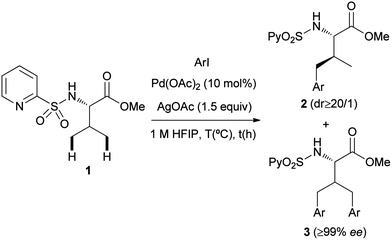
Palladium-catalyzed N-(2-pyridyl)sulfonyl-directed C(sp3)–H γ-arylation of amino acid derivatives†‡
Nuria
Rodríguez
,
Jose A.
Romero-Revilla
,
M. Ángeles
Fernández-Ibáñez
* and
Juan C.
Carretero
*
Departamento de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain. E-mail: tati.fernandez@uam.es; juancarlos.carretero@uam.es; Fax: +34 914973966; Tel: +34 914973925
First published on 14th September 2012
Abstract
The direct Pd-catalyzed γ-arylation of amino acid esters bearing a removable N-(2-pyridyl)sulfonyl directing group is described. A variety of N-(2-pyridyl)sulfonamide amino acid derivatives, including α-quaternary amino acid and β-amino acid substrates, react with iodoarenes in the presence of Pd(OAc)2 to provide γ-arylated products in synthetically useful yields. An unprecedented remote C(sp3)–H arylation of dipeptides is presented, illustrating the compatibility of the method with the presence of the peptidic bond. The process occurs without racemization at the Cα center and the auxiliary controlling group can be easily installed and removed in the amino acid backbone. A bimetallic PdII γ-metalated complex has been isolated and characterized showing the key role exerted by the (2-pyridyl)sulfonyl unit.
Introduction
Development of efficient methods for the catalytic functionalization of C–H bonds is a fundamental challenge in current organometallic and synthetic organic chemistry.1 In recent years, a great progress has been made in the direct and selective metal-catalyzed transformation of arene C(sp2)–H bonds into a variety of functional groups through the formation of new C(sp2)–C or C(sp2)–X bonds.1,2 However, the activation of the less reactive C(sp3)–H bonds has now entered the mainstream and it is still at its infancy.3 In many cases this type of process deals with the functionalization of C(sp3)–H bonds at benzylic positions or those next to heteroatoms.4 To date, catalytic processes for the functionalization of unactivated simple C(sp3)–H bonds are relatively rare and, consequently, any progress in this field is of great synthetic interest.5In 2005, Daugulis et al. described the Pd-catalyzed β-arylation of carboxylic acid and the γ-arylation of amine derivatives by using the 8-aminoquinoline and the picolamide auxiliary directing groups.6,7 This type of Pd-catalyzed chelating strategy, although at an early stage, has been also applied to the synthesis of non-natural amino acids. The development of methods for the synthesis of amino acids, especially from native amino acids, is highly important due to their broad use in drug discovery and biotechnology.8 In this regard, Corey et al. described the first pioneering examples of β- and γ-functionalization of N-phthaloyl-α-amino amides by employing the 8-aminoquinoline auxiliary.9 This coordinating group and the 2-thiomethylaniline group have been recently applied by Daugulis et al. in the efficient β-arylation of N-phthaloylalanine derivatives with iodoarenes.10 On the other hand, in the elegant study of He and Chen on the Pd-catalyzed picolinamide-directed C(sp3)–H bond functionalization, some examples of γ-arylation of natural amino acids were described, although with moderate chemical yields.11
In spite of this progress in the β- and γ-functionalization of natural amino acids, there is plenty of room for further improvements both regarding the overall chemical efficiency of the process and the scope at the amino acid structure, as well as the study of the presumed palladacycle intermediates12 and the final removal of the key directing group. In the last few years, our research group has demonstrated the efficiency of the easily removable N-(2-pyridyl)sulfonyl auxiliary in the selective functionalization of C(sp2)–H bonds.13 We report herein the application of this auxiliary group to the efficient γ-arylation of the readily available N-(2-pyridyl)sulfonamide derivatives of simple amino acid methyl esters, and the first examples of remote C(sp3)–H arylation in dipeptide derivatives. Some mechanistic studies dealing with the palladation step, including the isolation of a palladacycle species, are also described.
Results and discussion
After screening a wide variety of reaction conditions, we found that the reaction of the N-(2-pyridyl)sulfonamide of L-valine methyl ester (1) with 4-iodotoluene (2.5 equiv.), Pd(OAc)2 (10 mol%) and AgOAc (1.5 equiv.) in HFIP (1,1,1,3,3,3-hexafluoro-2-propanol)14 at 150 °C, resulted in 95% conversion of the starting chiral amino acid derivative, providing a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the γ-monoarylated derivative 2a and the γ-bisarylated product 3a. After chromatographic purification both products were isolated in 44% and 42% yield, respectively (Table 1, entry 1). In addition to the chemical efficiency of this C(sp3)–H functionalization reaction, three issues are worth noting: (a) the γ-monoarylated product 2a was obtained with very high diastereoselectivity (dr ≥ 20:1), with the arylation occurring exclusively at the pro-S methyl valine group. (b) The Pd-catalyzed process takes place preserving the chiral integrity at the α position, as demonstrated by the very high enantiopurity of product 3a (≥99% ee determined by HPLC). (c) No gem-diarylated products were observed, presumably because of steric factors.
:1 mixture of the γ-monoarylated derivative 2a and the γ-bisarylated product 3a. After chromatographic purification both products were isolated in 44% and 42% yield, respectively (Table 1, entry 1). In addition to the chemical efficiency of this C(sp3)–H functionalization reaction, three issues are worth noting: (a) the γ-monoarylated product 2a was obtained with very high diastereoselectivity (dr ≥ 20:1), with the arylation occurring exclusively at the pro-S methyl valine group. (b) The Pd-catalyzed process takes place preserving the chiral integrity at the α position, as demonstrated by the very high enantiopurity of product 3a (≥99% ee determined by HPLC). (c) No gem-diarylated products were observed, presumably because of steric factors.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | ArI (equiv.) | t/h | T/°C | Conv.b/% (ratio 2:3) |
Yieldc | |
| 2/% | 3/% | |||||
| a Reaction conditions: 1 (0.25 mmol), 10 mol% Pd(OAc)2, 1.5 equiv. AgOAc, HFIP (1 M). b Conversions determined by 1H NMR on the crude mixtures. c Isolated yields after chromatographic purification. d 3.0 equiv. of AgOAc. | ||||||
| 1 | 4-Me–C6H4–I (2.5) | 4 | 150 | 95 (53:47) |
44 | 42 |
| 2d | 4-Me–C6H4–I (5.0) | 4 | 150 | 100 (8:92) |
6 | 92 |
| 3 | 4-Me–C6H4–I (1.5) | 4 | 150 | 70 (67:33) |
— | — |
| 4 | 4-Me–C6H4–I (2.5) | 4 | 140 | 90 (80:20) |
70 | 18 |
| 5 | 2,4-MeO–C6H3–I (2.5) | 3 | 150 | 88 (80:20) |
68 | 17 |
With this result in hand, we next tried to improve the mono/bisarylation selectivity. As expected, the bisarylated product 3a could be isolated in excellent yield (92%, entry 2) by simply increasing the amount of 4-iodotoluene (5 equiv.) and AgOAc (3 equiv.). On the other hand, the use of only a slight excess of 4-iodotoluene (1.5 equiv.) resulted in incomplete conversion (70% after 4 h) and moderate mono/bisarylation selectivity (2a/3a = 67/33). The γ-monoarylated product 2a could be isolated in 70% yield by performing the reaction at a slightly lower temperature (140 °C for 4 h,15 entry 4) using 2.5 equiv. of 4-iodotoluene. According with the importance of steric effects, the arylation of 1 with the more sterically demanding 2,4-dimethoxyiodobenzene provided the monoarylated product 2b with reasonably good selectivity (2b/3b = 80:20, 68% yield 2b, entry 5). The relative and absolute configuration of 2b was determined by single-crystal X-ray diffraction analysis.16
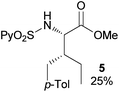
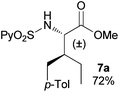
In agreement with the highly stereoselective monoarylation of the L-valine derivative 1, a very different reactivity profile was observed in the reaction of the isoleucine derivative 4 and the allo-isoleucine diastereomer 6 (Table 2, entries 1 and 2). Thus, while substrate 4 afforded only 25% of the γ-monoarylated isoleucine derivative 5,17 the diastereomeric allo-isoleucine derivative 6, having the methyl group at the same stereochemical position as the pro-S methyl in the valine substrate 1, provided 7a in 72% yield under standard reaction conditions. Similar stereochemical effects, likely based on the accessibility of the C(sp3)–H bond in the palladation step and stability of the resulting palladacycle, have been previously described.9a It is also worth mentioning that the reaction takes place exclusively over the methyl γ C–H bond and not at the methylene γ′ C–H bond.

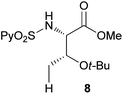

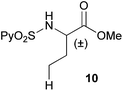
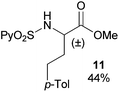
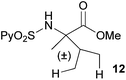
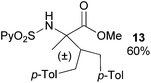
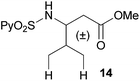
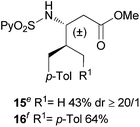
Extending the scope at the amino acid structure, the reaction of the protected threonine derivative 8 provided the γ-monoarylated product 9 in 70% yield (entry 3). The more conformationally flexible homoalanine derivative 10 led to the corresponding γ-arylated product 11 in 44% yield (entry 4), providing a direct method for the synthesis of homoarylalanines, important intermediates in medicinal chemistry.18 Additional examples include the arylation of the γ-C(sp3)–H bond at a α-quaternary amino acid derivative, the α-methylvaline substrate 12, and the β-amino acid derivative 14 (entries 5 and 6). While the α-methylvaline substrate 12 revealed a strong trend to provide the corresponding bisarylated product 13 (60% yield), the β-amino acid derivative 14 furnished the γ-monoarylated product 15 in 43% yield under standard reaction conditions or the bisarylated product 16 (64% yield) by increasing the amount of 4-iodotoluene and AgOAc.
Next, we evaluated the scope of the process with regard to the substitution at the aryl iodide and using the allo-isoleucine derivative 6 (Scheme 1). Electron-donating and electron-withdrawing substituents were well tolerated (71–75% yield), including fluorinated aryl iodides that have an important role in agrochemicals and pharmaceuticals.19 Interestingly, bromide is also tolerated opening the access to scaffolds that may be later functionalized using standard Pd0 coupling processes. The ortho substituted 2,4-dimethoxyiodobenzene also proved to be a suitable substrate (72% yield for 7g).
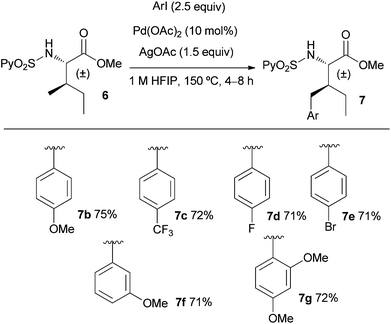 | ||
| Scheme 1 Scope of the γ-CH3 arylation of the allo-isoleucine derivative 6. | ||
To highlight the synthetic utility of this process in non-natural amino acid synthesis, the efficient three-step sequence for the diarylation of L-valine methyl ester is shown in Scheme 2: introduction of the key 2-pyridylsulfonyl directing group, the Pd-catalyzed bisarylation and the final reductive removal of the sulfonyl moiety.20 Importantly, the reductive elimination of the sulfonyl moiety took place with less than 2% of racemization as determined by HPLC (98% ee for 17a). Although the Pd-catalyzed reactions were typically run on a 0.25 mmol scale, a 2.00 mmol scale reaction provided 3a with similar high yield (98%), thus demonstrating the robustness of the reaction.
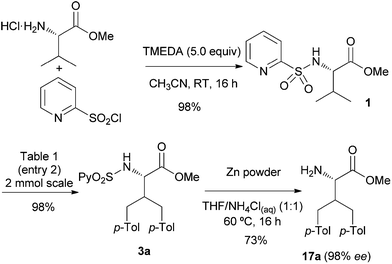 | ||
| Scheme 2 Introduction and removal of the N-(2-pyridyl)sulfonyl group. | ||
Having thus succeeded in the arylation of different amino acid esters, we next undertook the remote C(sp3)–H arylation of dipeptides, which as far as we know it is unprecedented.21 Satisfyingly, both the arylation of the valine–glycine 18 and the allo-isoleucine-glycine dipeptide 20 occurred with good selectivity at the γ-methyl group providing the monoarylated dipeptides 19 and 21 in synthetically useful yields (60% and 43% yield, respectively) under the usual reaction conditions (Scheme 3). This result shows the compatibility of the Pd-catalyzed arylation conditions with the presence of the peptidic bond.
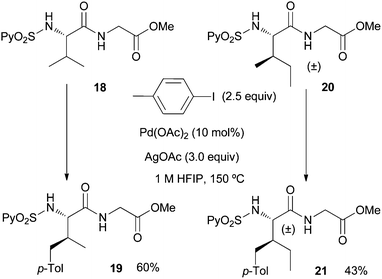 | ||
| Scheme 3 Palladium-catalyzed C(sp3)–H γ-arylation of dipeptides. | ||
To obtain some mechanistic insights into the course of this C(sp3)–H activation process and the coordination mode of the palladium catalyst, we focused our attention on the stoichiometric reaction of the N-(2-pyridyl)sulfonyl tert-leucine derivative 22 with Pd(OAc)2 (Scheme 4). To our delight, the reaction in acetonitrile at 60 °C cleanly provided a palladium containing compound, which after recrystallization and X-ray diffraction analysis proved to be the bimetallic five-membered ring palladacycle complex 23, metalated at one carbon of the methyl tert-leucine groups (Fig. 1) (see ESI† for crystallographic details).22 The Pd(1)–Pd(2) distance is 3.1276 (12) Å, which is within the sum of van der Waals radii (3.26 Å),23 and the Pd–C distance (1.997 (11) Å) are similar to the distances found in five-membered ring metalated complexes.6a,12 Interestingly, in this dimeric complex the slightly distorted square planar PdII atom is bonded to the sulfonamide nitrogen and the γ-palladated carbon, while the other two coordination positions are occupied by the pyridine nitrogen and one of the sulfonylic oxygens. In agreement with this type of palladium coordination mode, either the p-tolylsulfonamide derivative 26 (lacking the key pyridine nitrogen), or the N-methyl sulphonamide 27 (lacking the easily palladated acidic NH group), or the alanine and leucine derivatives 28 and 29 (both lacking the γ-methyl group required in the formation of a five-membered palladacycle) proved to be completely unreactive under the standard arylation conditions (Scheme 5). Finally, we confirmed that palladacycle 23 was able to react with 4-iodotoluene (60 °C in HFIP) leading to a nearly equimolar mixture of the monoarylated and bisarylated products 24 and 25 (79% yield).
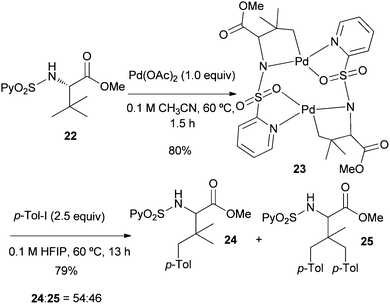 | ||
| Scheme 4 Synthesis and reactivity of the bimetallic complex 23. | ||
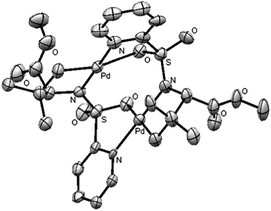 | ||
| Fig. 1 ORTEP view of 23, hydrogen atoms have been removed for simplicity. | ||
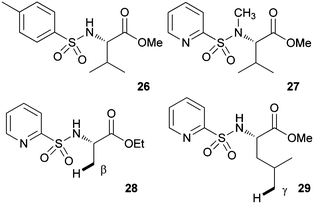 | ||
| Scheme 5 Unreactive substrates. | ||
Conclusions
In summary, the N-(2-pyridyl)sulfonyl group behaves as an efficient controlling auxiliary for the Pd-catalyzed γ-C(sp3)–H methyl activation in amino acid esters. A variety of N-(2-pyridyl)sulfonamide amino acid derivatives, including α-quaternary amino acids, β-amino acids, and dipeptide substrates, react with iodoarenes in the presence of Pd(OAc)2 to provide γ-arylated products in synthetically useful yields. The process occurs without racemization at the Cα center and the auxiliary controlling group can be easily installed and removed in the amino acid backbone. A bimetallic PdII γ-metalated complex has been isolated and characterized showing the key role exerted by the (2-pyridyl)sulfonyl unit. Further synthetic applications and mechanistic work are in progress.Acknowledgements
Financial support from the Ministerio de Economía y Competitividad (MINECO, project CTQ2009-07791) and the Consejería de Educación de la Comunidad de Madrid (programme AVANCAT; S2009/PPQ-1634) are gratefully acknowledged. M.A.F.-I., N.R. and J.R.R. thank the MINECO for a Juan de la Cierva contract, a Ramón y Cajal contract and a predoctoral fellowship, respectively.Notes and references
- For general reviews on C–H functionalization, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (d) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (e) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC. For a recent special issue dedicated to this topic, see: (f) Acc. Chem. Res., 2012, Issue 6, 777–958 Search PubMed.
- For a specific review on C(sp2)–H functionalization, see: D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS.
- For specific reviews on C(sp3)–H functionalization, see: (a) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS; (b) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191 RSC.
- Selected examples based on Pd-catalysis: (a) G. Dyker, Angew. Chem., Int. Ed. Engl., 1992, 31, 1023 CrossRef; (b) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS; (c) C.-G. Dong and Q.-S. Hu, Angew. Chem., Int. Ed., 2006, 45, 2289 CrossRef CAS; (d) L.-C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266 CrossRef CAS; (e) P. Novák, A. Correa, J. Gallardo-Donaire and R. Martin, Angew. Chem., Int. Ed., 2011, 50, 12236 CrossRef; (f) P. Xie, Y. Xie, B. Qian, H. Zhou, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 9902 CrossRef CAS. Ru-catalysis: (g) C.-H. Jun, D.-C. Hwang and S.-J. Na, Chem. Commun., 1998, 1405 RSC. Ir-catalysis: (h) B. DeBoef, S. J. Pastine and D. Sames, J. Am. Chem. Soc., 2004, 126, 6556 CrossRef CAS; (i) K. Tsuchikama, M. Kasagawa, K. Endo and T. Shibata, Org. Lett., 2009, 11, 1821 CrossRef CAS.
- For selected recent references on remote C(sp3)–H bond activation based on Pd-catalysis: (a) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS; (b) X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634 CrossRef CAS; (c) J. J. Neumann, S. Rakshit, T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 6892 CrossRef CAS; (d) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 CrossRef CAS; (e) E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378 CrossRef CAS; (f) S. Rousseaux, M. Davi, J. Sofack-Kreutzer, C. Pierre, C. E. Kefalidis, E. Clot, K. Fagnou and O. Baudoin, J. Am. Chem. Soc., 2010, 132, 10706 CrossRef CAS; (g) J. Pan, M. Su and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 8647 CrossRef CAS; (h) see also ref. 7. Ru-catalysis: (i) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070 CrossRef CAS; (j) M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündig, Angew. Chem., Int. Ed., 2011, 50, 7438 CrossRef CAS. Ir-catalysis: (k) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70 CrossRef CAS.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS.
- For other references on remote C(sp3)–H bond activation using 8-aminoquinoline, picolinic acid or 2-thiomethylaniline auxiliaries, see: (a) Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414 CrossRef CAS; (b) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS; (c) W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076 CrossRef CAS; (d) Y. Xie, Y. Yang, L. Huang, X. Zhang and Y. Zhang, Org. Lett., 2012, 14, 1238 CrossRef CAS; (e) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS; (f) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7 CrossRef CAS.
- Chemistry of Natural Products, ed. S. V. Bhat,B. A. Nagasampagi and M. Sivakumar, Springer: Narosa, 2005, p. 317 Search PubMed.
- (a) B. V. Subba Reddy, L. Rajender Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef. For the application in the total synthesis of Celogentin C, see: (b) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958 CrossRef CAS.
- L. Dieu Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef.
- G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192 CrossRef CAS.
- Palladacycles, ed. J. Dupont and M. Pfeffer, Willey-VCH, Weinheim, 2008 Search PubMed.
- For the use of the N-(2-pyridyl)sulfonyl group in C(sp2)–H activation processes, see: (a) A. García-Rubia, R. Gómez Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511 CrossRef; (b) A. García-Rubia, B. Urones, R. Gómez Arrayás and J. C. Carretero, Chem.–Eur. J., 2010, 16, 9676 CrossRef; (c) A. García-Rubia, B. Urones, R. Gómez Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2011, 50, 10927 CrossRef.
- For examples of the use of HFIP in C–H activation processes, see ref. 7c and M. Ochiai, K. Miyamoto, T. Kaneaki, S. Hayashi and W. Nakanishi, Science, 2011, 332, 448 CrossRef CAS.
- Longer reaction times resulted in the formation of greater amounts of the bisarylated product.
- CCDC 887848 contains the supplementary crystallographic data for compound 2b†.
- Low conversion (44%) and formation of byproducts were detected by 1H NMR of the crude reaction mixture.
- For instance, L-homophenylalanine is a key intermediate in the synthesis of Enalapril (antihypertensive agent).
- For the importance of fluorinated compounds, see: X. J. Zhang, T. B. Lai and R. Y. Kong, Top. Curr. Chem., 2012, 308, 365 CrossRef CAS.
- For Zn-mediated reductive desulfonylations, see: (a) R. A. Holton, R. M. Kenedy, H.-B. Kim and M. E. Krafft, J. Am. Chem. Soc., 1987, 109, 1597 CrossRef CAS; (b) J. Adrio, M. Rodríguez Rivero and J. C. Carretero, Angew. Chem., Int. Ed., 2000, 39, 2906 CrossRef CAS; (c) O. García Mancheño, R. Gómez Arrayás, J. Adrio and J. C. Carretero, J. Org. Chem., 2007, 72, 10294 CrossRef.
- For the C(sp2)–H arylation of peptides, see: J. Ruiz-Rodríguez, F. Albericio and R. Lavilla, Chem.–Eur. J., 2010, 16, 1124 CrossRef.
- CCDC 887847contains the supplementary crystallographic data for compound 23†.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: data for new compounds, experimental procedures, crystallographic details, copies of HPLC chromatograms and 1H and 13C NMR spectra. CCDC 887847 and 887848. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21162a |
| ‡ In memory of Dr. Christian G. Claessens. |
| This journal is © The Royal Society of Chemistry 2013 |