Single-electron oxidation of N-heterocyclic carbene-supported nickel amides yielding benzylic C–H activation†
C. A.
Laskowski
a,
G. R.
Morello‡
b,
C. T.
Saouma
c,
T. R.
Cundari
*b and
G. L.
Hillhouse
*a
aGordon Center for Integrative Science, Department of Chemistry, The University of Chicago, 929 E. 57th St., Chicago, IL 60637, USA. E-mail: g-hillhouse@uchicago.edu
bCenter for Advanced Computing and Modeling, Department of Chemistry, University of North Texas, Denton, TX 76203, USA. E-mail: t@unt.edu
cDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 24th September 2012
Abstract
The dimeric Ni(I)–Ni(I) N-heterocyclic carbene complex {(IPr)Ni(μ-Cl)}2 (3; IPr = 1,3-(2,6-iPr2C6H3)2imidazolin-2-ylidene)) reacts with the lithium terphenylamides LiNHdmp and LiNHdippp (dmp = 2,6-di(mesityl)phenyl; dippp = 2,6-bis(2,6-di-iso-propylphenyl)phenyl) to give the monomeric Ni(I) amides (IPr)Ni(NHdmp) (4) and (IPr)Ni(NHdippp) (5), respectively. These nickel amides are 1-electron paramagnets, and crystallographic characterization indicates both are stabilized by Ni–C(ipso) interactions with a flanking aryl group of the terphenyl fragment. This results in significant deviation from the linear CNHC–Ni–N geometry typical for a two-coordinate transition-metal complex (112.17(9)° in 4, 116.41(9)° in 5). One-electron oxidation of 4 by ferrocenium results in intramolecular deprotonation at a terphenyl benzylic position by the amide, giving the diamagnetic Ni(II) complex [(IPr)Ni(κ2-C,N:NH2C6H3(Mes)C10H9)][B(ArF)4] (7). DFT calculations on oxidized 4 (i.e., 4+) indicate short amide N⋯CH3 interactions. One-electron oxidation of 5 by ferrocenium gives a new high-spin Ni(II) amide complex salt, [(IPr)Ni(NHdippp)][B(ArF)4] (9). The solid-state structure of 9 indicates it maintains the bent CNHC–Ni–N core. Unlike three-coordinate cationic Ni(II) amides, 9 has not been observed to undergo smooth deprotonation (at N) to afford a two-coordinate imido complex.
Introduction
Two- and 3-coordinate nickel complexes containing terminal imide groups (RN2−) and supported by neutral, ancillary trialkylphosphine or N-heterocyclic carbene (NHC) ligands have been shown to exhibit fascinating nitrene-transfer reactivity with a variety of unsaturated small molecules like carbon monoxide, aryl- and alkylisocyanides, and olefins.1 For example, carbon monoxide reacts with either 2-coordinate (IPr*)Ni![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(dmp)1d (1) (IPr* = 1,3-bis(2,6-bis(diphenylmethyl)-4-methylphenyl)imidazol-2-ylidene; dmp = 2,6-di(mesityl)phenyl) or 3-coordinate (dtbpe)NiNAr (2; dtbpe = 1,2-bis(di-tert-butylphosphino)ethane; Ar = 2,6-di-iso-propylphenyl) to effect RN-group transfer and give the corresponding arylisocyanates.1a,2 In contrast, reaction of ethene with either 1 or 2 results in very different RN-transfer products, forming the vinylamine CH2CHN(dmp) from 1, via net C–H amination, and the aziridine cyclo-CH2CH2NAr from 2.
N(dmp)1d (1) (IPr* = 1,3-bis(2,6-bis(diphenylmethyl)-4-methylphenyl)imidazol-2-ylidene; dmp = 2,6-di(mesityl)phenyl) or 3-coordinate (dtbpe)NiNAr (2; dtbpe = 1,2-bis(di-tert-butylphosphino)ethane; Ar = 2,6-di-iso-propylphenyl) to effect RN-group transfer and give the corresponding arylisocyanates.1a,2 In contrast, reaction of ethene with either 1 or 2 results in very different RN-transfer products, forming the vinylamine CH2CHN(dmp) from 1, via net C–H amination, and the aziridine cyclo-CH2CH2NAr from 2.
The chemistry of 3-coordinate Ni imides like 2 is reasonably well-developed, and they can be prepared in good yield by three routes: (i) directly by reaction of Ni(0) derivatives with organoazides; (ii) by sequential oxidation and deprotonation of Ni(I) amides such as (dtbpe)Ni(NHAr); or (iii) by H-atom abstraction from Ni(I) amides (Scheme 1).2–4 The chemistry of 2-coordinate imides is much less developed, with 1 being the only example, and the organoazide route is thus far the only successful method for its synthesis (from the Ni(0) toluene adduct (IPr*)Ni(η6-C7H8)). Since 2 was originally prepared via sequential oxidation and deprotonation of (dtbpe)Ni(NHAr), we were curious if this method could be applied to generating 2-coordinate imides. We have found that the analogous transformation, requiring a cationic, 2-coordinate, 12-e− amide intermediate, presents unique challenges.
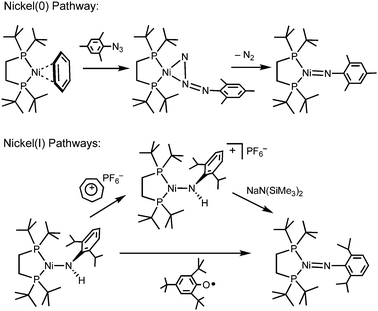 | ||
| Scheme 1 Three independent routes to 3-coordinate Ni imides. | ||
Results and discussion
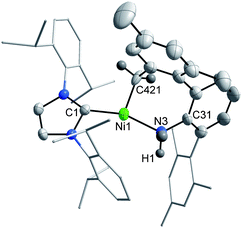
The NHC-supported Ni(I) dimer {(IPr)Ni(μ-Cl)}2 (3; IPr = 1,3-(2,6-iPr2C6H3)2imidazolin-2-ylidene) serves as a precursor for two-coordinate, Ni(I) complexes.5 We have reported that oxidation of (IPr)Ni(NHAr) or (IPr)Ni{N(SiMe3)2} results in η3-heterobenzylic coordination of the aryl amide or Si–CH3 bond cleavage, respectively, in the cationic Ni(II) products.5b In an attempt to generate simple single-electron oxidation reactivity from two-coordinate precursors, bulkier (and presumably less reactive) amide substituents are now considered. Large, terphenyl-substituted organic fragments have been widely used to stabilize reactive species through steric protection.6 Here we describe the oxidation chemistry of two new terphenyl-substituted Ni(I) amides that afford either C–H activation or a simple amide in the isolated cationic Ni(II) products.7Combination of LiNH(dmp) with 3 in Et2O solution results in elimination of LiCl with formation of the paramagnetic d9 amide (IPr)Ni(NHdmp) (4) that can be isolated in 84% yield as a rust-red solid (μeff = 1.88μB in C6D6; Scheme 2). Single crystal X-ray studies of 4 indicate significant steric congestion about Ni with Ni–N = 1.936(2) Å, greatly increased from the Ni–N distance of 1.831(4) Å found in (IPr)Ni(NHAr) (Fig. 1).5b Planes defined by the NHC five-membered core and the central terphenyl ring of the amide are twisted ∼7.3° from coplanarity. The C(1)–Ni–N(3) angle of 112.17(9)° deviates dramatically from linearity, allowing for a short, stabilizing contact with the ipso-carbon on a flanking mesityl group of the dmp ligand (Ni–C(51) = 2.137(2) Å), with longer Ni distances of 2.502(2) and 2.396(3) Å to the ortho carbons of that mesityl. Similar interactions are seen in the Ni(I) thiolate complex (IMes)Ni{S(dmp)}, where the metal center engages in an η2 Ni-aryl bonding mode with Ni–C distances of ∼2.1 Å.8
 | ||
| Scheme 2 Formation of IPr-supported terphenyl amides via chloride substitution from 3. Oxidation of the resulting Ni amides leads to either C–H activation or simple cation formation. A = B(ArF)4 anion. | ||
 | ||
| Fig. 1 X-ray structure of 4 (50% ellipsoids). The amide H-atom was located in the difference map and refined isotropically. Selected metrical parameters for 4: Ni–N(3) = 1.936(2), Ni–C(1) = 1.949(2), Ni–C(51) = 2.137(2), Ni–C(52) = 2.502(2), Ni–C(56) = 2.396(3) Å; C(1)–Ni–N(3) = 112.17(9), C(1)–Ni–C(51) = 165.81(9), C(51)–Ni–N(3) = 81.99(9)°. | ||
The bulkier amide LiNHdippp (dippp = 2,6-bis(2,6-di-iso-propylphenyl)phenyl) affords (IPr)Ni(NHdippp) (5) in 79% yield upon reaction with 3. The solid-state structure of 5 (Fig. 2) shows Ni–N(3) = 1.953(2), Ni–C(41) = 2.114(2) Å and C(1)–Ni–N(3) = 116.41(9)°. In contrast with the rigorously linear (N–Ni–N) geometry found in Ni(NHdippp)2 (6), 5 has a much shorter interaction with the ipso carbon of the dippp subunit (cf. 2.680 Å in 6).9 Similar bonding motifs have been observed by Power et al. in 2-coordinate terphenylamide complexes of other 3d metals.10
 | ||
| Fig. 2 X-ray structure of 5 (50% ellipsoids). The amide H-atom was located in the difference map and refined isotropically. Ni–N(3) = 1.953(2), Ni–C(1) = 1.959(2), Ni–C(41) = 2.114(2), Ni–C(42) = 2.385(2), Ni–C(46) = 2.380(2) Å; C(1)–Ni–N(3) = 116.41(9), C(1)–Ni–C(41) = 161.90(9), C(41)–Ni–N(3) = 81.62(9)°. | ||
A cyclic voltammogram of 4 exhibits an irreversible oxidation event at −0.63 V (vs. Cp2Fe/Cp2Fe+, THF/TBAH), attributed to oxidation of 4 to a Ni(II) species. Chemical oxidation can be accomplished with ferrocenium-based oxidants, and combination of Et2O solutions of 4 with [Cp2Fe][B(ArF)4] (ArF = 3,5-(CF3)2C6H3) allows for the subsequent isolation of a highly unsymmetric, diamagnetic species (Scheme 2). 1H-NMR spectra of the blue product show five singlets for inequivalent alkyl groups of the dmp fragment and four doublets for the isopropyl methyl groups in the IPr ligand.
X-ray studies revealed that the oxidation product, [(IPr)Ni(κ2-C,N:NH2C6H3(Mes)C10H9)][B(ArF)4] (7, Scheme 2; Fig. 3), contains an unusual 7-membered metallacycle featuring new Ni-κ1-benzyl and Ni-κ1-amine bonds. Thus, 1-electron oxidation of 4 results in formal deprotonation of a dmp ortho-methyl by the basic amide to give a 3-coordinate, 14-e− T-shaped Ni(II) benzyl amine complex salt. The neutral IPr and NH2R ligands occupy mutually trans positions (C(1)–Ni–N(3) = 159.8(2)°), with the strongly trans-directing benzyl moiety oriented cis to them (C(1)–Ni–C(421) = 102.3(2), C(421)–Ni–N(3) = 97.8(2)°). The amine ligand has a Ni–N(3) distance of 1.972(4) Å, similar to other σ-donor nickel amines,11 and its two hydrogen atoms were located in a difference map in the X-ray structure. The short Ni–C(421) distance (1.866(6) Å)12 is similar to the Ni–CH3 distance of 1.907(7) Å observed in T-shaped [(IPr)Ni(CH3){N(SiMe3) = SiMe2·Et2O}][B(ArF)4], in which the coordination site trans to the methyl ligand is also vacant.5b
 | ||
| Fig. 3 X-ray structure of 7 (50% ellipsoids). H-atoms except on N(3) and C(421) and the entire B(ArF)4 counterion have been omitted for clarity. Selected metrical parameters for 7: Ni–N(3) = 1.972(4), Ni–C(421) = 1.866(6), Ni–C(1) = 1.858(5) Å; C(1)–Ni–C(421) = 102.3(2), C(1)–Ni–N(3) = 159.8(2), N(3)–Ni–C(421) = 97.8(2)°. | ||
Carrying out the oxidation of 4 in THF, or addition of THF to solutions of 7, results in solvent coordination to give a 4-coordinate complex [(IPr)Ni(OC4H8)(κ2-C,N:NH2C6H3(Mes)C10H9)][B(ArF)4] (8) as a dark-red solid that has been characterized by NMR, X-ray crystallography (Fig. 4), and elemental analysis. Ligation of THF results in a slight elongation of both the trans carbyl group and the Ni–N bond (Ni–N(3) = 2.001(2), Ni–C(421) = 1.931(3), Fig. 4). Removal of the bound THF can be accomplished by precipitation of 8 from Et2O or CH2Cl2.
 | ||
| Fig. 4 X-ray structure of 8 (50% ellipsoids). H-atoms except on N(3) and C(421) and the entire B(ArF)4 counterion, have been omitted for clarity. Selected metrical parameters: Ni–N(3) = 2.001(2), Ni–C(421) = 1.931(3), Ni–C(1) = 1.904(3), Ni–O = 2.057(2) Å; C(1)–Ni–O = 95.05(10), O–Ni–N(3) = 84.86(9), N(3)–Ni–C(421) = 88.40(12), C(1)–Ni–C(421) = 91.74(12), C(1)–Ni–N(3) = 177.44(12), O–Ni–C(421) = 173.17(11)°. | ||
Unlike 4, a cyclic voltammogram of 5 indicates a quasi-reversible oxidation potential at E1/2 = −0.63 V (vs. Cp2Fe/Cp2Fe+, THF/TBAH). 5 can be cleanly oxidized with [Cp2Fe][B(ArF)4] to give a new paramagnetic Ni(II) complex, [(IPr)Ni(NHdippp)][B(ArF)4] (9, 94% yield, Scheme 2). Reaction of 9 with KC8 results in 1-e− reduction with reformation of 5 in 72% isolated yield. Electronically, replacing the strong-field alkyl ligand in 7 with a weaker-field amide in 9, and replacing an amine ligand in 7 with a weak Cipso–π interaction in 9 shifts the ground-state of these molecules from singlet (in 7) to triplet (in 9) configurations.
The solid-state structure of the cationic Ni core of 9 (Fig. 5) shows subtle differences from the parent neutral Ni(I) complex, with Ni–N = 1.854(2) and Ni–C(41) = 2.240(2) Å in 9. Despite the stabilizing contact between Ni and the flanking mesityl ring, 9 is high-spin d8 and with a magnetic moment of 3.2μB (SQUID) at room temperature, somewhat higher than the spin-only value of 2.83μB and similar in its magnetic profile to that seen for the bent, 2-coordinate Ni(II) amide Ni(NHdmp)2.10a Presumably, the increased steric bulk of the iso-propyl groups on the dippp ligand prevents C–H activation of an ortho-methine hydrogen as seen with the dmp derivative. A DFT analysis of the complex cation of 9 (B3LYP/6-31G(d) level) indicates a triplet ground state (ΔGST = 12 kcal mol−1) in which the optimized structure closely mimics that observed in the solid-state (Fig. 6). Interestingly, the optimized singlet and triplet DFT structures show similar Ni–N interactions, but an η6-coordination of one of the 2,6-diisopropylphenyl groups of the dippp ligand makes the singlet an 18-electron species. We have thus far been unsuccessful in cleanly deprotonating the amide ligand of 9 to give a neutral Ni(II) imide, possibly due to steric issues.13
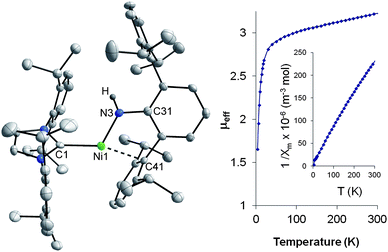 | ||
| Fig. 5 X-ray structure of the cation of 9 (left, 50% ellipsoids) and solid-state magnetic data at 5000 G (right). Most H-atoms and B(ArF)4 counterion omitted for clarity; selected metrical parameters: Ni–N(3) = 1.854(2), Ni–C(41) = 2.240(2), Ni–C(1) = 2.003(2) Å; C(1)–Ni–N(3) = 116.57(9), Ni–N(3)–C(31) = 122.0(2), N(3)–Ni–C(41) = 81.16(8), C(1)–Ni–C(41) = 162.17(8)°. | ||
 | ||
| Fig. 6 Optimized computed DFT structures of the complex cation of 9 for the triplet (left) and singlet (right) states, with calculated Ni–N distances of 1.846 and 1.872 Å, and angles of Ni–N–C (121.8 and 117.6°) and CNHC–Ni–N (117.0 and 99.1°), for the triplet and singlet structures, respectively. Ni is in green and N is in blue. | ||
The mechanism by which 4 undergoes benzylic activation of the NHdmp ligand is difficult to probe experimentally due to its paramagnetism. Cationic Ni(II) alkyls with γ-C–H agostic interactions, like the trimethylsilylmethyl complex cation [(dtbpe)Ni{CH2Si(CH3)3}][PF6], undergo smooth deprotonation by exogenous base to give neutral metallacyclobutane products (Scheme 3).14 It seemed reasonable that direct intramolecular deprotonation of a weak benzylic C–H bond (perhaps further weakened by an agostic interaction) in a cationic Ni(II) amide might be operative.
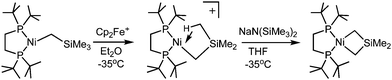 | ||
| Scheme 3 Deprotonation with metallation of a –CH3 group in a cationic Ni(II) complex cation to give a new Ni-alkyl bond.13 | ||
DFT calculations were carried out at the B3LYP/6-31G(d) level to elucidate the transformation of 4 to 7. While the optimized structure of the cation generated by removal of an electron from DFT-optimized 5 (5+ singlet) shows no protons in the vicinity of the amide nitrogen (all >3 Å except that directly bonded to N), the optimized structure of 4+ (singlet generated from DFT optimized 4) has a benzylic proton very close to the amide (∼2.61 Å; Fig. 7). Moreover, the benzylic C–H should be more acidic for aryl-CH3versus aryl-CMe2H since carbanions are less stable for more substituted carbons. Finally, the free energy to convert 4+ to 7 is calculated to be exergonic by 9 kcal mol−1. Taken together, synthetic, crystallographic, and DFT studies implicate an intramolecular deprotonation mechanism for the conversion of 4 to 7 upon ferrocenium oxidation.
 | ||
| Fig. 7 Optimized computed DFT structure of 4+, the (experimentally unobserved) complex cation formed on 1-e− oxidation of 4 that ultimately gives the observed metallated product 7. ΔG4+/7 (calculated) = −9 kcal mol−1. | ||
Conclusions
We have demonstrated that addition of terphenyl-substituted amides to 3 results in formation of pseudo two-coordinate amide/NHC complexes that display short, stabilizing contacts between nickel and flanking aryl ipso-carbons of the terphenyl amide in the solid state. Oxidation of these amide complexes with the 1-electron oxidant Cp2Fe+ results in C–H activation with dmp to give diamagnetic 7, or formation of a paramagnetic d8 amide cation 9 with the dippp amide. The latter salt has not yet been successfully deprotonated to yield a 2-coordinate imide.Acknowledgements
This work was supported by the US National Science Foundation through grant CHE-0957816 to G.L.H., and by the U. S. Department of Energy (Basic Energy Sciences) to T.R.C. through grant DE-FG02-03ER15387. T.R.C. acknowledges the Chemical Computing Group for their generous gift of the MOE program. C.T.S. is grateful for an NSF predoctoral fellowship.Notes and references
- (a) D. J. Mindiola and G. L. Hillhouse, Chem. Commun., 2002, 1840 RSC; (b) R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350 CrossRef CAS; (c) C. A. Laskowski and G. L. Hillhouse, Organometallics, 2009, 28, 6114 CrossRef CAS; (d) C. A. Laskowski, A. J. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2011, 133, 771 CAS.
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2001, 123, 4623 CrossRef CAS.
- (a) E. Kogut, H. L. Wiencko, L. Zhang, D. E. Cordeau and T. H. Warren, J. Am. Chem. Soc., 2005, 127, 11248 CrossRef CAS; (b) R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2008, 130, 12628 CrossRef CAS.
- V. M. Iluc and G. L. Hillhouse, J. Am. Chem. Soc., 2010, 132, 15148 CrossRef CAS.
- (a) B. R. Dibble, M. S. Sigman and A. M. Arif, Inorg. Chem., 2005, 44, 3774 CrossRef CAS; (b) C. A. Laskowski and G. L. Hillhouse, J. Am. Chem. Soc., 2008, 130, 13846 Search PubMed.
- Representative examples: (a) R. Melenkivitz and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 3486 CrossRef CAS; (b) T. Nguyen, A. Sutton, M. Brynda, J. Fettinger, G. Long and P. P. Power, Science, 2005, 310, 844 CrossRef CAS; (c) E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047 CrossRef CAS.
- Examples of terminal Ni amides: (a) P. L. Holland, R. A. Andersen, R. G. Bergman, J. Huang and S. P. Nolan, J. Am. Chem. Soc., 1997, 119, 12800 CrossRef CAS; (b) J. Campora, I. Matas, P. Palma, E. Alvarez, C. Graiff and A. Tiripicchio, Organometallics, 2007, 26, 3840 CrossRef CAS; (c) L.-C. Liang, P.-S. Chien, P.-Y. Lee, J.-M. Lin and Y.-L. Huang, Dalton Trans., 2008, 3320 RSC.
- M. Ito, T. Matsumoto and K. Tatsumi, Inorg. Chem., 2009, 48, 2215 CrossRef CAS.
- J. Li, H. Song, C. Cui and J. Cheng, Inorg. Chem., 2008, 47, 3468 CrossRef CAS.
- (a) A. M. Bryan, W. A. Merrill, W. M. Reiff, J. C. Fettinger and P. P. Power, Inorg. Chem., 2012, 51, 3366 CrossRef CAS; (b) P. P. Power, Chem. Rev., 2012, 112, 3482 Search PubMed; (c) C. Ni, J. C. Fettinger, G. J. Long and P. P. Power, Inorg. Chem., 2009, 48, 2443 CrossRef CAS.
- Representative examples: (a) C. E. Barclay, G. Deeble, R. J. Doyle, S. A. Elix, G. Salem, T. L. Jones, S. B. Wild and A. C. Willis, J. Chem. Soc., Dalton Trans., 1995, 57 RSC; (b) L. F. Groux and D. Zargarian, Organometallics, 2003, 22, 4759 CrossRef CAS; (c) P. J. Desrochers, D. S. Duong, A. S. Marshall, S. A. Lelievre, B. Hong, J. R. Brown, R. M. Tarkka, J. M. Manion, G. Holman, J. W. Merkert and D. A. Vicic, Inorg. Chem., 2007, 46, 9221 CrossRef CAS.
- For Ni alkyls: (a) C. Eaborn, M. S. Hill, P. B. Hitchcock and D. J. Smith, Chem. Commun., 2000, 691 RSC; (b) P. L. Holland, T. R. Cundari, L. L. Perez, N. A. Eckert and R. J. Lachiotte, J. Am. Chem. Soc., 2002, 124, 14416 CrossRef CAS; (c) H. L. Wiencko, E. Kogut and T. H. Warren, Inorg. Chim. Acta, 2003, 345, 199 CrossRef CAS; (d) K. Fujita, R. Schenker, W. Gu, T. C. Brunold, S. P. Cramer and C. G. Riordan, Inorg. Chem., 2004, 43, 3324 CrossRef CAS; (e) T. J. Anderson, G. D. Jones and D. A. Vicic, J. Am. Chem. Soc., 2004, 126, 8100 CrossRef CAS.
- Attempted deprotonation of 9 with NaN(SiMe3)2, KOtBu, or nBuLi resulted in the detection of multiple products inconsistent with the formation of a two-coordinate Ni imido complex. Single crystals isolated from one such reaction indicated (X-ray) a product consistent with deprotonation of the NHC backbone, as has been observed by others,13a,b but this could not be satisfactorily corroborated by additional spectroscopic methods. (a) Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2010, 132, 14370 CrossRef CAS; (b) P. K. Majhi, S. Sauerbrey, G. Schnakenburg, A. J. Arduengo III and R. Streubel, Inorg. Chem., 2012 DOI:10.1021/ic301641z , ASAP.
- K. D. Kitiachvili, D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2004, 126, 10554 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic details for 4, 5, 7, 8 and 9, data regarding the crystallographic refinements, and experimental and computational details (PDF). CCDC 896901–896905 contain the supplementary crystallographic data for this paper. See DOI: 10.1039/c2sc21345a |
| ‡ Current address: PPG Industries, 440 College Park Drive, Monroeville, PA, 15146, USA. |
| This journal is © The Royal Society of Chemistry 2013 |