranium oxide on zirconia likely undergoes partial re-dispersion during reaction. This explains the disappearance of the α-U3O8 peaks in the XRD pattern of the used sample (Fig. 3). α-U3O8 on SiO2 appears to be carried as nanoparticles of ca. 5 nm in the fresh catalyst (Fig. 4b). Upon use in HCl oxidation, the average particle size was reduced to ca. 2.5 nm (Fig. 4c, inset in b), supporting a certain degree of re-dispersion of the uranium phase. This agrees with the XRD results (Fig. 3). The origin of the active phase re-dispersion phenomenon, apparently common to both the zirconia- and silica-supported catalysts, is not fully understood. It is proposed that disaggregation of the uranium oxide structures might be induced by HCl and Cl2. The latter has been reported to produce such an effect on supported noble metal particles by generation of chlorides which readsorb on the solid carrier and are then reduced by the reaction environment.37,38 In our case, it is possible that uranium oxychloride species (UO2Cl2, melting point = 843 K)39 are formed to some extent. As they are highly unstable and readily re-oxidise under conditions similar to those applied in HCl oxidation,40 uranium will not be lost, but a certain degree of metal migration could be possible. This will ultimately improve the dispersion of the supported phase. Thus, based on the XRD and EM results, the activity differences seem to mainly depend on the uranium oxide dispersion. Still, the possibly different intrinsic activity of the chemical forms of uranium stabilised by the carriers might also play a role.
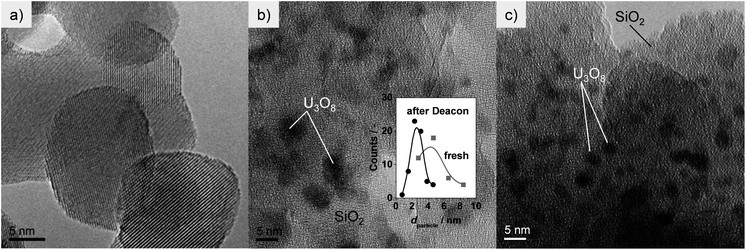 | ||
| Fig. 4 HRTEM of fresh U3O8/ZrO2 (a), U3O8/SiO2 in fresh form (b) and after Deacon (c). Inset in (b) shows the particle size distribution of the fresh and used U3O8/SiO2 sample. | ||
 | ||
| Fig. 5 HAADF-STEM of U3O8/ZrO2 in fresh form (a and b) and after Deacon reaction for 100 h (c). Bright spots (some of which are encircled) in b and c corresponds to atomically dispersed UOx. | ||
In view of its potential practical application, the U3O8/ZrO2 system was further studied in terms of optimisation of the active phase content as well as durability. Thus, catalysts with U loading comprised between 1 and 20 wt% were prepared and tested at 773 K and O2/HCl = 2 (Fig. 2c). The HCl conversion was found to raise with increasing U contents up to 10 wt%, while a loading of 20 wt% resulted in slightly lower activity. On the contrary, the U specific activity (i.e. reaction rate per mol of U) was the highest for the 1 wt% U catalyst and progressively diminished at increased U loadings. Hence, as a compromise between these parameters, a 5–10 wt% U content turns out to be optimal.
The robustness of U3O8(10 wt% U)/ZrO2 in HCl oxidation was tested in a long catalytic run (Fig. 2d). The HCl conversion moderately increased from 27 to 35% in the first 85 h on stream, remaining then stable up to a reaction time of ca. 100 h. Overall, this result evidences outstanding longevity, offering bright perspectives for an industrial application of zirconia-supported uranium catalysts in chlorine production. Still, the progressive catalyst activation indicates an alteration of the material's properties upon use. According to the above discussion of the characterisation data, this might originate from an increase in the dispersion of the active phase induced by the exposure to the reaction mixture. To further explore this point, samples after 5, 10, and 100 h on stream were collected and characterised by HAADF-STEM and H2-TPR (Fig. 5 and 6). While an increase in the uranium dispersion to certain extent has been already discussed for the sample after 5 h reaction (vide supra), HAADF-STEM of the sample after 100 h reaction evidenced that the uranium on ZrO2 carrier is mainly present as atomically dispersed UOx (Fig. 5c). The latter would be characterised by the highest dispersion of uranium oxide. This result provides a direct evidence for the dependence of activity on degree of dispersion.
Additional support was derived from the H2-TPR analysis. The reduction profile of fresh U3O8/ZrO2 features two main peaks at ca. 710 and 800 K (Fig. 6), which could be consistent with the presence of uranium oxide structures of different size (Fig. 5a and b), namely, thin layer (high-temperature signal) and atomic dispersion (low-temperature signal). For the sample collected after 5 h, a broad and more intense reduction peak centred at ca. 730 K with low- (695 K) and high-temperature (775 K) shoulders was visualised, while that taken after 10 h of reaction produced a single, symmetric, and sharper signal with maximum at 740 K. The curve of the catalyst unloaded at the end of the run displays an even narrower and more intense peak, slightly shifted to lower temperature (710 K). The depletion of the high-temperature signals with reaction time and the strengthening of a single peak at lower temperature supports a change in the morphology of α-U3O8 phase towards the formation of more uniformly-sized atomically dispersed nanostructures (UOx), in line with the HAADF-STEM results. The latter actually represents the predominant uranium distribution after 100 h on stream (Fig. 5c). Still, considering the modifications in peak position and shape, along with the significant increase in H2 consumption, the presence of more oxidic uranium in UOx than in the original α-U3O8 phase cannot be excluded. Based on the structural equivalence between the zirconia support and β-UO3 (both monoclinic, the latter having about double cell parameters with respect to the former),1 it could be possible that α-U3O8 undergoes transformation into this oxide during reaction. Although α-U3O8 is the most stable bulk oxide under HCl oxidation conditions and γ-UO3 is converted into it during reaction, it is plausible that, when the incipient uranium oxychloride is oxidised by the O2 excess, the structural matching offered by the support could stabilise β-UO3 as an oxidation product rather than α-U3O8. However, this phase is not detected by XRD owing to its very small size. Thus, from increased H2 uptake and development of atomically dispersed UOx with reaction time, it can only be suggested that uranium generated in situ as UOx is in higher oxidation state than in the original α-U3O8 and the transformation of α-U3O8 to UOx is accompanied by an enhancement of the dispersion. Since the presence of some UOx is evidenced already for the fresh catalyst (Fig. 5b and 6), it could even be possible that a part of the uranium is already stabilised as UOx during calcination and the atomic dispersion (Fig. 5b) rendering it undetectable by XRD (Fig. 3). An increase of uranium oxide dispersion during HCl oxidation was also evidenced for U3O8/SiO2 (Fig. 3, 4b and c). However, from a similar H2 consumption of fresh and used catalyst (not shown), it appears that in situ oxidation of the uranium phase does not occur on silica. This could be related to a specific property of the carrier and its interaction with the active phase. Thus, it seems that the support determines the degree of redispersion and reoxidation characteristics of the uranium phase. An in-depth understanding of these complex phenomena will require deeper characterisation studies, which are beyond the scope of this paper.
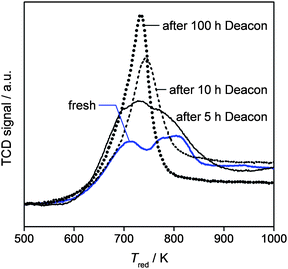 | ||
| Fig. 6 H2-TPR profiles of U3O8/ZrO2 in fresh form and after Deacon reaction for different times. | ||
Finally, we tested under the same HCl oxidation conditions an as-impregnated catalyst sample with equal U loading (i.e. no calcination applied after impregnating the U-precursor). This material reached a similar HCl conversion level (ca. 36%) to U3O8/ZrO2 after only 3 h on stream (Fig. 2d). On the basis of this outcome and of the resemblance of the HAADF-STEM images and XRD pattern of the two catalysts after use (not shown), it is suggested that UOx can be directly created in situ from the uranium precursor and with much faster kinetics. The latter is probably related to the ease of altering an amorphous and unstable deposit rather than a well-crystallised and stable phase.
Comparison with other systems
The performance of U3O8/ZrO2 was contrasted with other known supported HCl oxidation catalysts, namely, RuO2(2 wt% Ru)/SnO2–Al2O3,20 CeO2(9 wt% Ce)/ZrO2,29 and CuO(15 wt% Cu)/SiO2 (synthesised by dry impregnation, followed by calcination at 823 K for 10 h). Fig. 7a displays the dependence of the HCl conversion level on temperature for these materials. The equilibrium HCl conversion (dashed line) is reported as a reference. The activity of RuO2/SnO2–Al2O3 increases with the temperature and reaches a HCl conversion close to the equilibrium value at 673 K. Beyond this temperature the active phase of this catalyst starts to form volatile RuO4.16 This indicates that the optimal high temperature boundary for RuO2-based catalysts is 673 K. CuO/SiO2 possesses a volcano-shaped activity profile. A strong deactivation above 723 K is due to huge copper loss in the form of CuCl and CuCl2.41 Differently, U3O8/ZrO2 and CeO2/ZrO2 show a steady increase of the HCl conversion with temperature. The difference of activity between these two systems is relatively low (ca. 30 K). Cl2 co-feeding at comparable initial HCl conversion levels (attained by adjusting Tbed, Experimental section) also displays a very similar inhibition of HCl oxidation activity (Fig. 7b). Still, CeO2/ZrO2 was observed to undergo bulk chlorination and, thus, deactivation at low O2 excess.29 Accordingly, U3O8/ZrO2 stands as the most robust catalyst among all and belongs to the category of high-temperature catalysts, similar to CeO2/ZrO2. However, the former offers a superior resistance to bulk chlorination.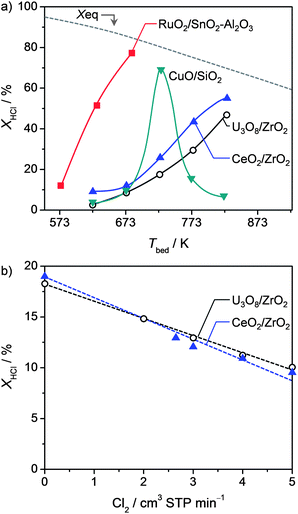 | ||
| Fig. 7 Steady-state HCl conversion versus bed temperature (a) and amount of Cl2 co-fed (b) at O2/HCl = 2. Other conditions are detailed in the Experimental section. | ||
Conclusions
Uranium catalysts have been successfully evaluated for HCl oxidation to Cl2. Extraordinary resistance of bulk uranium oxides against chlorination demonstrates their suitability as a stable active phase for this reaction. While α-U3O8 maintains its oxidation state, UO2 and γ-UO3 tend to transform into α-U3O8 under reaction conditions. The support of the uranium phase plays a very important role on its performance. ZrO2 allows depositing of the oxidic uranium phase in the form of film-like nanostructures and atomic dispersion, thus leading to a superior catalyst. U3O8/ZrO2 activates under reaction conditions before reaching a stable performance after ca. 85 h on stream. The catalyst activation is related to in situ re-dispersion and gradual transformation of the original α-U3O8 phase into a more oxidic and atomically dispersed UOx. An uncalcined sample allows faster generation of this highly dispersed UOx. The unique robustness of ZrO2 supported uranium oxide under the harsh reaction conditions and stable Cl2 production for more than 100 h on stream justifies its consideration as a high-temperature HCl oxidation catalytic technology. Uranium materials are less sensitive to metal loss and sintering than other known catalysts and are cost-effective since they can be prepared from waste produced in the uranium-enrichment processes.Experimental
Materials
ZrO2-monoclinic (Saint-Gobain NorPro, 99.8%), γ-Al2O3 (Alfa Aesar, catalyst support, 43855), SiO2 (ABCR, 99%), and TiO2-anatase (Aldrich, nanopowder, 99.7%) were calcined at 773 K (10 K min−1) for 5 h prior to their use. The starting uranium compounds UO2 and UO2(NO3)2·6H2O (International Bio-Analytical Industries) derive from depleted uranium sources and were used as received. The most important precaution for the safe handling of uranium compounds is to avoid their access to the body, through direct contact with the skin and/or inhalation, and dispersal in the environment. In the present case, personal protective equipment such as impervious gloves, boots, and an apron were worn to prevent skin contact. U3O8 and UO3 were prepared by thermal decomposition of UO2(NO3)2·6H2O in static air following existing protocols.8,42 U3O8 was obtained by two-step calcination of UO2(NO3)2·6H2O. The uranyl nitrate was treated at 573 K (5 K min−1) for 1 h and then, without intermediate cooling, at 1073 K (5 K min−1) for another 3 h. UO3 was synthesised by calcination of the uranyl nitrate at 723 K (5 K min−1) for 3 h. Supported catalysts were prepared by dry impregnation of the carriers with an aqueous solution of uranyl nitrate (nominal 1–20 wt% U), followed by drying at 338 K for 12 h and calcination, according to the same protocol applied for the synthesis of bulk U3O8. Unless stated otherwise, the supported catalysts, denoted as U3O8/support, contain 10 wt% U.Characterisation techniques
Powder X-ray diffraction (XRD) was measured in a PANalytical X'Pert PRO-MPD diffractometer. Data were recorded in the 10–70° 2θ range with an angular step size of 0.017° and a counting time of 0.26 s per step. N2 sorption at 77 K was performed in a Quantachrome Quadrasorb-SI gas adsorption analyser. Prior to the measurement, the samples were evacuated at 473 K for 12 h. Temperature-programmed reduction with hydrogen (H2-TPR) was measured in a Thermo TPDRO 1100 unit. The samples were loaded in a quartz micro-reactor (11 mm i.d.), pre-treated in He (20 cm3 STP min−1) at 473 K for 30 min, and cooled to 323 K in He. The analysis was carried out in 5 vol% H2/N2 (20 cm3 STP min−1), ramping the temperature from 323 to 1173 K at 10 K min−1. High-resolution transmission electron microscopy (HRTEM) measurements were undertaken on a FEI Tecnai F30 microscope (field emission gun), operated at 300 kV. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) investigations were performed on an aberration-corrected Hitachi HD-2700CS microscope, operated at 200 kV and equipped with an energy-dispersive X-ray spectrometer (EDXS, EDAX) for elemental analysis. The incorporated probe correction system (CEOS) enables a resolution of below 0.1 nm to be achieved.43Catalytic tests
The gas-phase oxidation of hydrogen chloride was studied at ambient pressure in a continuous-flow set up44 composed of mass-flow controllers to feed HCl (Messer, purity 2.8, anhydrous), O2 (Pan Gas, purity 5.0), Cl2 (Messer, purity 2.8, anhydrous), and N2 (Pan Gas, purity 5.0), a home-made electrically heated oven hosting a 8 mm i.d. quartz micro-reactor, and a Mettler Toledo G20 Compact Titrator for quantitative Cl2 analysis in the product stream. The catalysts were loaded in the tubular reactor and pre-treated in N2 at 673 K for 30 min. Thereafter, steady-state experiments at variable bed temperatures (Tbed = 673–823 K), inlet O2/HCl ratios (0.5–7), and catalyst amounts (W = 0.25 or 0.5 g for supported or bulk catalysts, respectively) were carried out. The inlet HCl concentration and total volumetric flow (FT) were fixed at 10 vol% and 166 cm3 STP min−1, respectively. The O2/HCl dependence was measured by increasing the O2 content in the inlet mixture from 5 to 70 vol% with N2 as balance gas. The influence of Cl2 co-feeding on the rate of HCl oxidation was studied by introducing variable amounts (2–5 cm3 STP min−1) of Cl2 to the inlet feed with O2/HCl = 2 at 733 K and 703 K over U3O8/ZrO2 and CeO2/ZrO2, respectively. Used samples were collected for characterisation after rapidly cooling down the reactor to room temperature in N2 flow. The percentage of HCl conversion was determined as XHCl = (2 × mole Cl2 at the reactor outlet/mole HCl at the reactor inlet) × 100.Acknowledgements
We thank Bayer MaterialScience AG for permission to publish these results. Dr Martin Badertscher of the Radiochemistry Laboratory of the ETH Zurich is thanked for granting access to the facility and for the training on the safe handling of uranium-based materials. The Electron Microscopy Centre of the ETH Zurich is acknowledged for the use of their facilities.Notes and references
- S. T. Taylor, in Metal Oxide Catalysis, Wiley-VCH, Weinheim, 2009, ch. 13, p. 539 Search PubMed.
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341 CrossRef CAS.
- H. Idriss, Surf. Sci. Rep., 2010, 65, 67 CrossRef CAS.
- W. J. Hale, US1 595 299, assigned to Dow Chemical Company, 1926.
- A. E. Craver, US1 636 854, assigned to Barrett Company, 1927.
- Z. R. Ismagilov, S. V. Kuntsevich, N. V. Shikina, V. V. Kuznetsov, M. A. Kerzhentsev, V. A. Ushakov, V. A. Rogov, A. I. Boronin and V. I. Zaikovsky, Catal. Today, 2010, 157, 217 CrossRef CAS.
- G. J. Hutchings, C. S. Heneghan, I. D. Hudson and S. H. Taylor, Nature, 1996, 384, 341 CrossRef CAS.
- S. H. Taylor and S. R. O'Leary, Appl. Catal., B, 2000, 25, 137 CrossRef CAS.
- T. Campbell, M. A. Newton, V. Boyd, D. F. Lee and J. Evans, J. Phys. Chem. B, 2005, 109, 2885 CrossRef CAS.
- P. L. Cottingham and L. K. Barker, Ind. Eng. Chem. Prod. Res. Dev., 1973, 12, 41 CAS.
- J. L. Callahan and B. Gertisser, US3 198 750, assigned to The Standard Oil Company, 1965.
- J. L. Callahan and B. Gertisser, US3 308 151, assigned to The Standard Oil Company, 1967.
- R. K. Grasselli and J. L. Callahan, J. Catal., 1969, 14, 93 CrossRef CAS.
- I. Grenthe, J. Drozdzynski, T. Fujino, E. C. Buck, T. E. Albrecht-Schmitt, S. F. Wolf, in The Chemistry of the Actinide and Transactinide Elements, Springer, Dordrecht, 2006, vol. 1, ch. 5, p. 253 Search PubMed.
- H. Deacon, US85 370, assigned to Gaskell, Deacon and Co., 1868.
- J. Pérez-Ramírez, C. Mondelli, T. Schmidt, O. F.-K. Schlüter, A. Wolf, L. Mleczko and T. Dreier, Energy Environ. Sci., 2011, 4, 4786 Search PubMed.
- K. Seki, Catal. Surv. Asia, 2010, 14, 168 CrossRef CAS.
- H. Over, J. Phys. Chem. C, 2012, 116, 6779 CAS.
- A. Wolf, L. Mleczko, O. F. Schlüter and S. Schubert, EP2026905, assigned to Bayer MaterialScience, 2006.
- C. Mondelli, A. P. Amrute, F. Krumeich, T. Schmidt and J. Pérez-Ramírez, ChemCatChem, 2011, 3, 657 CrossRef CAS.
- D. Crihan, M. Knapp, S. Zweidinger, E. Lundgren, C. J. Weststrate, J. N. Andersen, A. P. Seitsonen and H. Over, Angew. Chem., Int. Ed., 2008, 47, 2131 CrossRef CAS.
- N. López, J. Gómez-Segura, R. P. Marín and J. Pérez-Ramírez, J. Catal., 2008, 255, 29 CrossRef.
- M. A. G. Hevia, A. P. Amrute, T. Schmidt and J. Pérez-Ramírez, J. Catal., 2010, 276, 141 CrossRef CAS.
- D. Teschner, R. Farra, L.-D. Yao, R. Schlögl, H. Soerijanto, R. Schomaecker, T. Schmidt, L. Szentmiklósi, A. P. Amrute, C. Mondelli, J. Pérez-Ramírez, G. Novell-Leruth and N. López, J. Catal., 2012, 285, 273 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, H. A. Hansen, I. C. Man, J. Rossmeisl and T. Bligaard, ChemCatChem, 2010, 2, 98 CrossRef CAS.
- T. Hibi, H. Nishida and H. Abekawa, US5 871 707, assigned to Sumitomo Chemical Company, 1999.
- D. Teschner, G. Novell-Leruth, R. Farra, A. Knop-Gericke, R. Schlögl, L. Szentmiklósi, M. G. Hevia, H. Soerijanto, R. Schomäcker, J. Pérez-Ramírez and N. López, Nat. Chem., 2012, 4, 739 CrossRef CAS.
- A. P. Amrute, C. Mondelli, M. Moser, G. Novell-Leruth, N. López, D. Rosenthal, R. Farra, M. E. Schuster, D. Teschner, T. Schmidt and J. Perez-Ramírez, J. Catal., 2012, 286, 287 CrossRef CAS.
- M. Moser, C. Mondelli, T. Schmidt, F. Girgsdies, M. E. Schuster, R. Farra, L. Szentmiklósi, D. Teschner and J. Pérez-Ramírez, Appl. Catal., B, 2013, 132–133, 123 CrossRef CAS.
- A. Wolf, L. Mleczko, S. Schubert and O. F. Schlüter, US20100202959, assigned to Bayer Technology Services, 2010.
- A. Wolf, L. Mleczko, O. F. Schlüter and S. Schubert, US20110180419, assigned to Bayer Technology Services, 2011.
- K. J. Notz and M. G. Mendel, J. Inorg. Nucl. Chem., 1960, 14, 55 CrossRef CAS.
- Y.-S. Yang, Y.-H. Kang and H.-K. Lee, Mater. Chem. Phys., 1997, 50, 243 CrossRef CAS.
- A. P. Amrute, C. Mondelli, T. Schmidt, R. Hauert and J. Pérez-Ramírez, ChemCatChem, 2013, 5, 748 CrossRef CAS.
- R. H. Marsuall and H. R. Hoekstra, J. Inorg. Nucl. Chem., 1965, 27, 1947 CrossRef.
- C. Colliex, A. Gloter, K. March, C. Mory, O. Stéphan, K. Suenaga and M. Tencé, Ultramicroscopy, 2012, 123, 80 CrossRef CAS.
- K. Foger and H. Jaeger, J. Catal., 1985, 92, 64 CrossRef CAS.
- P. Birke, S. Engels, K. Becker and H. D. Neubauer, Chem. Technol., 1980, 32, 253 CAS.
- T. Subramanian, B. P. Reddy, P. Venkatesh, R. Kandan, T. Vandarkuzhali, K. Nagarajan and P. R. V. Rao, in Pyrochemical Separations: Workshop Proceeding, Avignon, France, OECD Publishing, Paris, 2000, p. 190 Search PubMed.
- T. Sato, S. Shiota, S. Ikoma and F. Ozawa, J. Appl. Chem. Biotechnol., 1977, 27, 275 CAS.
- C. Mondelli, A. P. Amrute, T. Schmidt and J. Pérez-Ramírez, Chem. Commun., 2011, 47, 7173 RSC.
- V. J. Wheeler, R. M. Dell and E. Wait, J. Inorg. Nucl. Chem., 1964, 26, 1829 CrossRef CAS.
- H. Inada, L. Wu, J. Wall, D. Su and Y. Zhu, J. Electron Microsc. Tech., 2009, 58, 111 CrossRef CAS.
- A. P. Amrute, C. Mondelli and J. Pérez-Ramírez, Catal. Sci. Technol., 2012, 2, 2057 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, H2-TPR profiles, and TEM images of differently treated uranium oxides. See DOI: 10.1039/c3sc22067b |
| This journal is © The Royal Society of Chemistry 2013 |