Sc2S@C2(7892)–C70: a metallic sulfide cluster inside a non-IPR C70 cage†
Ning Chen‡a, Marc Mulet-Gasb, Yu-Yang Lia, Riane E. Stenea, Curtis W. Athertona, Antonio Rodríguez-Fortea*b, Josep M. Poblet*b and Luis Echegoyen*a
aDepartment of Chemistry, University of Texas at El Paso, El Paso, Texas 79968, USA. E-mail: echegoyen@utep.edu; Fax: +1-915-747-8807; Tel: +1-915-747-7573
bDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, c/Marcel·lí Domingo s/n, 43007 Tarragona, Spain. E-mail: josepmaria.poblet@urv.cat; antonio.rodriguezf@urv.cat; Fax: +34-977-559-563; Tel: +34-977-559-569
First published on 8th October 2012
Abstract
A new cage isomer of C70, Sc2S@C2(7892)–C70, has been isolated and characterized by mass spectrometry, UV-Vis-NIR absorption spectroscopy, cyclic voltammetry and DFT calculations. The combined experimental and computational studies lead to the unambiguous assignment of the cage symmetry to C2(7892)–C70. The comparison between Sc2S@C2(7892)–C70 and related endohedral structures has been discussed. A close structural resemblance between Sc2S@C2(7892)–C70 and Sc2S@Cs(10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 528)–C72 suggests that the conversion of these two molecules may be the result of a simple insertion of C2 and the structural difference between Sc2S@C2(7892)–C70 and Sc3N@C2v(7854)–C70 shows that the nature and geometry of the encaged cluster plays an important role on the selection of the non-IPR cage.
528)–C72 suggests that the conversion of these two molecules may be the result of a simple insertion of C2 and the structural difference between Sc2S@C2(7892)–C70 and Sc3N@C2v(7854)–C70 shows that the nature and geometry of the encaged cluster plays an important role on the selection of the non-IPR cage.
Introduction
Fullerenes are spherical molecules composed of pentagons and hexagons.1,2 Endohedral fullerenes, which possess outer surface available for chemical modification and inner space capable of encapsulation of variable clusters and molecules, have generated considerable recent research interest.3,4 As a special class of endohedral fullerenes, clusterfullerenes (CFs) are known for their exceptionally high yield and fascinating structural versatility of the encapsulated clusters and carbon cages.5–7 After the discovery of the first clusterfullerene in 1999, Sc3N@Ih–C80,8 nitride cluster fullerenes (NCFs),9 carbide cluster fullerenes (CCFs),10,11 oxide cluster fullerenes (OCFs)12–14 and sulfide cluster fullerenes (SCFs)15–17 were discovered and the encapsulated clusters were found to have a significant influence on the cage structure and their chemical reactivities.18 With the remarkable versatility to alter their physical and chemical properties based on the structural variety of both cages and clusters, these molecules are attracting widespread interest in applications such as MRI agents and in molecular electronic devices and solar cells.19–27Sulfide cluster fullerenes (SCFs) are the newest member in the CF families.15,16 Different methods have been utilized to synthesize these compounds. Dunsch and co-workers reported the formation of one isomer of M2S@C82 (M = Sc, Lu, Dy) by introducing CH5N3·HSCN as a solid sulfur source.15 We demonstrated that an extensive family of novel scandium sulfide cluster fullerenes with cages ranging from C68 to C100 can be obtained in macroscopic quantities by introducing SO2 into the arc reactor.16 Further isolation and characterization of some of these new species revealed novel structures and properties which are unique to this family. Two isomers of Sc2S@C82, Sc2S@Cs(6)–C82 and Sc2S@C3v(8)–C82, were identified as the most abundant products in this family and were recently characterized by single crystal X-ray crystallography.17 Sc2S@Cs(6)–C82, in particular, was found to possess completely ordered cage and cluster in its single crystal structure, the first in the clusterfullerene series.17 Very recently, we reported the isolation of a metallic clusterfullerene in a non-IPR (Isolated Pentagon Rule) C72 cage, Sc2S@C72. Crystallography clearly established that the C72 cage has an unusual Cs(10528) symmetry.28 A combined computational and experimental study revealed that the unique geometry of the Sc2S cluster together with a formal charge transfer of four electrons between the cluster and the cage play an important role in the stabilization of this new cage which had never been detected experimentally before in any of the CF families. These results indicate that more novel endohedral structures are likely to be found in the SCF family and systematic studies of these structures might provide a better understanding of the fundamental aspects of SCFs, and useful design principles to guide the preparation of high yielding endohedral compounds with unique and useful optoelectronic properties.
Herein we report a new sulfide cluster fullerene, Sc2S@C2(7892)–C70, in which the sulfide cluster is encapsulated inside a non-IPR C70 cage. This represents the third isomer of a C70 cage discovered to date. Combined computational and experimental studies lead to the unambiguous assignment of the cage symmetry to C2(7892)–C70.
Results and discussion
Preparation and characterization of Sc2S@C70
The SCFs were synthesized in a conventional Krätschmer–Huffman reactor using an atmosphere of helium and SO2.16,29 The as-produced soot was Soxhlet-extracted with CS2 and a multi stage high-performance-liquid chromatography (HPLC) procedure was utilized to isolate and purify Sc2S@C70.Sc2S@C70 is the second smallest fullerene in the SCF family after Sc2S@C68. The HPLC-MALDI TOF analyses show that on a 5PYE column, the Sc2S@C70 fraction overlaps with those of C76, C78 and Sc2S@C72 (see Fig. 1). The retention time of Sc2S@C70 is shorter than that of Sc2S@C72, which agrees with the relatively smaller size of this compound. This fraction was further separated by a two-stage recycling HPLC procedure using a Buckyprep column, that resulted in the isolation of pure Sc2S@C70 (see ESI†). However, compared to the previously reported Sc2S@Cs(10528)–C72, a very similar non-IPR SCF in a small cage, the yield of this compound is much lower than that of Sc2S@Cs(10528)–C72. Out of the arcing processes of 60 packed graphite rods, along with 2.0 mg of Sc2S@Cs(10528)–C72, only 0.4 mg of Sc2S@C70 were obtained after HPLC purification.
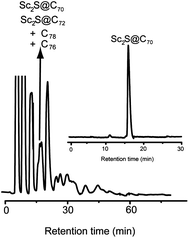 | ||
| Fig. 1 HPLC Chromatograms of the fullerene extract obtained on a 10 mm × 250 mm 5PYE column using λ = 320 nm, a flow rate of 4 mL min−1, and toluene as the eluent at 25 °C. Inset: HPLC of purified Sc2S@C70 obtained on a 10 mm × 250 mm 5PYE column using λ = 320 nm, a flow rate of 4 mL min−1, and toluene as the eluent at 25 °C. | ||
The MALDI-TOF spectrum (Fig. 2) of the isolated fraction shows a single peak at 961.904. The isotopic distribution of the experimental MALDI spectrum shows excellent agreement with the corresponding theoretical spectrum (see Fig. 2). The purity of this sample was further checked by HPLC as shown in Fig. 1.
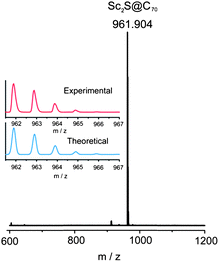 | ||
| Fig. 2 Mass spectrum of the purified Sc2S@C70. Inset: the experimental and theoretical isotopic distribution for Sc2S@C70. | ||
The purified Sc2S@C70 has a yellow-brown color in toluene solution. The UV-Vis-NIR absorption of Sc2S@C70 is shown in Fig. 3. A relatively strong absorption occurs at 1102 nm along with other absorptions at 461, 634, 699 and 952 nm. The UV-Vis-NIR absorption spectrum of Sc2S@C70 shows some similarities to that of Sc2S@Cs(10528)–C72, which also features a strong absorption at 1076 nm around the absorption onset region, but is rather featureless in other spectral regions. The characteristic features of this spectrum are also substantially different from those of empty C70 and from those of the previously reported Sc3N@C2v(7854)–C70, which presents the strongest absorption at 696 nm along with several shoulder peaks at 468, 558, 807 and 894 nm.30 Since the absorption spectra of fullerenes in the visible and NIR region are dominated by the π–π* transitions of the carbon cages and the spectra are very sensitive to the carbon cage symmetries,3 these differences clearly indicate that Sc2S@C70 has a non-IPR C70 cage and the cage symmetry is likely different from that of Sc3N@C2v(7854)–C70.
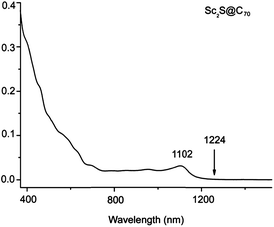 | ||
| Fig. 3 UV-Vis-NIR absorption of Sc2S@C70 in CS2 solution. | ||
The cyclic voltammogram (CV) of Sc2S@C2(7892)–C70 was recorded in o-dichlorobenzene (o-DCB) containing 0.05 M tetra(n-butyl)ammonium-hexafluorophosphate, (n-Bu4NPF6), as the supporting electrolyte using a scan rate of 100 mV s−1 (Fig. 4). The CV of Sc2S@C2(7892)–C70 shows some similarities and differences when compared to those of the other reported SCFs. The CV shows a reversible first oxidation followed by an irreversible second oxidation step, which is very similar to the oxidative behavior of Sc2S@Cs(10528)–C72,23 but different from those of the two isomers of Sc2S@C82, which exhibit two reversible oxidative steps.16 On the other hand, while Sc2S@Cs(10528)–C72 shows all-reversible reductive processes, the reductive behavior of Sc2S@C2(7892)–C70 shows similarities to those of the two isomers of Sc2S@C82 as well as to most of the cluster fullerenes, which typically exhibit irreversible reductive processes.31
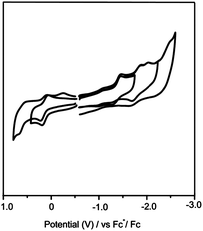 | ||
| Fig. 4 Cyclic voltammograms of Sc2S@Cs(7892)–C70 in n-Bu4NPF6/o-DCB with ferrocene as the internal standard; scan rate 100 mV s−1. | ||
The redox potentials of Sc2S@C2(7892)–C70 also show major differences from those of other reported SCFs. The first oxidation potential is shifted dramatically from 0.64 V for Sc2S@Cs(10528)–C72, to 0.14 V for Sc2S@C2(7892)–C70 (see Table 2).28 This shows that Sc2S@C2(7892)–C70 is much easier to oxidize even though the cage sizes and topologies are very similar (see below). The electrochemical gap of Sc2S@C2(7892)–C70 is 1.57 V, which is smaller than that of Sc2S@Cs(10528)–C72 (1.78 V) but somewhat larger than those of the corresponding Sc2S@C82 isomers (1.56 eV for Sc2S@C3v(8)–C82 and 1.47 eV for Sc2S@Cs(6)–C82).
Computational studies
Besides the experimental characterization of Sc2S@C70, we also performed an exhaustive computational study to assign the C70 cage symmetry among the 8149 possible isomers.§ From the electronic structure of the different computed isomers of Sc2S@C70, we verified that there is a formal transfer of four electrons, (Sc2S)4+@(C70)4−, as for the other Sc2S@C2n (2n = 72 and 82) known so far (see orbital interaction diagram, Fig. 5).32,33 We have computed the energies of the tetraanions using DFT at BP86/TZP level for all 111 C70 cages with three or less adjacent pentagon pairs (APP): 1 IPR structure, 1 APP1, 18 APP2 and 91 APP3 (see ESI†). The SCF corresponding to the lowest-energy tetraanionic cages were also computed (a total of 17 isomers, Table 1).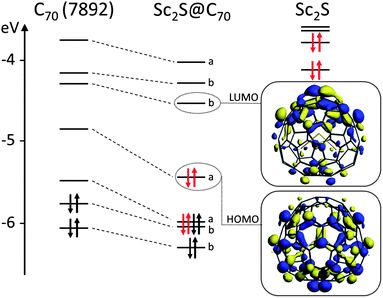 | ||
| Fig. 5 Orbital interaction diagram for Sc2S@C2(7892)–C70. The fragments, Sc2S and C2(7892)–C70, were calculated with the same geometry they have in the clusterfullerene. | ||
| Isomer | APP | C704− | Sc2S@C70 |
|---|---|---|---|
| a Isomer number according to the spiral algorithm of Fowler and Manolopoulos.35b APP: number of adjacent pentagon pairs. | |||
| 8149 | 0 | 0.0 | 20.6 |
| 7892 | 2 | 3.9 | 0.0 |
| 7957 | 2 | 4.7 | 19.1 |
| 7851 | 3 | 6.2 | 20.3 |
| 7852 | 3 | 6.3 | 23.5 |
| 7887 | 3 | 7.0 | 21.6 |
| 7854 | 3 | 8.5 | 26.3 |
| 7893 | 3 | 9.4 | 19.7 |
| 7886 | 3 | 10.8 | 21.5 |
| 7924 | 2 | 11.6 | 18.6 |
| 7846 | 3 | 13.2 | 29.3 |
| 7960 | 2 | 14.1 | 21.9 |
| 7922 | 3 | 14.2 | 27.7 |
| 7921 | 3 | 14.7 | 29.1 |
| 7850 | 3 | 14.7 | 33.2 |
| 7847 | 3 | 15.6 | 28.8 |
| 8094 | 1 | 16.7 | 26.5 |
Interestingly, the IPR cage, D5h–C70(8149), was found to be the lowest-energy tetraanion. Other cage isomers with two and three APPs show relative energies within 10 kcal mol−1. In particular, cage C2(7892)–C70 exhibits the second most-stable tetraanion and is only 4 kcal mol−1 higher in energy than the IPR cage tetraanion (see Table 1). Among the 18 isomers with two pairs of fused pentagons, cage C2(7892)–C70 shows the lowest number of pyracylene motifs and maximally separated pentagonal units (see ESI†).34
To predict the stability of a given clusterfullerene, it is necessary to consider electron transfer from the cluster to the cage but also the stability provided by the interaction between the metal atoms of the trapped cluster and the carbon cage. These interactions are not so critical for IPR cages; however, the cluster–cage interactions are crucial for those CFs that have one or more APPs. The systems with non-IPR cages are stabilized by the proximity of the metal cations of the cluster to the pentalene motifs, as in, for example, Sc3N@D3(6140)–C68,36 Gd3N@Cs(39663)–C82,37 M3N@Cs(51365)–C84 (M = Gd, Tm)38 and the recently reported Sc2S@Cs(10528)–C72.28 Thus, appropriate localization of the APPs on the fullerene cage that provide optimized interactions between the metal cations and the pentalene motifs is a key factor in the stabilization of non-IPR clusterfullerenes. In the present case, which resembles that of Sc2S@Cs(10528)–C72,28 it is only after the encapsulation of Sc2S that the 7892–C70 cage becomes by far (∼20 kcal mol−1) the most stable endohedral isomer (Table 1). The most characteristic structural parameters, 2.352 Å for the Sc–S distance and 97.8° for the Sc–S–Sc angle, resemble those previously found for the two Sc2S@C82 isomers19 and for Sc2S@Cs(10528)–C72.28 In particular, the value of the Sc–S–Sc angle is smaller than those for Sc2S@Cs(6)–C82 (114°) and Sc2S@Cs(10528)–C72 (124°), but similar to that found for Sc2S@C3v(8)–C82 (97°). The IPR Sc2S@D5h(8149)–C70 is destabilized due to the lack of the metal cation–pentalene interactions. The other non-IPR cages do not possess optimal positioning of the APPs to maximize the metal–pentalene interactions as observed for Sc2S@C2(7892)–C70 (see Fig. 6).
 | ||
| Fig. 6 DFT-optimized structure for Sc2S@C2(7892)–C70. | ||
The molar fractions of the lowest-energy Sc2S@C70 isomers as a function of the temperature were also computed using the rigid rotor and harmonic oscillator (RRHO) approximation and the related free-encapsulating model (FEM) proposed by Slanina (see Fig. 7).39,40 Somewhat different results are obtained for the FEM and RRHO approximations. For the FEM, the SCF with cage 7892 is the most abundant isomer in the whole temperature range (to 4000 K). Within the RRHO approximation, there is an isomer preference crossing at around 1700 K, with the IPR isomer predominating at higher temperatures. So far, the predictions derived from the FEM model agree better with experiments than the ones made using the RRHO approximation. In the FEM model we consider that if at high temperature the cluster is rotating freely inside the carbon cages, its contribution to the partition function will be similar for the different cages and will cancel out. In the present case, the Sc2S will probably rotate inside the spherical IPR cage at sufficiently high temperatures, but it is not so clear that the Sc2S cluster will freely rotate inside the non-IPR APP2 carbon cage 7892 since the interaction of the Sc ions with the pentalene motifs is significant and there is not a lot of space to rotate inside this cage. The FEM approximation would be the most suitable for the IPR clusterfullerene, but it is not so clear that it would also be the best for the non-IPR 7892 cage, so reality probably lies somewhere between these two approximations. Consequently, we predict Sc2S@C2(7892)–C70 to be the most abundant isomer produced within the temperature range present in the arc.
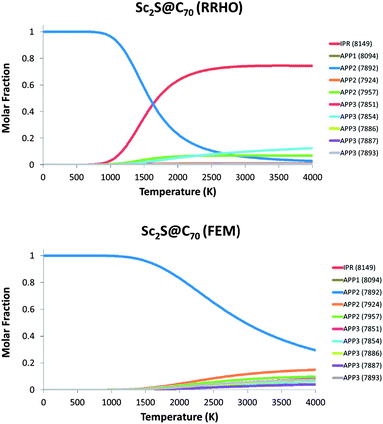 | ||
| Fig. 7 Predicted molar fractions within the RRHO (top) and FEM (bottom) models as a function of temperature for ten different isomers of Sc2S@C70. | ||
Good agreement of the computed first and second oxidation and reduction potentials of Sc2S@C2(7892)–C70 with the peaks measured experimentally by cyclic voltammograms reaffirms the assignment, see Table 2. The first anodic potential is predicted to appear at +0.04 V, in good agreement with the experimental half-wave potential (+0.14 V).41 The predicted first cathodic potential is also close to the experimental value with a difference of around 100 mV (Table 2). The computed electrochemical (EC) gap, 1.37 V, compares reasonably well with experiment, 1.58 V, although the error (210 mV) is somewhat larger than for nitride clusterfullerenes.41 Inclusion of thermal and entropic effects does not improve the computed gap, as for the family of nitrides.41 It is worth noting that, for irreversible processes, the comparison between experimental and theoretical electrochemical gaps is difficult, because computations predict standard potentials, and not experimental peak potentials. The predictions for the second anodic and cathodic potentials are in excellent agreement with experiment (with an error of only 50 mV). The HOMO–LUMO gap for the IPR clusterfullerene (0.56 eV) is much smaller than for the 7892 cage (0.95 eV). We have computed the first oxidation potential for Sc2S@D5h(8149)–C70 and have found that it is more easily oxidized than Sc2S@C2(7892)–C70 (−0.16 V vs. +0.04 V, respectively). We have had problems computing the first reduction potential for Sc2S@D5h(8149)–C70 because the LUMO and LUMO+1 orbitals are almost degenerate and the DFT mono-determinant approach is not suitable to describe such a reduced state (multi-determinant wavefunctions would be needed, but the computational cost is too high for such large systems). According to the energy of the LUMOs (Sc2S@D5h(8149)–C70: −4.04 eV; Sc2S@C2(7892)–C70: −3.84 eV) the first reduction potential for Sc2S@D5h(8149)–C70 should be lower (around 200 mV) than for Sc2S@C2(7892)–C70, leading to a much smaller EC gap (around 1 eV or even smaller). So, the comparison between computed and experimental electrochemical properties discards Sc2S@D5h(8149)–C70 and suggests Sc2S@C2(7892)–C70 as the isomer formed in the arc.
The definitive experiment that confirms isomer 7892 as the carbon cage that encapsulates Sc2S is the UV-Vis-NIR spectrum. We have computed the spectra using time-dependent (TD) DFT for the two possible SCFs, Sc2S@D5h(8149)–C70 and Sc2S@C2(7892)–C70, in the Vis-NIR region, for wavelengths larger than 500 nm. Despite the systematic underestimation of excitation energies by TD-DFT, this methodology provides a reasonable agreement with experiments for the family of SCFs.28 From the computations we can clearly discard Sc2S@D5h(8149)–C70 because its spectrum shows transitions at large wavelengths (around 1800 and 1600 nm, see Table 3 and ESI†), in total contrast with experimental observations. The spectral onset appears at 1224 nm, see Fig. 3. In addition, the spectrum predicted for Sc2S@C2(7892)–C70 agrees rather well with the experimental one, with the first transition (corresponding to the HOMO–LUMO transition) at 1158 nm (to be compared with the experimental 1102 nm). Therefore, we feel confident that the isolated Sc2S@C70 clusterfullerene corresponds to Sc2S@C2(7892)–C70.
| E (eV) | λ (nm) | fa | Leading configurationsb (%) |
|---|---|---|---|
| a Only excitations with f (oscillator strength) > 0.001 are listed.b Contributions less than 10% are omitted. | |||
| Sc2S@C2(7892)–C70 | |||
| 1.071 | 1158 | 0.00650 | HOMO → LUMO (98) |
| 1.212 | 1023 | 0.00356 | HOMO → LUMO+1 (98) |
| 1.664 | 745 | 0.00104 | HOMO → LUMO+3 (99) |
| 1.681 | 738 | 0.00519 | HOMO → LUMO+4 (96) |
| 1.853 | 669 | 0.00721 | HOMO-1 → LUMO (89) |
| Sc2S@D5h(8149)–C70 | |||
| 0.688 | 1802 | 0.00365 | HOMO → LUMO (63); HOMO → LUMO+1 (36) |
| 0.763 | 1625 | 0.00103 | HOMO-1 → LUMO+1 (63); HOMO-1 → LUMO (26) |
| 0.802 | 1546 | 0.00715 | HOMO-1 → LUMO (71); HOMO-1 → LUMO+1 (28) |
| 0.980 | 1265 | 0.00138 | HOMO → LUMO+2 (50); HOMO-1 → LUMO+3 (47) |
| 1.295 | 958 | 0.05558 | HOMO-1 → LUMO+3 (46); HOMO → LUMO+2 (35) |
At this point, it is worth remarking that C2(7892)–C70, the proposed cage for Sc2S@C70, and Cs(10528)–C72, the one found for Sc2S@C72, are intimately related as the Schlegel diagrams in Fig. 8 show. In fact, cage Cs(10528)–C72 can be obtained by a single C2 addition to a hexagon of C2(7892)–C70 (see ESI†). Therefore, conversion of these two SFCs may happen by single addition/extrusion of a C2 molecule without further atomic rearrangements. Moreover, it is possible that Sc2S@Cs(10528)–C72 could form by C2 addition to Sc2S@C2(7892)–C70 through the recently proposed closed network growth (CNG) mechanism.42,43
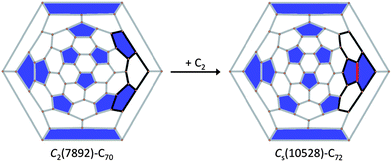 | ||
| Fig. 8 Schlegel diagrams for cages C2(7892)–C70 and Cs(10528)–C72. The difference between the two cages is highlighted in black with the added C2 unit in red. The larger distance between the two APPs in cage Cs(10528)–C72 compared to that for C2(7892)–C70 makes the Sc–S–Sc angle in Sc2S@Cs(10528)–C72 (124°) larger than that found for Sc2S@C2(7892)–C70 (98°). | ||
Beside Sc2S@C2(7892)–C70, only one non-IPR C70 endohedral fullerene, Sc3N@C2v(7854)–C70, has been reported so far. Very recently, Nagase, Zhao and co-workers proposed, using density functional theory, that Sc2@C2v(7854)–C70 and not the corresponding carbide, Sc2C2@C2v(6073)–C68, is the metallofullerene detected as Sc2C70.44 These two isomers, C2(7892)–C70 and C2v(7854)–C70, are rather close in energy when computed as tetraanions (Table 4). On the other hand, the hexaanionic state is much more favored for C2v(7854)–C70. If the trapped cluster is Sc3N, a six electron transfer combined with the optimal interaction between the three Sc atoms and the three pentalenes on cage C2v(7854)–C70 (Fig. 9) lead to the preferential stabilization of Sc3N@C2v(7854)–C70, as proposed by Yang et al.30 (Table 4). However, with the Sc2S cluster inside the Sc–pentalene interactions are optimized for Sc2S@C2(7892)–C70, which has, by far, the lowest energy (Tables 1 and 4). Although sharing the same cage size, the number and the position of the pentalene motifs are significantly different in these two isomers. This shows that the nature and geometry of the encaged cluster plays an important role on the selection of the cage isomer, along with the electronic stabilization resulting from cluster–cage electron transfer.
| C2(7892)–C70 | C2v(7854)–C70 | |
|---|---|---|
| a The stabilization of cage C2v(7854)–C70vs. C2(7892)–C70 (10.5 kcal mol−1) is smaller than for the corresponding hexaanions (16.2 kcal mol−1) due to larger structural distortions of the Sc3N cluster inside the C2v(7854)–C70 cage (see Fig. 9 and ESI†). | ||
| C704− | 0.0 | 4.6 |
| C706− | 16.2 | 0.0 |
| Sc2S@C70 | 0.0 | 26.3 |
| Sc3N@C70a | 10.5 | 0.0 |
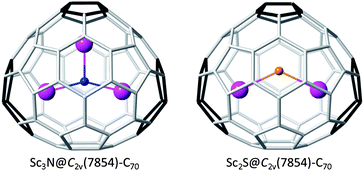 | ||
| Fig. 9 DFT-optimized structures for Sc3N@C2v(7854)–C70 and Sc2S@C2v(7854)–C70. | ||
Conclusions
In summary, a new isomer of C70, Sc2S@C2(7892)–C70, has been isolated and characterized by mass spectrometry, UV-Vis-NIR absorption spectroscopy, cyclic voltammetry and DFT calculations. The combined experimental and computational studies led to the unambiguous assignment of the cage symmetry to C2(7892)–C70, the second non-IPR isomer of C70 to be detected experimentally. Sc2S@C2(7892)–C70 is by far the lowest-energy isomer and the major candidate to be the experimentally detected Sc2S@C70. At high temperatures, isomer Sc2S@D5h(8149)–C70 could also be formed, but comparison of the experimental and theoretical electrochemical properties and UV-Vis-NIR spectra clearly discards the IPR Sc2S@D5h(8149)–C70. Cage C2(7892)–C70 is structurally intimately related to cage Cs(10528)–C72, the one that has been recently detected by single crystal X-ray crystallography for Sc2S@C72. Thus, besides Sc2S@Cs(10528)–C72, Sc2S@C2(7892)–C70 provides an additional example of stabilization of a new non-IPR cage resulting from the combined effect of a four-electron transfer and the unique geometrical fit between the Sc2S cluster and the cage. Many more structures and interesting properties are yet to be explored in the SCF family.Acknowledgements
We thank the Robert A. Welch Foundation for an endowed chair, grant #AH-0033 and the US NSF for grant CHE-1110967, which provided generous support for this work. This work is also supported by the Spanish Ministry of Science and Innovation (Project no. CTQ2011-29054-C02-01) and by the Generalitat de Catalunya (2009SGR462 and XRQTC). Additional support from the US NSF, grant CHE-1124075 and from the Spanish Ministry of Science and Innovation (PRI-PIBUS-2011-0995) for a joint US-Spain collaboration is also acknowledged.Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- A. Rodriguez-Fortea, S. Irle and J. M. Poblet, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 350–367 CrossRef CAS.
- H. Shinohara, Rep. Prog. Phys., 2000, 63, 843–892 CrossRef CAS.
- T. Akasaka and S. Nagase, Endofullerenes: A New Family of Carbon Clusters, Kluwer Academic Publishers, Dordrecht, Boston, 2002 Search PubMed.
- S. Yang, F. Liu, C. Chen, M. Jiao and T. Wei, Chem. Commun., 2011, 47, 11822–11839 RSC.
- L. Dunsch and S. Yang, Phys. Chem. Chem. Phys., 2007, 9, 3067–3081 RSC.
- A. Rodriguez-Fortea, A. L. Balch and J. M. Poblet, Chem. Soc. Rev., 2011, 40, 3551–3563 RSC.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55–57 CrossRef CAS.
- L. Dunsch and S. Yang, Small, 2007, 3, 1298–1320 CrossRef CAS.
- C. R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Angew. Chem., Int. Ed., 2001, 40, 397–399 CrossRef CAS.
- T. S. Wang, N. Chen, J. F. Xiang, B. Li, J. Y. Wu, W. Xu, L. Jiang, K. Tan, C. Y. Shu, X. Lu and C. R. Wang, J. Am. Chem. Soc., 2009, 131, 16646–16647 CrossRef CAS.
- S. Stevenson, M. A. Mackey, M. A. Stuart, J. P. Phillips, M. L. Easterling, C. J. Chancellor, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2008, 130, 11844–11845 CrossRef CAS.
- R. Valencia, A. Rodríguez-Fortea, S. Stevenson, A. L. Balch and J. M. Poblet, Inorg. Chem., 2009, 48, 5957–5961 CrossRef CAS.
- B. Q. Mercado, M. M. Olmstead, C. M. Beavers, M. L. Easterling, S. Stevenson, M. A. Mackey, C. E. Coumbe, J. D. Phillips, J. P. Phillips, J. M. Poblet and A. L. Balch, Chem. Commun., 2010, 46, 279–281 RSC.
- L. Dunsch, S. Yang, L. Zhang, A. Svitova, S. Oswald and A. A. Popov, J. Am. Chem. Soc., 2010, 132, 5413–5421 CrossRef CAS.
- N. Chen, M. N. Chaur, C. Moore, J. R. Pinzon, R. Valencia, A. Rodriguez-Fortea, J. M. Poblet and L. Echegoyen, Chem. Commun., 2010, 46, 4818–4820 RSC.
- B. Q. Mercado, N. Chen, A. Rodríguez-Fortea, M. A. Mackey, S. Stevenson, L. Echegoyen, J. M. Poblet, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2011, 133, 6752–6760 CrossRef CAS.
- N. Martin, Chem. Commun., 2006, 2093–2104 RSC.
- S. Laus, B. Sitharaman, É. Tóth, R. D. Bolskar, L. Helm, S. Asokan, M. S. Wong, L. J. Wilson and A. E. Merbach, J. Am. Chem. Soc., 2005, 127, 9368–9369 CrossRef CAS.
- E. Toth, R. D. Bolskar, A. Borel, G. Gonzalez, L. Helm, A. E. Merbach, B. Sitharaman and L. J. Wilson, J. Am. Chem. Soc., 2005, 127, 799–805 CrossRef CAS.
- S. Sato, S. Seki, G. Luo, M. Suzuki, J. Lu, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 11681–11686 CrossRef CAS.
- L. Feng, M. Rudolf, S. Wolfrum, A. Troeger, Z. Slanina, T. Akasaka, S. Nagase, N. Martín, T. Ameri, C. J. Brabec and D. M. Guldi, J. Am. Chem. Soc., 2012, 134, 12190–12197 CrossRef CAS.
- S. Sato, S. Seki, Y. Honsho, L. Wang, H. Nikawa, G. Luo, J. Lu, M. Haranaka, T. Tsuchiya, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2011, 133, 2766–2771 CrossRef CAS.
- J. R. Pinzon, M. E. Plonska-Brzezinska, C. M. Cardona, A. J. Athans, S. S. Gayathri, D. M. Guldi, M. A. Herranz, N. Martin, T. Torres and L. Echegoyen, Angew. Chem., Int. Ed., 2008, 47, 4173–4176 CrossRef CAS.
- R. B. Ross, C. M. Cardona, D. M. Guldi, S. G. Sankaranarayanan, M. O. Reese, N. Kopidakis, J. Peet, B. Walker, G. C. Bazan, E. Van Keuren, B. C. Holloway and M. Drees, Nat. Mater., 2009, 8, 208–212 CrossRef CAS.
- R. B. Ross, C. M. Cardona, F. B. Swain, D. M. Guldi, S. G. Sankaranarayanan, E. V. Keuren, B. C. Holloway and M. Drees, Adv. Funct. Mater., 2009, 19, 2332–2337 CrossRef CAS.
- E.-Y. Zhang, C.-Y. Shu, L. Feng and C.-R. Wang, J. Phys. Chem. B, 2007, 111, 14223–14226 CrossRef CAS.
- N. Chen, C. M. Beavers, M. Mulet-Gas, A. Rodríguez-Fortea, E. J. Munoz, Y.-Y. Li, M. M. Olmstead, A. L. Balch, J. M. Poblet and L. Echegoyen, J. Am. Chem. Soc., 2012, 134, 7851–7860 CrossRef CAS.
- W. Kratschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354–358 CrossRef.
- S. F. Yang, A. A. Popov and L. Dunsch, Angew. Chem., Int. Ed., 2007, 46, 1256–1259 CrossRef CAS.
- M. N. Chaur, F. Melin, A. L. Ortiz and L. Echegoyen, Angew. Chem., Int. Ed., 2009, 48, 7514–7538 CrossRef CAS.
- B. Q. Mercado, M. A. Stuart, M. A. Mackey, J. E. Pickens, B. S. Confait, S. Stevenson, M. L. Easterling, R. Valencia, A. Rodriguez-Fortea, J. M. Poblet, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2010, 132, 12098–12105 CrossRef CAS.
- B. Q. Mercado, N. Chen, A. Rodriguez-Fortea, M. A. Mackey, S. Stevenson, L. Echegoyen, J. M. Poblet, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2011, 133, 6752–6760 CrossRef CAS.
- A. Rodriguez-Fortea, N. Alegret, A. L. Balch and J. M. Poblet, Nat. Chem., 2010, 2, 955–961 CrossRef CAS.
- P. W. Fowler and D. E. Manolopoulos, An Atlas of Fullerenes, Oxford University Press, Oxford, 1995 Search PubMed.
- S. Stevenson, P. W. Fowler, T. Heine, J. C. Duchamp, G. Rice, T. Glass, K. Harich, E. Hajdu, R. Bible and H. C. Dorn, Nature, 2000, 408, 427–428 CrossRef CAS.
- B. Q. Mercado, C. M. Beavers, M. M. Olmstead, M. N. Chaur, K. Walker, B. C. Holloway, L. Echegoyen and A. L. Balch, J. Am. Chem. Soc., 2008, 130, 7854 CrossRef CAS.
- T. Zuo, K. Walker, M. M. Olmstead, F. Melin, B. C. Holloway, L. Echegoyen, H. C. Dorn, M. N. Chaur, C. J. Chancellor, C. M. Beavers, A. L. Balch and A. J. Athans, Chem. Commun., 2008, 1067–1069 RSC.
- Z. Slanina, S. L. Lee, F. Uhlik, L. Adamowicz and S. Nagase, Theor. Chem. Acc., 2007, 117, 315–322 CrossRef CAS.
- Z. Slanina and S. Nagase, ChemPhysChem, 2005, 6, 2060–2063 CrossRef CAS.
- R. Valencia, A. Rodriguez-Fortea, A. Clotet, C. de Graaf, M. N. Chaur, L. Echegoyen and J. M. Poblet, Chem.–Eur. J., 2009, 15, 10997–11009 CrossRef CAS.
- P. W. Dunk, N. K. Kaiser, C. L. Hendrickson, J. P. Quinn, C. P. Ewels, Y. Nakanishi, Y. Sasaki, H. Shinohara, A. G. Marshall and H. W. Kroto, Nat. Commun., 2012, 3, 855 CrossRef.
- P. W. Dunk, N. K. Kaiser, M. Mulet-Gas, A. Rodriguez-Fortea, J. M. Poblet, H. Shinohara, C. L. Hendrickson, A. G. Marshall and H. W. Kroto, J. Am. Chem. Soc., 2012, 134, 9380–9389 CrossRef CAS.
- H. Zheng, X. Zhao, W.-W. Wang, T. Yang and S. Nagase, J. Chem. Phys., 2012, 137, 014308 CrossRef.
- A. Osterholm, A. Petr, C. Kvarnstrom, A. Ivaska and L. Dunsch, J. Phys. Chem. B, 2008, 112, 14149–14157 CrossRef CAS.
- G. T. Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC chromatograms and computational results (relative energies and plots of the structures for all the isomers computed in this work, schematic representation showing the relationship between C2(7892)–C70 and Cs(10528)–C72 cages, and xyz coordinates for the lowest-energy computed isomers). See DOI: 10.1039/c2sc21101g |
| ‡ Current address: College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu, 212163, China. Email : E-mail: chenning@suda.edu.cn |
| § The calculations were carried out by using DFT methodology with the ADF 2010 package.45,46 The exchange-correlation functionals of Becke and Perdew were used.47,48 Relativistic corrections were included by means of the ZORA formalism. Slater triple-zeta polarization basis sets were employed to describe the valence electrons of C, S and Sc. Frozen cores consisting of the 1s shell for C and the 1s and 2p shells for S and Sc were described by means of single Slater functions. The computational study of the UV-Vis-NIR spectra has been performed using TDDFT methodology (also at BP86/TZP level). |
| This journal is © The Royal Society of Chemistry 2013 |