Conjugation of PEG and gold nanoparticles to increase the accessibility and valency of tethered RNA splicing enhancers†
Andrew J. Perretta, Rachel L. Dickinsonb, Željka Krpetićc, Mathias Brustc, Helen Lewisa, Ian C. Eperon*b and Glenn A. Burley*d
aDepartment of Chemistry, University of Leicester, University Road, Leicester, LE1 7RH, UK
bDepartment of Biochemistry, University of Leicester, Lancaster Road, Leicester, LE1 9HN, UK. E-mail: eci@le.ac.uk
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7 ZD, UK
dDepartment of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: glenn.burley@strath.ac.uk; Web: www.burleylabs.co.uk
First published on 22nd October 2012
Abstract
Since alternative splicing patterns are exhibited by most mammalian genes, the functional properties of a gene may be changed for therapeutic purposes by nudging splicing from one pattern to another. Splicing to a refractory exon can be enhanced using bifunctional targeted oligonucleotide enhancers of splicing (TOES), which anneal to a target exon and carry additional sequence motifs to recruit activator proteins. We have tested here the effectiveness of increasing the availability of the activating domain by using click chemistry to insert spacers of hexaethylene glycol (HEG) or abasic DNA between the activating and annealing domains of the oligonucleotide. We show that the presence of one or two HEG spacers can increase the effectiveness of an oligonucleotide, but the inclusion of 11 or 20 reduced the activity. To increase the valency of the oligonucleotides further, the annealing and activating domains were conjugated to gold nanoparticles (GNPs). We show that activity of these GNP–RNA conjugates in splicing assays depended on the size of the GNP conjugates, the RNA sequence and the directionality (i.e. attachment to GNP at either the 5′ or 3′-ends) of the RNA strands conjugated to the GNP surface. Strikingly, only the activating domains were required.
Introduction
The splicing of mRNA from nascent transcripts in higher eukaryotes is an essential process that generates diversity but is tightly regulated. Numerous RNA-binding proteins and small nuclear RNAs are involved in identifying splice sites and then splicing out the introns from the pre-mRNA precursor. Most genes contain a large number of optional splice sites, use of which can generate mature mRNA sequences that differ in specific regions and thus encode different proteins. This source of genetic diversity enables different tissues to express different proteins from the same gene. However, such flexibility can result in disease when compromised. An example of this is spinal muscular atrophy (SMA), where a single nucleotide (nt) difference at position +6 in exon 7 between the closely similar genes SMN1 and SMN2 causes exons 6 and 8 to splice together in SMN2, skipping exon 7. In persons lacking SMN1 genes, the failure of the SMN2 gene to express a viable SMN protein leads to SMA.1,2 Therefore, molecules that can re-direct pathological splicing patterns are likely to become important therapeutic strategies.The earliest strategies for re-directing splicing involved the use of oligonucleotides (ONs) complementary to splice sites or positively acting regulatory sequences in exons (enhancers or ESEs). These prevent the binding of splicing factors and therefore can cause an exon to be skipped.3 These have been developed with particular success as potential therapeutics for muscular dystrophy. The most recent variant of this strategy is the use of 2′-fluoro/phosphorothioate ON derivatives, which were shown to re-direct splicing by recruiting splicing repressors when hybridized to an RNA target.4
The development of methods for enhancing splicing is more recent. The first strategy developed was to use bifunctional ONs, which hybridize to a target exon that is being skipped and carry either a peptide or an RNA enhancer sequence to which activating proteins2,5 will bind (in effect, a portable ESE; Fig. 1a and b). This strategy is referred to as TOES, for targeted oligonucleotide enhancers of splicing. It can be adapted to recruit repressors, with the opposite effect.7 The second strategy is to design ONs that will block the binding of repressor proteins, thereby enabling splicing.6
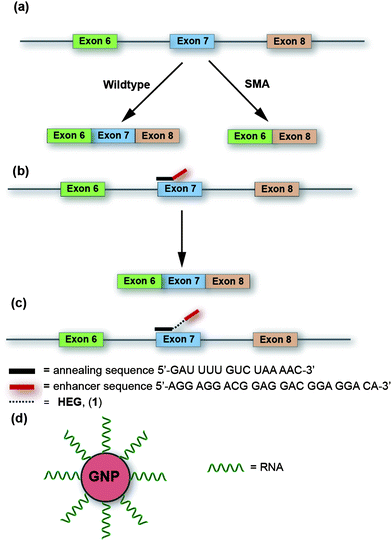 | ||
| Fig. 1 (a) Splicing of pre-mRNA SMN in wildtype and in Spinal Muscular Atrophy (SMA). (b) Mechanism of action of TOES in alternative RNA splicing of SMN pre-mRNA. (c) Our proposed model system is to insert flexible linkers between the hybridizing and enhancer sequences of TOES candidates. (d) Polyconjugated RNA-coated GNPs used in this study to investigate whether multi-valent display of TOES domains on a GNP surface increases splicing activity. | ||
Increasing the level of splicing of exon 7 of SMN2 would increase the expression of the correct SMN protein and is a major goal of therapies for SMA (Fig. 1b).7 We have shown previously that TOES ONs stimulate exon 7 inclusion strongly. The best ON contained three tandem repeats of a sequence (GGAGGAC) that was known to act as a strong enhancer when present in an exon, and it clearly worked effectively when recruited to SMN2 exon 7 by the part of the ON that was complementary to this exon. Systematic tests showed that the level of activity was reduced if there were fewer repeats. What was not clear, however was whether the higher activity observed with more repeats should be attributed to an increase in the number of potential binding sites for activator proteins or to an increase in the length of the sequence between the hybridizing and the protein-binding domains of the TOES (increased length might make the protein-binding domains more accessible, either for activator proteins to bind when the ON is hybridized to a protein-rich exon or for the bound proteins to make contact with targets at the splice sites7). Distinguishing between these two explanations can only be done if the ON is lengthened without introducing additional RNA sequences, which are likely to be bound themselves by proteins that affect splicing.
We describe here a strategy in which the ONs are lengthened by inserting non-RNA elements between the hybridizing and protein-binding domains of the ON (Fig. 1c, Scheme 1). In addition, we show that the number of binding sites for the activator proteins could be increased by conjugating the ONs to gold nanoparticles (GNPs, Fig. 1d). Our results indicate that both the length and functional valency stimulate splicing activity.
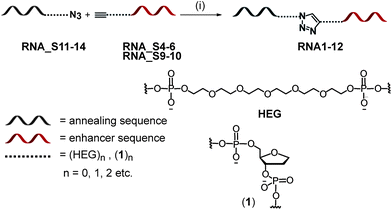 | ||
| Scheme 1 Synthetic strategy for the preparation of bifunctional 2′ OMe RNA ONs. Reagents and conditions: (i) CuBr, TBTA, DMSO–H2O. | ||
Results
Design of spacer units in candidate TOES constructs
To test whether the benefit of incorporating extra potential binding sites for activator proteins such as SRSF1 resulted from an increase in length and, perhaps, availability rather than from extra binding sites per se, we introduced two different types of spacer in between the annealing and protein-binding domains of a bifunctional ON. The linkages investigated were: (i) various numbers of hexaethyleneglycol (HEG) units, and (ii) abasic DNA (1, Scheme 1). A HEG tether provides a highly flexible linkage connecting the annealing region to an ESE and crucially bears no resemblance to the parent RNA structure. The HEG linker is commercially available and each unit corresponds in length to approximately three nucleotides (nts).The addition of HEG also reduces the density of charge in the RNA backbone as only a single charge is present every 1.2 nm relative to ∼0.4 nm in a typical RNA backbone. The abasic site building block (1) comprises a DNA phosphodiester unit, thereby maintaining the equivalent density of negative charge along an ON as in naturally occurring RNA ONs. The lack of both a 2′ OH group and a nitrogenous base differentiates (1) from a naturally occurring nt. The model construct used in this study was based on our original TOES design (Fig. 1b).2 The TOES construct consists of an artificial ESE sequence, derived from three repeats of a known splicing enhancer sequence, and a juxtaposed annealing sequence complementary to positions +2 to +16 of exon 7 in SMN2.8
In the most active TOES, termed GGA, the ESE domain consisted of 2′ OH RNA with phosphorothioate linkages at the 5′ end and the annealing sequence consisted of a mixture of 2′ OMe and phosphorothioate (pS) units.7 The phosphorothioate linkages make a major contribution to the activity, presumably because they reduce degradation. However, phosphorothioates were expected to create problems if the ONs were coupled to gold nanoparticles (GNPs) as each phosphorothioate functional group could act as a competing ligand for the metal nanoparticle surface.9 An alternative approach was to base our investigations on wholly 2′ OMe ON (termed GGA-O), which are less efficient,7 but would present no difficulties for testing the importance of multi-valency in splicing.
Synthesis of RNA bifunctional ONs containing spacers
The enhancer (RNA_S1–S3) and annealing (RNA_S4–S6) domains of the TOES series were prepared by solid phase chemical synthesis (Table 1). Coupling of NHS–azide (S7)10 to RNA_S1–S3 and RNA_S7 and S8 provided the azide-modified versions of these enhancer sequences (RNA_S11–S15). 5′ Alkyne-modified annealer sequences (RNA_S4–S6) were prepared by terminating the synthesis with the known alkyne phosphoramidite (S5).11| Sequence name | 2′OMe RNA ON sequence 5′ → 3′ |
|---|---|
a  . . | |
| RNA_S1 | AGGAGGACGGAGGACGGAGGACAX |
| RNA_S2 | AGGAGGACGGAGGACGGAGGACAWX |
| RNA_S3 | AGGAGGACGGAGGACGGAGGACAW WWW WWW WWWX |
| RNA_S4 | YGAUUUUGUCUAAAAC |
| RNA_S5 | YWGAUUUUGUCUAAAAC |
| RNA_S6 | YWW WWW WWW WWGAUUUUGUCUAAAAC |
| RNA_S7 | AGGAGGACGGAGGACGGAGGACAV VVX |
| RNA_S8 | AsGsGsAsGsGsAsCsGsGsAsGsGsAsCsGsGsAsGsGsAsCsAsWX |
| RNA_S9 | YGsAsUsUsUsUsGsUsCsUsAsAsAsAsC |
| RNA_10 | YWGsAsUsUsUsUsGsUsCsUsAsAsAsAsC |
| RNA_S11 | AGGAGGACGGAGGACGGAGGACAZ |
| RNA_S12 | AGGAGGACGGAGGACGGAGGACAWZ |
| RNA_S13 | AGGAGGACGGAGGACGGAGGACAW WWW WWW WWWZ |
| RNA_S14 | AGGAGGACGGAGGACGGAGGACAVVVZ |
| RNA_S15 | AsGsGsAsGsGsAsCsGsGsAsGsGsAsCsGsGsAsGsGsAsCsAsWZ |
All spacer-containing constructs were prepared as 2′ OMe RNA, which have demonstrated stability and efficacy in RNA splicing.7 Ligation of the azide-modified enhancer sequences (RNA_S11–S14) with alkyne-modified annealer sequences (RNA_S4–S6) by “click chemistry”12 provided the final RNA series RNA1–12 (Table 2). Click chemistry ligation results in the formation of a triazole linkage between the annealer and enhancer sequences. This reaction was chosen by virtue of its chemoselectivity, regiospecificity the minimal steric bulk imparted into the resulting tripartite constructs.13
| Sequence name | Precursor strands | RNA ON sequence 5′ → 3′ |
|---|---|---|
a Underline denotes 2′ OH RNA building blocks; s denotes a phosphorothioate backbone.b  . . | ||
| RNA1 | RNA_S4, RNA_S11 | AGGAGGACGGAGGACGGAGGACAZGAUUUUGUCUAAAAC |
| RNA2 | RNA_S5, RNA_S11 | AGGAGGACGGAGGACGGAGGACAZXGAUUUUGUCUAAAAC |
| RNA3 | RNA_S6, RNA_S11 | AGGAGGACGGAGGACGGAGGACAZXXXXXXXXXXGAUUUUGUCUAAAAC |
| RNA4 | RNA_S4, RNA_S12 | AGGAGGACGGAGGACGGAGGACAXZGAUUUUGUCUAAAAC |
| RNA5 | RNA_S5, RNA_S12 | AGGAGGACGGAGGACGGAGGACAXZXGAUUUUGUCUAAAAC |
| RNA6 | RNA_S6, RNA_S12 | AGGAGGACGGAGGACGGAGGACAXZXXXXXXXXXXGAUUUUGUCUAAAAC |
| RNA7 | RNA_S4, RNA_S13 | AGGAGGACGGAGGACGGAGGACAXXXXXXXXXXZGAUUUUGUCUAAAAC |
| RNA8 | RNA_S5, RNA_S13 | AGGAGGACGGAGGACGGAGGACAXXXXXXXXXXZXGAUUUUGUCUAAAAC |
| RNA9 | RNA_S6, RNA_S13 | AGGAGGACGGAGGACGGAGGACAXXXXXXXXXXZXXXXXXXXXXGAUUUUGUCUAAAAC |
| RNA10 | RNA_S9, RNA_S15 | AsGsGsAsGsGsAsCsGsGsAsGsGsAsCsGsGsAsGsGsAsCsAsXsZGsAsUsUsUsUsGsUsCsUsAsAsAsAsC |
| RNA11 | RNA_S10, RNA_S15 | AsGsGsAsGsGsAsCsGsGsAsGsGsAsCsGsGsAsGsGsAsCsAsXsZXsGsAsUsUsUsUsGsUsCsUsAsAsAsAsC |
| RNA12 | RNA_S4, RNA_S14 | AGGAGGACGGAGGACGGAGGACAYYYZGAUUUUGUCUAAAAC |
| RNA13 | N/A | AGGAGGACGGAGGACGGAGGACAYYYYYYGAUUUUGUCUAAAAC |
| RNA14 | N/A | ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) s s![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ssss ssss![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) YYYYYYGAUsUsUsUsGsUCUAAsAsAsC YYYYYYGAUsUsUsUsGsUCUAAsAsAsC |
| GGA | N/A | sssssGAUsUsUsUsGsUCUAAsAsAsC |
| GGA-O | N/A | AGGAGGACGGAGGACGGAGGACAGAUUUUGUCUAAAAC |
RNA ONs incorporating HEG units
Several structural features were surveyed in this bifunctional RNA series: (i) the location of the triazole group. The series incorporated exemplars where the triazole group is located close to the enhancer sequence (RNA1–3), towards the centre of both the bifunctional construct (RNA5, RNA9, RNA11) and close to the annealer sequence (RNA4, RNA7, RNA10); (ii) the number of HEG units (RNA1–11). This series investigated the direct ligation of the requisite halves lacking a HEG unit (RNA1) and incorporating one (RNA2, RNA4, RNA10), two (RNA5, RNA11), ten (RNA3, RNA7), eleven (RNA6, RNA8) or twenty (RNA9) HEG units; and (iii) the nature of the phosphodiester backbone. Both phosphodiester (RNA1–9) and phosphorothioate versions (RNA10 and 11) were prepared.RNA ONs incorporating units of (1)
RNA12 was prepared by solid phase chemical synthesis followed by click chemistry ligation between RNA_S4 and RNA_S14. RNA13 was prepared by solid phase synthesis using phosphoramidite (S4) in order to examine the influence of the triazole on splicing activity. Both RNA12 and 13 are of comparable dimensions to a single HEG unit (RNA4).Bifunctional ONs incorporating non-RNA linkers up to an equivalent length of 6nts stimulate splicing
The capacity of RNA1–RNA14 (Table 2) to stimulate specific splicing patterns was assessed in vitro using an established splicing assay. This assay utilizes an SMN2 pre-mRNA substrate, as described previously.2,7 The outcome measured is the proportion of spliced mRNA products that included exon 7 relative to those products which exclude exon 7 (Fig. 2).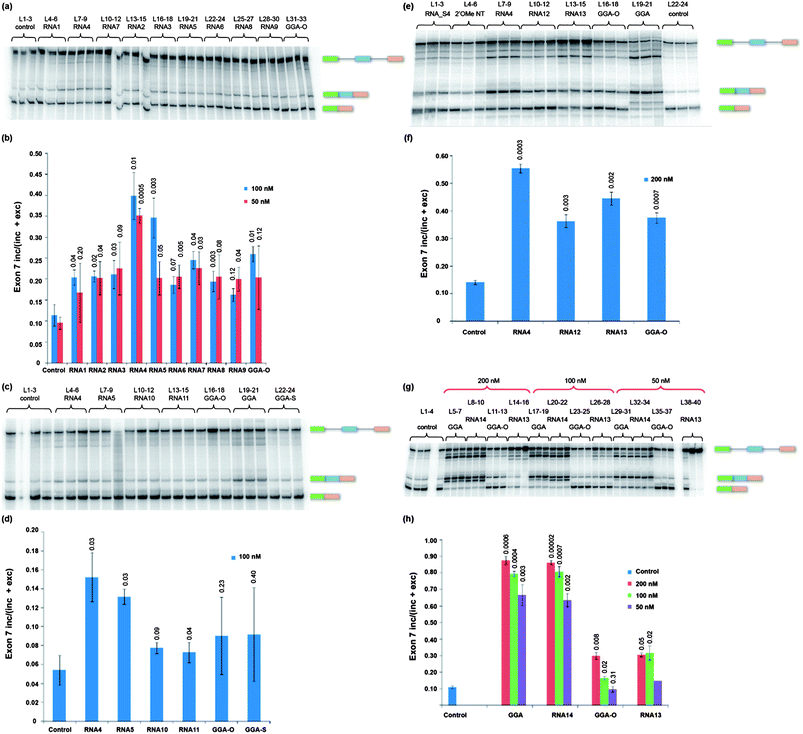 | ||
| Fig. 2 Assays in triplicate of splicing of SMN2 exon 7 in the presence of RNAs 1–14. Splicing reactions were incubated for 2 hours and products separated by denaturing gel electrophoresis. The ONs used are shown above the lanes (a, c, d and g). The proportion of exon 7 inclusion for each set of reactions is shown (b, e, f, and h). (a and b) RNAs 1–9 at 50 and 100 nM; (c and d), RNAs 10 and 11 at 100 nM; (e and f), RNAs 12 and 13 at 200 nM; (g and h), RNAs 13 and 14 at 50, 100 and 200 nM. The numbers above the bars in the charts show the probability by Student's t-test that the results could have been obtained from the same population as the control results. L = lane. | ||
RNA1–RNA9 series incorporating HEG linkers
Fig. 2a and b shows the amount of exon 7 inclusion observed in the presence of RNA1–RNA9 (lanes 4–30, Fig. 2a), a control lacking the addition of any ON (Fig. 2a, lanes 1–3) and a control done with the reference ON, GGA-O, which contained no spacer (Fig. 2a, lanes 31–33). Interestingly, RNA4 (lanes 7–9) retained most of its activity at 50 nM whereas at the same concentration RNA5 (lanes 19–21) was no more active than GGA-O (lanes 31–33). The results show that the inclusion of a triazole function alone (RNA1, lanes 4–6) or with the addition of one to twenty HEG groups (RNA2–RNA9) does not inhibit the enhancer action of GGA-O, but only spacers with one HEG group 5′ of the triazole (RNA4) or two HEG groups flanking the triazole (RNA5) increased splicing activity.Phosphorothioate modification of RNA reduces the enhancement of exon 7 inclusion
Using RNA4 and RNA5 as a benchmark, phosphorothioate versions of these two constructs (RNA10 and RNA11) were prepared (Table 2) and tested (Fig. 2c and d). Both RNA10 (lanes 10–12) and RNA11 (lanes 13–15) were inferior compared with RNA4 (lanes 7–9, Fig. 2a) and RNA5 (lanes 19–21, Fig. 2a) at 100 nM.Abasic sites also enhance exon 7 inclusion
An alternative way to increase the separation between the annealing and activating domains of the ON without introducing binding sites for RNA-binding proteins is to use abasic DNA. RNA12 and RNA13 contain 3 and 6 abasic sites, respectively (Table 2). The results of a splicing assay using these compounds are shown in Fig. 2e and f. The most notable structural difference between these two constructs is the presence of a triazole group in RNA12. Both RNA12 (lanes 10–12, Fig. 2e) and RNA13 (lanes 13–15, Fig. 2e) increased inclusion to approximately the same extent as GGA-O (lanes 16–18, Fig. 2e). The results confirm that the triazole linkage did not inhibit splicing activity. Nonetheless, RNA4 (L7–9, Fig. 2e), with its HEG linker, was superior (Fig. 2f). A comparison of activities in splicing between RNA13, RNA14, GGA-O and GGA over a range of ON concentrations confirmed that the abasic linker had no effect on the activity of the respective domains in either GGA-O or GGA ONs (Fig. 2g and h). The sequence of RNA14 incorporated six abasic linkers and was identical in sequence to GGA but for the inclusion of abasic nts between the activating and annealing domains (Table 2).Enhancement of splicing activity by RNA–GNP conjugates
The results obtained with ONs incorporating HEG and (1) spacers suggested that moderate extension of the domain containing the ESE motifs increases splicing activity. However, it is striking that no benefit was seen with 11 or 20 HEG units. One possible reason for this is that the probability that a bound activator protein coming in contact with a splicing factor would be reduced with longer HEG chains. Thus, a gain in accessibility might be counteracted by an effective dilution of the bound protein. This dilution effect could be addressed by increasing the effective valency of the TOES construct by attaching multiple copies of the ESE motifs to a nanoparticle, thereby increasing both accessibility and effective concentration. Gold nanoparticles (GNP) are readily conjugated to ONs, and conjugates of this type have well established gene-silencing activity with knock-down reported in both cell culture and animal tissues, without the need of co-carriers,14 but their utility to recruit splicing activators has not been tested. A series of RNA–GNP conjugates were prepared which varied in: (i) the size of the GNP (20 nm, 18 nm, 10 nm and 5 nm); (ii) the presence of RNA sequences presented on the surface of the GNP; corresponding to the annealing region of the TOES (RNA16 and 17) and the artificial ESE (RNA 15 and 18); and (iii) the 5′/3′ directionality of the ESE (RNA 15 and 18) when immobilised onto the GNP surface (Table 3).| Sequence name | 2′ OMe RNA ON sequence 5′ → 3′ |
|---|---|
a  . . | |
| RNA15 | YXXXAGGAGGACGGAGGACGGAGGACA |
| RNA16 | GAUUUUGUCUAAAACXXXY |
| RNA17 | YXXXGAUUUUGUCUAAAAC |
| RNA18 | AGGAGGACGGAGGACGGAGGACAXXXY |
Synthesis of RNA–lipoic acid conjugates (RNA15–18)
RNA ONs bearing lipoic acid linkages were prepared by coupling the amino-modified RNA strands (RNA_S16–S19, see ESI† for more details) with the NHS-ester of lipoic acid (S7) to form the requisite precursors RNA15–18 prior to their conjugation on the surface of GNPs (Table 3).15Synthesis of RNA–GNP conjugates (GNP1–18)
RNA–GNPs (GNP1–18) were prepared according to established ligand-exchange methodology (Table 4).16,18 This method of conjugation involved initial ligand exchange of citrate-capped GNPs with a phosphine ligand (BSPP), followed by ligand exchange with dithiol RNA15–18 formed in situ by the reduction of the disulfide with TCEP.19 Ageing of the RNA–GNPs afforded conjugates (GNP1–18) which were stable in aqueous buffered solutions up to 700 mM NaCl. A significant reduction in electrophoretic mobility was observed for RNA–GNP conjugates (GNP1–18) relative to a control conjugate coated with HEG (GNP9, GNP12, GNP15 and GNP18), thus confirming the coverage of the GNP conjugates with a polyanionic layer of RNA (Fig. S13†).20| Reference | GNP size (nm) | Strands conjugated | Reference | GNP size (nm) | Strands conjugated |
|---|---|---|---|---|---|
| a O-(2-Mercaptoethyl)-O′-methyl-hexa(ethylene glycol). | |||||
| GNP1 | 18 | RNA17 | GNP10 | 20 | RNA18 |
| GNP2 | 18 | RNA15 | GNP11 | 20 | RNA17, RNA18 |
| GNP3 | 18 | RNA16 | GNP12 | 20 | a |
| GNP4 | 18 | RNA18 | GNP13 | 10 | RNA18 |
| GNP5 | 18 | RNA15, RNA17 | GNP14 | 10 | RNA17, RNA18 |
| GNP6 | 18 | RNA17, RNA18 | GNP15 | 10 | a |
| GNP7 | 18 | RNA15, RNA16 | GNP16 | 5 | RNA18 |
| GNP8 | 18 | RNA16, RNA18 | GNP17 | 5 | RNA17, RNA18 |
| GNP9 | 18 | a | GNP18 | 5 | a |
Characterisation of RNA–GNP conjugates
Differential centrifugal sedimentation (CPS) measurements confirmed the attachment of the RNA ligands on the surface of GNPs (GNP16 and 17, Fig. S15†).21 1.9 and 1.6 nm shifts were observed with respect to the HEGylated GNPs (sample GNP18; dCPS = 5.4 nm) confirming the formation of a larger monolayer ligand shell as a consequence of passivating the GNP surface with the larger RNA ON ligands. Moreover, no aggregation of these GNP conjugates was detected in the examined size range (2–50 nm) confirming the formation of stable colloidal dispersions of RNA functionalized GNPs.17,21,22From the ICP-AES analysis, we estimate 400–420 RNA strands are present on the surface of GNP in GNP16 and 470–490 RNA ligands on the surface of GNP17. GNPs functionalized with the HEG–thiol ligand (GNP18) contained a higher density of ligands (880–900) on the GNP surface. TEM analysis confirmed a narrow size distribution of these particles with the following core sizes determined: GNP16 5.77 nm, GNP17 5.57 nm, GNP18 5.65 nm, counting a population of ca. 150 nanoparticles (Fig. S17†).
GNP conjugate (GNP16) bearing an ESE sequence stimulate exon 7 inclusion in SMN2
The capacity of the RNA–GNP conjugates to stimulate exon 7 inclusion was found to depend strongly on the size of the GNP. Strikingly, the 20 nm GNPs (GNPs 10, 11 and 12) inhibited splicing completely over all concentrations tested, regardless of the nature of the conjugated RNA strand (Fig. 3c and d). The 18 nm GNPs (GNPs 1–9) inhibited splicing strongly but not completely at 200 nM (lanes 3–11, Fig. 3a); at lower concentrations splicing continued and some conjugate-dependent effects were seen (Fig. 3a and b). In particular, GNP4 and, to a lesser extent, GNP6 (both conjugated to the 3′ end of RNA18, a 2′ OMe ESE sequence) appeared to preferentially inhibit exclusion of exon 7 and, at lower concentrations (50 nM and 100 nM), to stimulate inclusion (GNP4 is shown in lanes 6, 17 and 28 in Fig. 3a, whereas GNP6 is shown in lanes 8, 19 and 30). In contrast, GNP9 (lane 11, lane 22 and lane 33, Fig. 3a) which lacked an ON conjugate, suppressed inclusion of exon 7 at all concentrations.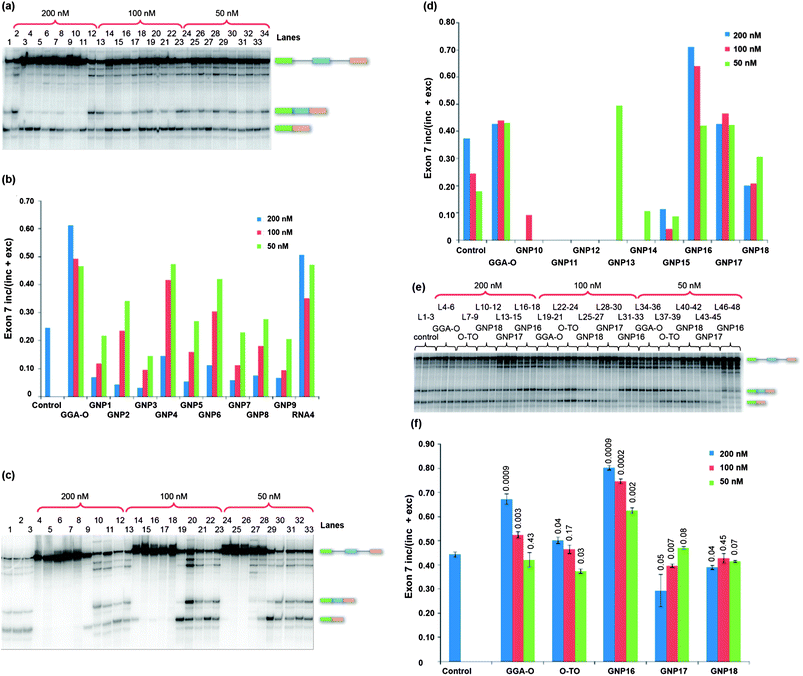 | ||
| Fig. 3 Splicing of SMN2 pre-mRNA for 2 hours in the presence of GNP conjugates. (a) 18 nm GNP–RNA conjugates. 200 nM series: control (no ON; L1); GGA-O (L2), GNP1–9 (L3–11 respectively) and RNA4 (L12). 100 nM series: GGA-O (L13), GNP1–9 (L14–22 respectively) and RNA4 (L23). 50 nM series: GGA-O (L24), GNP1–9 (L25–33 respectively) and RNA4 (L34). (b) Chart showing the proportions of exon 7 inclusion in (a). (c) 200 nM series: control (no GNP, L1), GNP10–18 (L4–12 respectively) and GGA-O (L13). 100 nM series: GNP10–18 (L14–22 respectively) and GGA-O (L23). 50 nM series: GNP10–18 (L24–32 respectively) and GGA-O (L33). (d) Chart showing proportions of exon 7 inclusion in (c). (e) Assays in triplicate with 5 nm GNP–RNA conjugates at the concentrations shown. GGA-O and O-TO (enhancer sequence only) refer to unconjugated ONs. (f) Chart showing proportions of exon 7 inclusion in (e). L = lane. | ||
The 10 nm GNPs with conjugated ONs (GNP13 and GNP14) inhibited splicing at all concentrations (lane 7, lane 17, lane 27 for GNP13, lane 8, lane 18, lane 28 for GNP14; Fig. 3c), whereas the unconjugated GNP15 (lane 9, lane 19, lane 29, Fig. 3c) was less inhibitory but suppressed inclusion of exon 7, much akin to 18 nm GNP9 (lane 11, Fig. 3a). In contrast, the 5 nm RNA–GNP conjugates (GNP16–18) caused considerably less splicing inhibition and GNP16 produced a substantial increase in exon 7 inclusion (lanes 16–18, lanes 31–33, lanes 46–48; Fig. 3e and f). Like the 18 nm GNP4, GNP16 consists of only the ESE conjugated to the GNP via a 3′-linkage. Surprisingly, a 5 nm RNA–GNP construct bearing both the annealing region and the ESE (GNP17) displayed poor inclusion of exon 7 (lanes 13–15, lanes 28–30, lanes 43–45 Fig. 3e and f).
Discussion
These experiments were designed to test whether accessibility and valency are important factors in determining the success of a TOES in splicing. We discuss here several conclusions that emerged from our results.The activity of TOES can be increased by the introduction of a flexible linker
Two ONs showed increased activity compared with the 2′ OMe reference ON, GGA-O. These are RNA4 (lanes 7–9, Fig. 2e and f), which has a single HEG tether on the enhancer side, and RNA5 (lanes 7–9, Fig. 2c and d), which has a single HEG unit either side of the triazole group. However, increasing the number of HEG units to 11 or 20 (RNA6–9) abolished the effect. Abasic DNA spacers (RNA12 and 13) maintained the splicing enhancement produced by GGA-O but did not increase it further. Although there have been no measurements of the rigidity and diffusional behaviour of abasic DNA, it seems likely that the negatively charged backbone and the deoxyribose groups will constrain freedom of movement compared with HEG. Future studies will focus on how rigidity of the linker as well as the density of charge influences splicing activity.We conclude that inserting a flexible spacer of moderate length can increase the activity of TOES. This is consistent with the expectation that accessibility amidst an exon crowded with proteins limits the efficiency of a bifunctional ON. The failure of the spacers with more HEG units could result from a reduction in the probability of interactions as a flexible chain lengthens, as with any three-dimensional diffusion.
Size of GNP conjugates is critical for splicing activity
The 10 nm, 18 nm and 20 nm GNP conjugates inhibit splicing (Fig. 3a–d) at high concentrations. One possible explanation for these observations is that the high concentration of ONs causes sequestration of splicing factors. However, we are inclined to exclude this because inhibition was seen also with the control GNP18, which had no conjugated ONs. Another possibility is that larger GNPs will have a higher surface area for a given particle concentration and that essential splicing factors might be adsorbed onto this. The most striking finding was the enhancement of exon 7 inclusion when the smaller 5 nm GNP conjugates were used.With GNP16 (Fig. 3c–f), the level of stimulation was better than our GGA-O benchmark and an ON containing only the enhancer sequence (O-TO). Levels of GNP16 stimulation were also comparable to the levels achieved by the HEG-containing RNA4. Since this GNP contained no sequences complementary to the target RNA, only the motifs required for binding by activator proteins, the enhancement was non-specific. It is probable that GNP16 binds to the splicing factor protein SRSF1 and that there is a high local concentration of the bound protein. SRSF1 has two RNA-binding domains as well as an auxiliary domain rich in positively charged arginine–serine dipeptide motifs. It seems likely that these domains at a high concentration enable the particle to bind RNA with low specificity and then to mediate the further interactions that are required for SRSF1 to promote the binding of splicing factors such as U1 snRNPs,23–25 U2AF23–25 and U2 snRNPs.26–28
Conclusions
We conclude that inserting a non-RNA spacer of one or two HEG units into the middle of a bifunctional ON improves splicing activity. In contrast, an abasic sequence has no effect, either when inserted between the domains of a bifunctional 2′ OMe or between the domains in the optimal RNA-based ON GGA. Flexible linkers are likely to become standard features in designs for bifunctional ONs that would work in any targeted exon. The stimulatory splicing effects observed when a 5 nm GNP carrying the activator domain of a TOES was added to our splicing assay suggest that small nanoparticles provide a promising generic method for stimulating splicing when, for example, there is an excessive expression of counteracting repressor proteins. This may be a feature of myc-expressing cancer cells.21 Since nanoparticles have the advantage that other tags, such as peptides facilitating uptake, can be readily attached, we envisage that this could provide a new multi-modal delivery platform for the development of new strategies to target diseases based on aberrant splicing pathways.Acknowledgements
This work was supported by the Leverhulme Trust. G.A.B. acknowledges the EPSRC for an Advanced Research Fellowship to G.A.B. (EP/E055095/1). We also thank Prof. Tom Brown and his group (Southampton) for their kind assistance in the mass spectrometric analysis of our RNA ONs.References
- I. Eperon, Nat. Chem. Biol., 2012, 8, 507–508 CrossRef CAS.
- L. A. Skordis, M. G. Dunckley, B. G. Yue, I. C. Eperon and F. Muntoni, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4114–4119 CrossRef CAS.
- Z. Dominski and R. Kole, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8673–8677 CrossRef CAS.
- M. G. Dunckley, M. Manoharan, P. Villiet, I. C. Eperon and G. Dickson, Hum. Mol. Genet., 1998, 7, 1083–1090 CrossRef CAS.
- F. Rigo, Y. Hua, S. J. Chun, T. P. Prakash, A. R. Krainer and C. F. Bennett, Nat. Chem. Biol., 2012, 8, 555–561 CrossRef CAS.
- L. Cartegni and A. R. Krainer, Nat. Struct. Biol., 2003, 10, 120–125 CrossRef CAS.
- Y. Hua, T. A. Vickers, B. F. Baker, C. F. Bennett and A. R. Krainer, PLoS Biol., 2007, 5, e73 CrossRef.
- Y. Hua, T. A. Vickers, H. L. Okunola, C. F. Bennett and A. R. Krainer, Am. J. Hum. Genet., 2008, 82, 834–848 CrossRef CAS.
- N. Owen, H. Zhou, A. A. Malygin, J. Sangha, L. D. Smith, F. Muntoni and I. C. Eperon, Nucleic Acids Res., 2011, 39, 7194–7208 CrossRef CAS.
- H. X. Liu, M. Zhang and A. R. Krainer, Genes Dev., 1998, 12, 1998–2012 CrossRef CAS.
- N. Ma, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2009, 4, 121–125 CrossRef CAS.
- R. Kumar, A. El-Sagheer, J. Tumpane, P. Lincoln, L. M. Wilhelmsson and T. Brown, J. Am. Chem. Soc., 2007, 129, 6859–6864 CrossRef CAS.
- M. Alvira and R. Eritja, Chem. Biodiversity, 2007, 4, 2798–2809 CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639–3642 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS.
- P. C. Patel, D. A. Giljohann, W. L. Daniel, D. Zheng, A. E. Prigodich and C. A. Mirkin, Bioconjugate Chem., 2010, 21, 2250–2256 CrossRef CAS.
- J. A. Dougan, C. Karlsson, W. E. Smith and D. Graham, Nucleic Acids Res., 2007, 35, 3668–3675 CrossRef CAS.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS.
- Z. Krpetic, P. Nativo, I. A. Prior and M. Brust, Small, 2011, 7, 1982–1986 CrossRef CAS.
- A. M. Cioran, A. D. Musteti, F. Teixidor, Z. Krpetic, I. A. Prior, Q. He, C. J. Kiely, M. Brust and C. Vinas, J. Am. Chem. Soc., 2012, 134, 212–221 CrossRef CAS.
- I. C. Eperon, O. V. Makarova, A. Mayeda, S. H. Munroe, J. F. Cáceres, D. G. Hayward and A. R. Krainer, Mol. Cell. Biol., 2000, 20, 8303–8318 CrossRef CAS.
- I. C. Eperon, D. C. Ireland, R. A. Smith, A. Mayeda and A. R. Krainer, EMBO J., 1993, 12, 3607–3617 CAS.
- S. Cho, A. Hoang, R. Sinha, X.-Y. Zhong, X.-D. Fu, A. R. Krainer and G. Ghosh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8233–8238 CrossRef CAS.
- Z. Wang, H. M. Hoffmann and P. J. Grabowski, RNA, 1995, 1, 21–35 CAS.
- H. Shen and M. R. Green, Genes Dev., 2006, 20, 1755–1765 CrossRef CAS.
- M. Chen and J. L. Manley, Nat. Rev. Mol. Cell Biol., 2009, 10, 741–754 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of RNA ONs, GNP conjugates and splicing assays. See DOI: 10.1039/c2sc20937c |
| This journal is © The Royal Society of Chemistry 2013 |