Synthetically modified Fc domains as building blocks for immunotherapy applications†
Chawita
Netirojjanakul
a,
Leah S.
Witus
a,
Christopher R.
Behrens
a,
Chih-Hisang
Weng
a,
Anthony T.
Iavarone
ab and
Matthew B.
Francis
*ac
aDepartment of Chemistry, University of California, Berkeley, California 94720-1460, USA. E-mail: mbfrancis@berkeley.edu
bQB3 Institute, University of California, Berkeley, CA 94720-3220, USA
cMaterials Sciences Division, Lawrence Berkeley National Laboratories, Berkeley, California 94720-1460, USA
First published on 1st November 2012
Abstract
Chemically based protein modification methods could provide useful strategies for the generation of antibody mimics. However, the highly complex structures of antibody domains make it exceptionally difficult to modify these proteins in a single or small number of locations. This complexity includes the presence of multiple polypeptide chains, extensive disulfide networks, and critically important glycosylation patterns, all of which must remain intact to obtain biological function. In this work, we have created novel antibody mimics by installing synthetic molecules at the N-termini of crystallizable fragment domains (Fc's) via a chemical modification approach. First, a pyridoxal 5′-phosphate (PLP) mediated N-terminal transamination reaction provided a compatible method for site-selectively installing ketones as reactive handles on Fc domains. High levels of conversion were achieved. For elaboration of the newly installed chemical handles, we used two strategies for the ligation of our desired compounds to the protein. In the first, we used alpha-effect amines to create oxime or hydrazone linkages. Alternatively, we used the ketone as a site to introduce a second reaction handle: an aniline group that can participate in a recently reported oxidative coupling reaction. The oxidative coupling provides a highly efficient ligation strategy requiring very short reaction times (two min or less) at room temperature. By combining the advantages of synthetic targeting agents (e.g. high stability, low cost, and facile and reproducible production and discovery) with the ability of Fc domains to mediate targeted cell death and extend plasma half-life, these new hybrid agents may possess the best qualities of both. As an initial proof of concept, Fc domains were functionalized with DNA aptamers. The specificity of the aptamers for binding their cellular targets was demonstrated, as was the ability of the modified Fc domains to bind to complement proteins. The full assessment of the immunological properties of these hybrid constructs is currently underway.
Introduction
Since the early 1980s proteins have emerged as a major new class of pharmaceuticals, with over 200 marketed products currently available for therapeutic, diagnostic, and vaccine use.1 Protein engineering has revolutionized this field by providing tools to customize existing proteins or to create new ones for specific clinical applications. A particularly important advance has been the generation of fusion proteins comprising segments derived from two or more different precursors. This approach allows multiple biological functions, such as binding and therapeutic activity, to be combined in a single entity. Arguably the most clinically and commercially successful fusion protein therapeutics to date contain the crystallizable fragment (Fc) region of immunoglobulins. The Fc fusions can endow attached peptides or proteins with the antibody-like property of long serum half-life (days to weeks) by binding to the FcRn salvage receptor.1,2 The abilities of the Fc to bind to Fc receptors and/or complement proteins can also provide the fusion protein with immunological cytotoxicity functions.1,3 Moreover, the smaller size of Fc domains, compared to full-size monoclonal antibodies (mAbs), may also improve tissue penetration4 and may allow for alternative routes of administration such as pulmonary delivery.5 They also only require a single gene for expression. However, using molecular biology alone, the Fc-fusions can only be made with polypeptide functional groups.Recently, Barbas and coworkers have devised a new methodology to conjugate a targeted small molecule and an RNA aptamer to aldol-catalyzing mAbs. The resulting construct exhibited significant increases in the plasma half-lives of the synthetic moieties.6 These results provide an exciting approach to the creation of antibody fusions using various types of targeting groups. However, their method required the use of full-size mAbs, with Fab domains that catalyzed the ligation reaction.6b Thus, the final hybrid products still retain the large size and complexity of full-sized antibodies. Currently there is no analogous way to conjugate other classes of therapeutics, such as small synthetic molecules, aptamers,7 peptoids,8 or chemically modified peptides, to only the Fc fragments in order to make Fc-fusions. This is largely due to the complex structure of Fc domains, which contain multiple polypeptide chains, extensive disulfide networks, and essential glycosylation patterns. These components make it very difficult to design chemical approaches that can modify Fc domains site-specifically.
To date the most promising methods for site-selective modification of complex molecules containing disulfide bonds and oligosaccharides have included the double alkylation of cysteines resulting from the reduction of interchain disulfide bonds,9 the alkylation of a site-specifically introduced cysteine residue,10 native chemical ligations at the C-termini,11 and the chemical modification of genetically encoded aldehyde tags.12 The Bertozzi aldehyde tagging method12 provides a particularly intriguing possibility for subsequent site-selective hydrazone and oxime formation, and could indeed be used as an alternative strategy for the generation of the conjugates described herein. As an additional possibility, artificial amino acids can also be incorporated for the site-selective modification of antibody. For example, Rader and co-workers have demonstrated the insertion of selenocysteine residue at the C-termini of Fc proteins for an attachment to LLP2A, an α4β1 integrin-binding small molecule.13 More recently, Schultz and co-workers also showed an incorporation of p-acetylphenylalanine to the antigen-binding fragments (Fab) for the attachment of the protein toxin saporin.14 The latter method could also potentially provide the carbonyl groups that are targeted in the experiments described below.
In this work, we present a new method to create antibody mimics by directly conjugating antibody Fc domains to synthetic targeting molecules using chemically based protein modification methods. This synthesis leads to the production of Fc-synthetic molecule hybrids, where the Fc domains serve as building blocks to improve the pharmacokinetic properties of synthetic agents and potentially to endow them with immunological activating properties (such as the ability to induce antibody dependent cell-mediated cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC)). These Fc-synthetic molecule hybrids are still much smaller in size compared to mAbs. Moreover, these chemical modification techniques should be extensible for the modification of many other complex biomolecules, including IgG itself.
Results and discussion
Recently, the potential of using pyridoxal 5′-phosphate (PLP)-mediated transamination has been demonstrated as a convenient way to install one new functional group in a single location without modifying native free cysteine or lysine residues.15 Taking advantage of this selectivity, the synthesis of Fc-synthetic molecule hybrids was designed around using PLP-mediated N-terminal transamination to provide a unique ketone group at the N-terminus of the Fc-domain for further functionalization (Fig. 1). The synthetic moiety can then be conjugated to the N-terminus using traditional oxime or hydrazone formation.16 We also explored the use of an oxidative coupling reaction as a new approach to ligate synthetic groups to the protein with significantly enhanced efficiency17—a useful feature for the installation of high-value cargo, such as complex drug molecules or the nucleic acid aptamers described herein.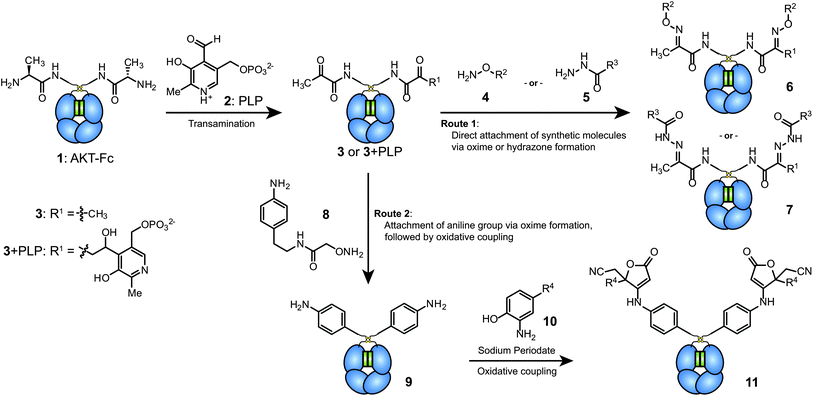 | ||
| Fig. 1 Modification scheme for Fc proteins. First, ketone functional groups are installed at the N-termini through PLP-mediated transamination. These groups can then be functionalized using two different approaches. The first involves the direct attachment of molecules of interest via oxime formation (with 4) or hydrazone formation (with 5). The second strategy uses a highly efficient oxidative coupling reaction. This approach involves the chemoselective coupling of aniline groups on the Fc proteins (9) with aminophenol-containing reagents (10). | ||
Since the efficiency of PLP-mediated transamination depends on the sequence of the N-terminus,15c the Fc protein was first mutated to contain a highly reactive alanine–lysine–threonine (AKT) sequence15d immediately following the IL2 signaling peptide (see sequence in ESI, Fig. S1†). This leader peptide was cleaved during the secretion of the AKT–Fc proteins from the host cells. The protein was expressed in glycosylated form in transfected HEK 293T cells (see mass spectrometry characterization in ESI, Fig. S3†) and purified using a protein G column.
The AKT–Fc proteins were then exposed to a freshly prepared 100 mM solution of PLP in pH 6.5 phosphate buffer at 37 °C for 1 h. Due to the small mass difference (1 Da) corresponding to the transamination of each terminus, the resulting protein was exposed to benzyl hydroxylamine (BnONH2, 4a) for 2 d at RT before characterization using mass spectrometry. To simplify the analysis, the carbohydrate domain was removed from the samples using PNGase F. As shown for the non-reduced Fc products in Fig. 2a and b, very high conversion was observed. Two major products were obtained (6a and 6a + PLP), both resulting from the desired oxime formation reaction at the two Fc termini. The higher mass product resulted from an aldol reaction between the aldehyde of PLP and the transaminated terminus during the first reaction step (shown as 3 + PLP in Fig. 1). The only other product that was evident was a small amount of Fc with a single oxime modification (species 12). In samples lacking PLP treatment, exposure to BnONH2 led to no observable modification. Oxime formation using AlexaFluor 488 alkoxyamine was also used to detect reaction progress via SDS-PAGE (Fig. 2c). Fluorescence was detected in the +PLP lanes using both reducing and non-reducing gel loading buffers, suggesting that AKT–Fc underwent transamination by PLP and that the Fc remained in dimeric state under the reaction conditions. In addition to the expected products, a small amount of residual Fc dimer (∼4 to 7% by densitometry) was observed in the reduced lane. We presume this resulted from an aldol reaction between the two terminal ketones, which are directly adjacent to one another in the dimeric Fc structure. Such a species would still possess a single remaining ketone group, thus allowing its labeling with the dye molecule. The presence of this minor species could also explain species 12 in Fig. 2b, but the identities of these two byproducts have not been confirmed further due to their very low abundance.
 | ||
| Fig. 2 Analysis of transamination efficiency for AKT–Fc domains. AKT–Fc (1) was exposed to 50 mM 4a for 40 h at pH 6.5, followed by treatment with PNGase F (a) without prior transamination with PLP (as a negative control), or (b) following transamination with 100 mM PLP at 37 °C for 1 h. The peak at 52315 Da corresponded to unmodified AKT–Fc (expected: 52315 Da). The double-oxime product (6a) appeared at 52525 Da (expected: 52523 Da), and the peak at 52772 Da corresponded to product 6a plus a single PLP addition (expected: 52770 Da). The peak at 52419 Da corresponded to the addition of one molecule of 4a to the AKT–Fc fragment (12, expected: 52419 Da). (c) Samples of AKT–Fc with and without transamination using PLP (100 mM PLP at 37 °C for 1 h) were exposed to 80 μM Alexa-Fluor 488 alkoxyamine (4b) for 43 h in the presence of 100 mM aniline as a catalyst. They were then analyzed by SDS-PAGE under reducing (Lanes 1 and 2) or nonreducing (lanes 3 and 4) conditions. The fluorescent images of the Fc–AlexaFluor 488 oxime products (top) were taken using a Typhoon imaging system. The bottom gel was stained using Coomassie blue. | ||
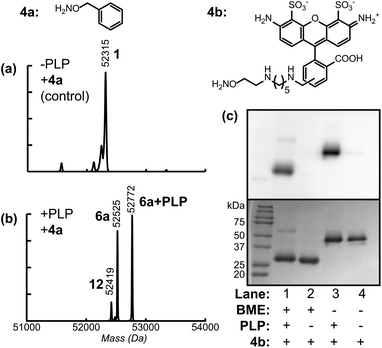
Due to slow kinetics of oxime and hydrazone formation, long reaction times and large excesses of reagent are typically used to reach full conversion. While this is fine for inexpensive small molecules, it poses a problem when the group to be attached is a large or expensive reagent (such as a drug). With these cases in mind, oxime formation was used to attach an aniline-containing small molecule to allow further ligation using oxidative coupling reactions. As reported recently, the periodate-mediated oxidative coupling18 between anilines and aminophenols can achieve chemoselective protein modification with very high levels of product conversion in less than 2 min. Using the transamination/oxime formation procedure described above with compound 8 produced Fc proteins bearing an aniline group at each of the two termini (species 9 and 9 + PLP in Fig. 3b).
 | ||
| Fig. 3 Modification of Fc domains via oxidative coupling (O.C). AKT–Fc (1) was exposed to 25mM 8 for 40 h at pH 4.5, followed by PNGase F treatment (a) without prior transamination with PLP (as a negative control), or (b) following transamination with 100 mM PLP at 37 °C for 1 h. The peak at 52315 Da corresponded to unmodified AKT–Fc (expected: 52315 Da). The double-oxime product (9) appeared at 52697 Da (expected: 52695 Da), and the peak at 52944 Da corresponded to product 9 plus a single PLP addition (expected: 52942 Da). The peak at 52506 Da corresponded to the addition of one molecule of 8 to the AKT–Fc fragment (13, expected: 52506 Da) (c) Samples of unmodified Fc (1) and Fc–aniline (9) were exposed to 100 μM 2k PEG–aminophenol (10a) and 1mM NaIO4 for 2 min at RT. lanes 1–5 display negative controls. Only in the presence of both aniline on the Fc and NaIO4 did the attachment of 2 k PEG–aminophenol occur (lane 6). In the presence of 10 mM mannose (lane 7), the O.C. still proceeded; however the yield suffered when the mannose concentration was increased to 100 mM (lane 8). (d) The extent of oxidation of the oligosaccharides on the Fc protein within the reaction time of the O.C. was analyzed. The Fc proteins were exposed to 1 mM NaIO4 for 2 and 60 min. The reaction was stopped upon addition of TCEP and any aldehydes formed from the oxidation were then detected by AlexaFluor 488–ONH2. The fluorescent images of Fc–AlexaFluor488 oxime products (top) were taken using a Typhoon imaging system. The oligosaccharides on the Fc were minimally oxidized under the O.C. reaction time of 2 min (lane 2) and this oxidation was lowered to background level upon addition of 10 mM mannose or higher concentration (lanes 3 and 4). The oxidation of oligosaccharides on Fc with NaIO4 for 1 h was shown as a positive control (lane 5). Lane 1 and 6 display the background level of oxidized sugar in the absence of NaIO4. | ||
Following successful installation of aniline groups on the Fc proteins, the oxidative coupling reaction was then investigated using aminophenol containing polyethyleneglycol 10a (MW ∼ 2000 Da).17b Aniline Fc 9 (10–40 μM) was combined with 100 μM 10a in pH 6.5 phosphate buffer. A stock solution of sodium periodate was added to a final concentration of 1 mM, and the reaction was incubated for 2 min. The resulting solution was then passed through a NAP5 gel filtration column to quench and remove the periodate. SDS-PAGE analysis under reducing conditions (and without the use of PNGase F) indicated that 50% of the individual Fc chains were converted to the singly PEGylated product (Fig. 3c, lane 6), as indicated by optical densitometry. This yield likely results from the close proximity of the two N-termini in the intact Fc domains, resulting in the attachment of only one PEG chain to each protein dimer. Nonetheless, a high degree of modification was observed using very short coupling times.
As one potential concern with this strategy, immunoglobulin proteins contain oligosaccharides, which could be cleaved to form aldehydes in the presence of sodium periodate. To determine to what degree this occurs, unmodified Fc domains were exposed to NaIO4 under the oxidative coupling conditions for 2 min and 1 h. The resulting aldehyde groups were then visualized by subsequent reaction with AlexaFluor 488 alkoxyamine (4b). As seen in Fig. 3d, the oligosaccharides on the Fc protein were only minimally oxidized by NaIO4 at the 2 min time point (compare lanes 2 and 5 to the background labeling for unmodified Fc in lane 1). Furthermore, we found that the addition of 10 mM to 100 mM mannose could suppress this oxidation of the carbohydrate groups completely. Interestingly, the oxidative coupling reaction still proceeded with similar conversion in the presence of 10 mM mannose (Fig. 3c, lane 7), and with somewhat reduced conversion in the presence of 100 mM mannose (lane 8). Thus, this strategy provides a method to protect glycoproteins from undesired oxidation with this procedure. Even though it should be noted that the Fc protein examined here does not contain sialic acids, which are the most susceptible to oxidation, we anticipate that oxidative coupling will still occur in a much shorter timescale in comparison to oxidation of oligosaccharides.
Since 1990s, small RNA and DNA aptamers have emerged as a new class of molecules for therapeutic and diagnostic purposes, owing to the successful development of the systematic evolution of ligands by an exponential enrichment process, known as SELEX.7,19 Using SELEX, new aptamers can be evolved to bind to cells with high specificity and affinity, often without prior knowledge of the specific molecular targets. These readily evolved binding groups could endow Fc domains with specific cell binding abilities, and, conversely, the Fc domains could improve the in vivo circulation times of the oligonucleotides, as shown by Barbas et al.6a Two aptamers were selected for attachment to the Fc proteins as a proof of principle for the production of Fc-synthetic molecule hybrids: (1) sgc8c, which targets protein tyrosine kinase 7 (PTK7)20 and (2) TD05.1, which targets membrane-bound IgM (mIgM, also known as the B-cell receptor).21 The sgc8c aptamer has been used in many applications22,17c and shown high specificity to its target. The use of TD05.1 could be advantageous because there is currently no antibody that is specific to binding mIgM without also binding to circulating IgM in the blood.21 For use as a negative control in cell binding experiments, a scrambled 41-nucleotide DNA sequence (M2M2) was also attached to Fc.
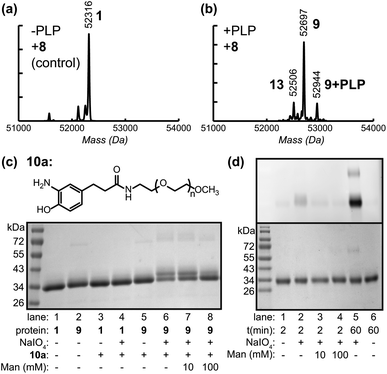
Using the hydrazone formation strategy with 100 μM hydrazide-oligonucleotides (5) and 100 mM aniline as a coupling catalyst23 over a 48 h period, the Fc–aptamer conjugates could be obtained as identified by SDS-PAGE analysis (Fig. 4b). Due to the high negative charge of the DNA portion, the Fc conjugates could also be separated using anion exchange chromatography (Fig. 4c and ESI, Fig. S8†), allowing more accurate determination of the product ratios. The major product (64%) possessed a single attachment of the oligonucleotide to one of the Fc N-termini, likely due to steric hindrance and electronic repulsion between two DNA molecules. The doubly modified product corresponded to 20% yield, with only 16% of unconjugated Fc protein remaining. The alternative method, the attachment of aminophenol–DNA oligonucleotides (10b, 100 μM) to Fc–aniline (9) via oxidative coupling reaction, was achieved at RT in 2 min. In this case, 58% of the protein product corresponded to the single aptamer conjugate and 39% corresponded to the double aptamer conjugate, representing a total of >95% of the protein species. The overall yields were therefore slightly higher than those achieved by direct hydrazone formation (Fig. 4a, lane 4–6), but they were obtained with drastically reduced coupling times. Anion exchange chromatography was again used to obtain pure conjugates for use in subsequent binding studies, Fig. 4d.
 | ||
| Fig. 4 Construction of Fc–aptamer conjugates. Two different aptamers, sgc8c targeting PTK7 and TD05.1 targeting membrane-bound IgM, were attached to keto-Fc using the two approaches shown in Fig. 1. (a) Structure of hydrazide– and aminophenol–DNA oligonucleotides used for hydrazone formation and the oxidative coupling reaction, respectively. (b) SDS-PAGE analysis under non-reducing conditions showed the formation of Fc hybrids using either hydrazone formation (labeled as ‘h’, lanes 1–3) or oxidative coupling (‘o’, lanes 4–6). Single aptamer conjugates were the major products, along with lesser amounts of doubly labeled conjugates species. (c) Anion exchange-HPLC analysis of the crude Fc–TD05.1 adduct following the oxidative coupling reaction, indicating the relative quantities of Fc, Fc–DNA, and Fc–(DNA)2. The shaded fraction was collected for further use. (d) SDS-PAGE analysis (non-reducing) of the purified Fc–aptamer conjugates used for cell binding analysis. | ||
The specificities of Fc–sgc8c, Fc–TD05.1, and Fc–M2M2 hybrid constructs generated from both the hydrazone and the oxidative coupling strategies were next evaluated for selective cell binding using flow cytometry. Jurkat cells, a T-cell leukemia cell line overexpressing PTK7, were used as the targeted cells for the Fc–sgc8c constructs. Ramos cells, a Burkitt's lymphoma cell line overexpressing mIgM, were used as target cells for Fc–TD05.1. U266 cells, a B-cell line overexpressing neither membrane protein, were chosen as a negative control sample. The binding assay was conducted as outlined in Fig. 5a, with detection of the cell-bound Fc conjugates using fluorescently labeled anti-human IgG1. Only Jurkat cells were bound by Fc–sgc8c, and only Ramos cells were recognized by Fc–TD05.1 (Fig. 5b). Neither cell line was recognized by Fc–M2M2. Moreover, the U266 negative control cell line did not bind to any of the Fc–aptamer constructs. In addition to confirming that the aptamers retained their specificity after attachment to the Fc domains, these results also indicated that the Fc region retained its proper folding and thus could still be recognized by the fluorescent secondary antibodies.
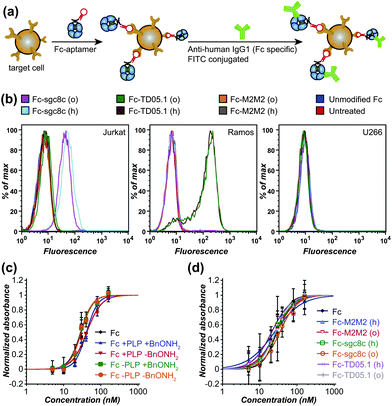 | ||
| Fig. 5 Cell binding specificity and C1q binding ability of Fc–aptamer conjugates. (a) The general cell binding analysis scheme is shown. The cells were incubated with Fc–aptamer samples, which were subsequently detected using FITC-labeled secondary antibodies specific for Fc of human IgG1. (b) Flow cytometry data are shown for the binding of Fc–aptamer conjugates to Jurkat cells (overexpressing PTK7, the target of the sgc8c aptamer, ref. 20), Romos cells (overexpressing membrane-bound IgM, the target for the TD05.1 aptamer, ref. 21), and U266 cells as a negative control. All Fc–sgc8c and Fc–TD05.1 conjugates retained their targeting specificity, whether they were generated using hydrazone formation (labeled ‘h’) or oxidative coupling (labeled ‘o’). Unmodified Fc proteins and an Fc–M2M2 conjugate (bearing a scrambled 41-base oligonucleotide) did not bind to any cell lines. (c) ELISA data are shown for C1q binding to unmodified Fc, PLP-treated Fc, and Fc after oxime formation with BnONH2. (d) ELISA data are shown for C1q binding to the Fc–aptamer conjugates used in (b). | ||
A key feature associated with the toxicity of many antibodies is the recruitment of complement proteins found in blood serum. We therefore evaluated the ability of the modified Fc proteins to bind to the C1q complement protein using ELISA.24 Briefly, varying concentrations of the Fc conjugates were bound to a polystyrene 96-well plate, after which a 2 μg mL−1 solution of human C1q was added. Binding ability was confirmed using an anti-C1q antibody conjugated to horseradish peroxidase (HRP). These results showed that the PLP-treated Fc and Fc–aptamer constructs still retained their ability to bind to C1q with similar affinities. This suggests that the bulk of the immunological activating properties of the Fc domains remained intact following the modification process (Fig. 5c and d).
Conclusions
Through these studies, we have developed two new approaches for the site-specific modification of antibody Fc domains. Although both strategies lead to the formation of well-defined conjugates with similar yields, the oxidative coupling strategy has the advantage of requiring much shorter reaction times for the attachment of larger molecules, and the use of much lower concentrations of molecules that are in short supply. The ability to produce Fc–aniline conjugates in large batches for the subsequent attachment of various synthetic molecules may also prove useful as a modular platform for discovering novel biologics for immunotherapy. We further demonstrated the utility of these strategies through the generation of a new class of Fc–aptamer conjugates. These hybrid agents still retain the binding specificity of the original aptamers while adding the ability of the Fc domain to be recognized by complement proteins. The strategy developed here could be readily adapted for the attachment of other classes of synthetic molecules, such as peptoids or small molecules, to Fc domains for in vivo applications. There are also a number of nanoscale delivery vehicles that could benefit from extended plasma circulation times, which these convenient Fc building blocks could convey. Finally, it is highly likely that these chemical strategies can be applied to full-sized IgG molecules as well. These synthetic targets are being pursued in current experiments, as is the more thorough immunological characterization of the Fc conjugates described herein.Acknowledgements
These studies were generously supported by the DOD Breast Cancer Research Program (BC016995). CN was supported by a HHMI International Student Research Fellowship and an Abramson Graduate Scholarship. CRB was supported by DOE CARE (California Alliance for Radiotracer Education) grant DESC0002061. The UC Berkeley Chemical Biology Graduate Program (Training Grant 1 T32 GMO66698) is also acknowledged for their support of CRB and LSW. We would also like to thank Dr Michelle Farkas and Samuel Sternberg for helpful discussion.Notes and references
- (a) G. Walsh, Nat. Biotechnol., 2010, 28, 917–924 CrossRef CAS; (b) P. Carter, Exp. Cell Res., 2011, 317, 1261–1269 CrossRef CAS; (c) C. Huang, Curr. Opin. Biotechnol., 2009, 20, 692–699 CrossRef CAS.
- (a) D. C. Roopenian and S. Akilesh, Nat. Rev. Immunol., 2007, 7, 715–725 CrossRef CAS; (b) V. Ghetie and E. S. Ward, Annu. Rev. Immunol., 2000, 18, 739–766 CrossRef CAS.
- (a) A. Iannello and A. Ahmad, Cancer Metastasis Rev., 2005, 24, 487–499 CrossRef CAS; (b) K. A. Gelderman, S. Tomlinson, G. D. Ross and A. Gorter, Trends Immunol., 2004, 25, 158–164 CrossRef CAS; (c) R. L. Shields, A. K. Namenuk, K. Hong, Y. G. Meng, J. Rae, J. Briggs, D. Xie, J. Lai, A. Stadlen, B. Li, J. A. Fox and L. G. Presta, J. Biol. Chem., 2001, 276, 6591–6604 CrossRef CAS; (d) R. A. Clynes, T. L. Towers, L. G. Presta and J. V. Ravetch, Nat. Med., 2000, 6, 443–446 CrossRef CAS.
- (a) T. Yokota, D. E. Milenic, M. Whitlow and J. Schlom, Cancer Res., 1992, 52, 3402–3408 CAS; (b) B. J. Hicke, A. W. Stephens, T. Gould, Y.-F. Chang, C. K. Lynott, J. Heil, S. Borkowski, C.-S. Hilger, G. Cook, S. Warren and P. G. Schmidt, J. Nucl. Med., 2006, 47, 668–678 CAS.
- (a) S. C. Low, S. L. Nunes, A. J. Bitonti and J. A. Dumont, Hum. Reprod., 2005, 20, 1805–1813 CrossRef CAS; (b) A. J. Bitonti, J. A. Dumont, S. C. Low, R. T. Peters, K. E. Kropp, V. J. Palombella, J. M. Stattel, Y. Lu, C. A. Tan, J. J. Song, A. M. Garcia, N. E. Simister, G. M. Spiekermann, W. I. Lencer and R. S. Blumberg, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 9763–9768 CrossRef CAS.
- (a) U. Wuellner, J. I. Gavrilyuk and C. F. Barbas III, Angew. Chem., Int. Ed., 2010, 49, 5934–5937 CAS; (b) C. Rader, S. C. Sinha, M. Popkov, R. A. Lerner and C. F. Barbas III, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5396–5400 CrossRef CAS.
- (a) A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS; (b) C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- R. N. Zuckermann and T. Kodadek, Curr. Opin. Mol. Ther., 2009, 11, 299–307 CAS.
- S. Shaunak, A. Godwin, J. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. L. Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS.
- J. Xiao, R. Chen, M. A. Pawlicki and T. J. Tolbert, J. Am. Chem. Soc., 2009, 131, 13616–13618 CrossRef CAS.
- P. Wu, W. Shui, B. L. Carlson, N. Hu, D. Rabuka, J. Lee and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3000–3005 CrossRef CAS.
- T. Hofer, J. D. Thomas, T. R. Burke and C. Rader, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12451–12456 CrossRef CAS.
- (a) B. M. Hutchins, S. A. Kazane, K. Staflin, J. S. Forsyth, B. Felding-Habermann, P. G. Schultz and V. V. Smider, J. Mol. Biol., 2011, 406, 595–603 CrossRef CAS; (b) B. M. Hutchins, S. A. Kazane, K. Staflin, J. S. Forsyth, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Chem. Biol., 2011, 18, 299–303 CrossRef CAS.
- (a) J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS; (b) R. A. Scheck and M. B. Francis, ACS Chem. Biol., 2007, 2, 247–251 CrossRef CAS; (c) R. A. Scheck, M. T. Dedeo, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 11762–11770 CrossRef CAS; (d) L. S. Witus, T. Moore, B. W. Thuronyi, A. P. Esser-Kahn, R. A. Scheck, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2010, 132, 16812–16817 CrossRef CAS . For previous studies of transamination with PLP, see: ; (e) E. E. Snell, J. Am. Chem. Soc., 1945, 67, 194–197 CrossRef CAS; (f) H. B. F. Dixon, J. Protein Chem., 1984, 3, 99–108 CrossRef CAS; (g) H. B. F. Dixon and R. Fields, Methods Enzymol., 1972, 25, 409–419 CrossRef CAS.
- (a) W. P. Jencks, J. Am. Chem. Soc., 1959, 81, 475–481 CrossRef CAS; (b) V. W. Cornish, K. M. Hahn and P. G. Schultz, J. Am. Chem. Soc., 1996, 118, 8150–8151 CrossRef CAS; (c) L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS; (d) J. Kalia and R. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS.
- (a) J. M. Hooker, A. P. Esser-Kahn and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 15558–15559 CrossRef CAS; (b) C. R. Behrens, J. M. Hooker, A. C. Obermeyer, D. W. Romanini, E. M. Katz and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 16398–16401 CrossRef CAS; (c) G. J. Tong, S. C. Hsiao, Z. M. Carrico and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 11174–11178 CrossRef CAS.
- For examples of other bioconjugation reactions involving sodium periodate, see: (a) K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS; (b) B. Liu, L. Burdine and T. Kodadek, J. Am. Chem. Soc., 2006, 128, 15228–15235 CrossRef CAS.
- (a) X. Fang and W. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS; (b) A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS; (c) S. D. Jayasena, Clin. Chem., 1999, 45, 1628–1650 CAS; (d) S. M. Nimjee, C. P. Rusconi and B. A. Sullenger, Annu. Rev. Med., 2005, 56, 555–583 CrossRef CAS; (e) P. R. Bouchard, R. M. Hutabarat and K. M. Thompson, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 237–257 CrossRef CAS.
- (a) D. Shangguan, Y. Li, Z. Tang, Z. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, C. J. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS; (b) D. Shangguan, Z. Tang, P. Mallikaratchy, Z. Xiao and W. Tan, ChemBioChem, 2007, 8, 603–606 CrossRef CAS; (c) D. Shangguan, Z. Cao, L. Meng, P. Mallikaratchy, K. Sefah, H. Wang, Y. Li and W. Tan, J. Proteome Res., 2008, 7, 2133–2139 CrossRef CAS.
- (a) P. Mallikaratchy, Z. Tang, S. Kwame, L. Meng, D. Shangguan and W. Tan, Mol. Cell. Proteomics, 2007, 6, 2230–2238 CrossRef CAS; (b) P. R. Mallikaratchy, A. Ruggiero, J. R. Gardner, V. Kuryavyi, W. F. Maguire, M. L. Heaney, M. R. McDevitt, D. J. Patel and D. A. Scheinberg, Nucleic Acids Res., 2011, 39, 2458–2469 CrossRef CAS.
- (a) H. Shi, X. He, K. Wang, X. Wu, X. Ye, Q. Guo, W. Tan, Z. Qing, X. Yang and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3900–3905 CrossRef CAS; (b) S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS; (c) Y.-L. Luo, Y.-S. Shiao and Y.-F. Huang, ACS Nano, 2011, 5, 7796–7804 CrossRef CAS; (d) N. Stephanopoulos, G. J. Tong, S. C. Hsiao and M. B. Francis, ACS Nano, 2010, 4, 6014–6020 CrossRef CAS.
- (a) A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS; (b) A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS.
- E. E. Idusogie, L. G. Presta, K. Totpal, P. Y. Wong, Y. G. Meng, M. G. Mulkerrin and E. Alerts, J. Immunol., 2000, 164, 4178–4184 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and additional characterization data. See DOI: 10.1039/c2sc21365f |
| This journal is © The Royal Society of Chemistry 2013 |