Intramolecular electron transfer in the photodimerisation product of a tetrathiafulvalene derivative in solution and on a surface†
Claudia
Simao
a,
Marta
Mas-Torrent
*a,
Vânia
André
b,
M. Teresa
Duarte
b,
Jaume
Veciana
a and
Concepció
Rovira
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC) and Networking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Campus Universitari de Bellaterra, Cerdanyola, E-08193, Barcelona, Spain. E-mail: mmas@icmab.es; cun@icmab.es; Fax: +34 935805729; Tel: +34 931888786
bCentro de Química Estrutural, Instituto Superior Técnico, Universidade Técnica de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal
First published on 10th September 2012
Abstract
Photodimerization of a TTF derivative gives rise to a dimeric molecule with a face-to-face structure that in its mixed-valence state exhibits a through space intramolecular electron transfer, both in solution and linked to a surface.
The electrical characteristics and supramolecular chemistry of tetrathiafulvalene (TTF) derivatives have made this family of molecules a key building block in organic electronics since its first discovery.1 A large variety of metallic and superconducting TTF complexes and salts have been reported2 and, more recently, TTFs have also been successfully employed as organic semiconductors in field-effect transistors.3 This molecular system has also shown great potential in molecular electronics, mainly acting as a π-electron donor in donor–acceptor (D–A) dyads4 and as molecular switch benefiting from its rich electrochemistry.5 Modifications of the TTFs have placed emphasis on obtaining dimeric molecules in which two TTF units are linked by one or more spacer groups.6 The appeal of these systems lies on their multi-stage redox behaviour and the possibility they offer to form a mixed-valence system exhibiting intramolecular electron transfer (IET) through bonds or through space.7 In addition, only very few linked donors have been obtained from TTF derivatives functionalised with electron-deficient groups that led to a [2 + 2] solid-state photocyclization.8,9 Solid-state reactions impose the formation of one isomer only and, therefore, constitute a powerful tool to synthesise new materials. Here we study the photodimerization product of a carboxymethyl derivatized TTF. In solution, an IET can be triggered by generating the mixed-valence state, but also self-assembled monolayers (SAMs) of a derivative of this material permit to investigate the formation of such a mixed-valence state in surface-grafted dimers, of significant importance for developing functional hybrid surfaces.
TTF derivative 1 bearing a carboxymethyl electro-deficient group and a benzene ring that promotes π–π interactions, was synthesised as previously reported.10 Crystals of 1 were grated to thin dust and kept in sunlight for 24 hours, resulting in the photodimer 2 in a 75% yield (Scheme 1). This reaction can be followed by the changes in IR and 1H NMR. A blue displacement in the IR spectra of the carbonyl stretch band was observed, from 1724 cm−1 in 1 to 1740 cm−1 in dimer 2. In the 1H NMR spectra, the aromatic benzene protons were shifted from 7.1–7.2 ppm to 6.8–6.9 ppm. Additionally, 2 exhibits a signal at 5.3 ppm attributable to the two protons of the cyclobutane ring.
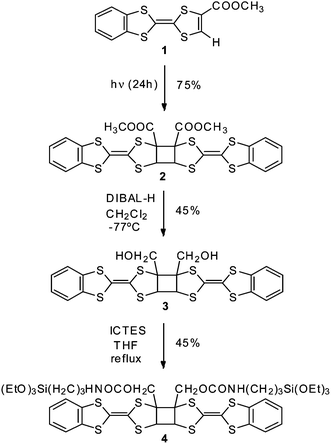 | ||
| Scheme 1 | ||
Single-crystals of 2 were obtained from crystallization in dichlorometane–ethanol. The molecule crystallizes in the monoclinic system. The structure shows unambiguously that the dihydrotetrathiafulvalene (TTFH)2 cores are constrained in a face-to-face structure and the esters moieties are in cis(Z) conformation (Fig. 1). The two TTFH core planes within the dimer are at a distance of 3.81(2) Å, slightly tilted between them, with an angle of 2.8(9)° (Fig. 1a). The crystal packing revealed dimerization of the photodimer molecules in a head-to-tail fashion along the longest molecular axis (c). These dimers based on short π–π interactions (3.77(3) Å) grow in a cisoid fashion and are isolated by the ethanol molecules (Fig. S1 and S2†). S⋯S interactions (dS⋯S = 3.60(2) Å) are established between molecules of adjacent layers. Due to the high reactivity under X-ray of the precursor TTF 1, it has not been possible to determine its crystal structure even at low temperature. This fact prevents to establish a correlation between the crystal packing of molecules of 1 in the crystals and the cisoid conformation obtained in dimer 2.
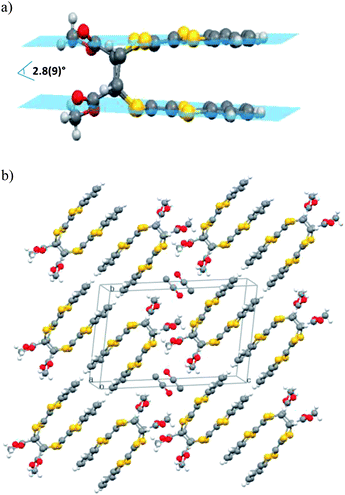 | ||
| Fig. 1 (a) Molecular structure of dimer 2 with a graphical representation of the TTFH core planes within the dimer, and (b) view of the packing in the single-crystal structure of compound 2. | ||
The electrochemical characteristics of 2 were investigated by cyclic voltammetry (CV) using as electrolyte a solution of 0.1 M tetrabutylammonium hexafluorophosphate (TBAHFP) in CH2Cl2. Two reversible redox waves were observed at E11/2 = +0.75 V and E21/2 = +1.00 V (vs. Ag/AgCl), which were attributed to the sequential one-electron oxidation of the two electroactive TTFH cores to the radical-cation species, as a consequence of the strong interaction between the two electroactive moieties in the close face-to-face configuration (Fig. 2).8c Further, the CV exhibited a third irreversible wave at E31/2 = +1.60 V corresponding to the formation of an unstable trication due to high electrostatic repulsion.
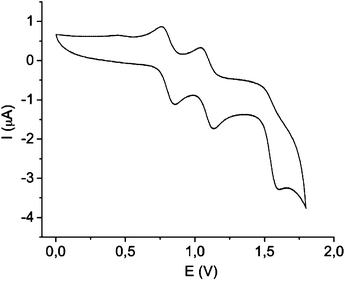 | ||
| Fig. 2 Cyclic voltammogram of 2 using as electrolyte a solution 0.1 M TBAHFP in dichloromethane, with platinum wires as working and counter electrodes and Ag/AgCl as the reference electrode. | ||
Taking into account the electrochemical results, the mixed-valence (MV) species was explored in order to study if an IET could take place in the radical-cation of the (TTFH)2. The controlled electrochemical oxidation of 2 in a mixture CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN (7:3) was carried out in a chronoamperometric experiment in steps of decimals of electrons (e−) and monitored in the UV-Vis-NIR spectrophotometer (Fig. 3). As soon as the oxidation of 2 started, the initial yellow solution immediately turned purple along with a charge transfer band appearing at 1786 nm, confirming the formation of the MV species and the existence of IET in this dyad. When the oxidation exceeded 1 electron per molecule, the decreasing of the charge-transfer band was observed at the same time that a new band at 867 nm appeared, corresponding to the formation of the diradical-dication [TTFH+˙]2. However, when the oxidation approaches 2 electrons per molecule, decomposition of the compound was observed, which is in accordance with the irreversible nature of the third wave in the CV (Fig. S3†). The solvent dependence of the charge transfer band position in the radical-cation of 2 generated chemically, with 1 equivalent of Fe(ClO4)3, was investigated employing solvents with different polarity. Generally, a blue-shift of the charge-transfer band with increasing polarity was observed, indicating a through space electron transfer (Fig. S4†). Following the Marcus–Hush theory,11 the donor–acceptor coupling parameter, HAB, which measures the electronic interaction between the two units, was estimated in CH2Cl2 to be approximately 1538 cm−1, and the reorganization parameter λT was found to be of 5534 cm−1. These values are in accordance with the values found in literature for TTF mixed valence encounter complexes12 and are in agreement with slow electron-transfer rates expected for a class II system of the Robin–Day classification.13
:CH3CN (7:3) was carried out in a chronoamperometric experiment in steps of decimals of electrons (e−) and monitored in the UV-Vis-NIR spectrophotometer (Fig. 3). As soon as the oxidation of 2 started, the initial yellow solution immediately turned purple along with a charge transfer band appearing at 1786 nm, confirming the formation of the MV species and the existence of IET in this dyad. When the oxidation exceeded 1 electron per molecule, the decreasing of the charge-transfer band was observed at the same time that a new band at 867 nm appeared, corresponding to the formation of the diradical-dication [TTFH+˙]2. However, when the oxidation approaches 2 electrons per molecule, decomposition of the compound was observed, which is in accordance with the irreversible nature of the third wave in the CV (Fig. S3†). The solvent dependence of the charge transfer band position in the radical-cation of 2 generated chemically, with 1 equivalent of Fe(ClO4)3, was investigated employing solvents with different polarity. Generally, a blue-shift of the charge-transfer band with increasing polarity was observed, indicating a through space electron transfer (Fig. S4†). Following the Marcus–Hush theory,11 the donor–acceptor coupling parameter, HAB, which measures the electronic interaction between the two units, was estimated in CH2Cl2 to be approximately 1538 cm−1, and the reorganization parameter λT was found to be of 5534 cm−1. These values are in accordance with the values found in literature for TTF mixed valence encounter complexes12 and are in agreement with slow electron-transfer rates expected for a class II system of the Robin–Day classification.13
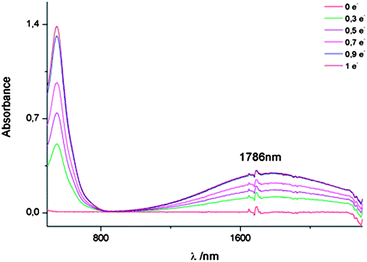 | ||
| Fig. 3 Vis-NIR spectra of compound 2 in its neutral state and at different stages of the chronoamperometric oxidation to reach the cation radical. | ||
The magnetic properties of the oxidised species of 2 were also investigated by Electron Paramagnetic Resonance (EPR). A series of EPR spectra were acquired whilst electrochemically oxidizing 2 within the EPR cell. When a voltage of +0.9 V (vs. Ag/AgCl) was applied one EPR line was observed at a g factor of 2.0060 and line-width 1.9 G in accordance with the formation of radical-cation species (Fig. S5†). Temperature variations showed a Curie–Weiss behaviour since the spectral line intensity increased with decreasing temperature. Nevertheless, since no hyperfine structure was resolved, the EPR technique did not give any further information concerning the IET process kinetics. Moreover, when a voltage of +1.3 V (vs. Ag/AgCl) was applied and the diradical-dication dimer was generated, the spectral line disappeared, pointing out an antiferromagnetic coupling of the two neighbour radicals within the dimer structure.
Considering the above, we can affirm that an IET can be promoted in compound 2 by a partial chemical oxidation or by the application of a specific electrochemical voltage that results in the formation of a MV species. In addition, at a longer stage of chemical oxidation or applying a higher voltage a stable diradical-dication species can be also generated. These results are very similar to the ones obtained when TTF face-to-face dimers are stabilized in solution inside “molecular flasks”.14
To go a step forward we propose to study if it is possible to preserve the same behaviour observed in solution in surface-anchored dimers. For this purpose, compound 2 was functionalised with a silane group in order to graft the molecule on an ITO substrate (Scheme 1). The conversion of esters groups in 2 to a primary alcohol was achieved by reduction with diisobutylaluminium hydride (DIBAL-H), obtaining derivative 3 in 45% yield. Finally, silane 4 was obtained by condensation of compound 3 with 3-isocyanatopropyltriethoxysilane (ICTES) using triethylamine in THF refluxing overnight (45% yield). Cyclic voltammetry of compound 4 is in accordance with the maintenance of the cis conformation of the dimeric core (Fig. S7a†).
Self-assembled monolayers (SAMs) of 4 on glass-coated with ITO were fabricated by immersion of the freshly cleaned substrates in a 1 mM solution of the molecule in dry toluene. SAM S-4 were fully characterized by contact angle, X-ray photoelectron spectroscopy and Time-of-flight Secondary Ion Mass Spectrometry (ToF-SIMS) (Fig. S6†). Similar to what was observed in solution, the CV using the functionalised ITO as a working electrode in a 0.02 M solution of TBAHFP in acetonitrile showed two reversible redox waves arising from the sequential oxidation to the radical-cation of the two TTFH cores in S-4 at 0.74 and 0.99 V (vs. Ag(s)). The voltage difference between the two oxidation processes is in agreement with the results obtained in solution (Fig. S7†). This indicates that the dimer in the cis configuration is preserved and, therefore, it is also possible to generate a MV state on surface. This has also been further demonstrated by the appearance of the charge transfer band corresponding to the MV state when the SAM S-4 was oxidized with an acetonitrile solution of Fe(ClO4)3 (Fig. S8†). To our knowledge, the observation of a MV state in TTF SAMs has only been achieved by the partial oxidation of a SAM upon the formation of a ionic salt or a donor–acceptor complex.15 In our case, the origin of the MV species is of purely intramolecular nature. This result is of high interest for the development of multistate switches16 or for the formation of a 2-dimensionally surface conducting SAM.
Further characterization of the different oxidation states of the cis dimer on surface was obtained by EPR. Thus, by applying the appropriate voltage (+0.9 and +1.5 V versus Ag(s)) to the functionalized surface for 2 minutes in the CV cell the cation-radical and diradical-dicationic species were formed, and then the EPR spectra of the SAM in air in each oxidation state were registered. As expected, a flat EPR baseline was observed in the neutral S-4 state that shows only the residual signal of the ITO substrate, while in the radical-cation MV SAM S-4a a broad EPR signal (ΔHpp = 9.5 Gauss) at g-factor 2.0062 was registered due to the paramagnetic character of the mixed-valence SAM (Fig. 4). Very importantly, such EPR signal disappeared when the diradical-dicationic state S-4b was formed, which demonstrated that on the surface the two TTF radical-cation moieties also interact antiferromagnetically between them, thus indicating that the short distance between the redox centers imposed by the cyclobutane ring is maintained. Subsequently, the starting neutral state of S-4 was recovered by applying a negative voltage of −0.2 V (versus Ag(s)). The switching between the three states could be then repeated for more than ten cycles (Fig. S9†). The EPR of the SAM changed according to the voltage profile applied recovering completely the line intensity of the MV cation radical state S-4a after each cycle, which elucidated that the SAMs of these dimeric molecules are fully stable as previously observed in similar TTF SAMs.10
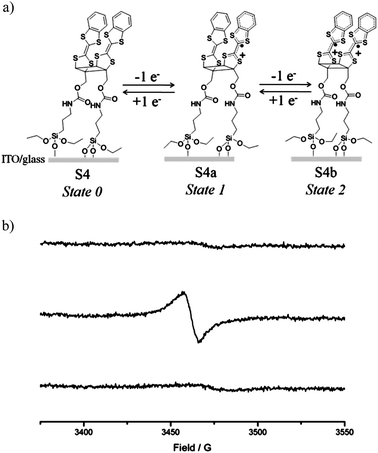 | ||
| Fig. 4 a) Schematic representation of the SAM of 4 in its three oxidation states and (b) EPR spectra of SAM S4 (top) and upon electrochemical oxidation applying potentials of +0.9 (middle) and +1.5 V (versus Ag(s)) (bottom). | ||
Conclusions
In summary, this work demonstrates that the dimer produced from a solid-state photodimerisation reaction of a TTF can lead to a MV state in solution that exhibits an intramolecular electron transfer. Further, synthesizing a derivative bearing an appropriate surface anchoring group, surface confined (TTFH)2 dimers have been prepared. These hybrid materials lead to charged states with the same spectroscopic characteristics as the ones observed in solution. Interestingly, they can be switched reversibly by applying the appropriate potential. We believe that the fabrication of functional surfaces exhibiting distinct redox states can provide the field of molecular electronics with novel perspectives.Acknowledgements
We thank the support of the EU by the EC FP7 ONE-P large-scale project (no. 212311) and the Marie Curie Est FuMASSEC, DGI, Spain (contract CTQ2010-195011/BQU), the Generalitat de Catalunya (2009SGR00516), the CIBER de Bioingeniera, Biomateriales y Nanomedicina (CIBER-BBN), promoted by ISCIII, Spain. We also thank Dr Vega Lloveras for the EPR measurements.Notes and references
- F. Wudl, G. M. Smith and E. J. Hufnagel, J. Chem. Soc. D, 1970, 1453 RSC.
- (a) J. M. Williams, J. R. Ferraro, R. J. Thorn, K. D. Carlson, U. Geiser, H. H. Wang, A. M. Kini and M.-H. Whangbo, Organic Superconductors (Including Fullerenes): Synthesis, Structure, Properties, and Theory, Prentice Hall, Englewood Cliffs, New Jersey, 1992 Search PubMed; (b) Special issue, Chem. Rev., 2004, 104, (Molecular Conductors); (c) J. S. Brooks, Chem. Soc. Rev., 2010, 39, 2667 CAS.
- (a) M. Mas-Torrent and C. Rovira, J. Mater. Chem., 2006, 16, 433 RSC; (b) M. Mas-Torrent and C. Rovira, Chem. Soc. Rev., 2008, 37, 827 RSC.
- J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS.
- D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC.
- (a) T. Otsubo, Y. Aso and K. Takimiya, Adv. Mater., 1996, 8, 203 CrossRef CAS; (b) J. Yamada and T. Sugimoto, TTF Chemistry, Kodanska Ltd. and Springer-Verlag, Tokyo, 2004 Search PubMed; (c) M. Iyoda, M. Hasegawa and Y. Miyake, Chem. Rev., 2004, 104, 5085 CrossRef CAS; (d) J. O. Jepessen and M. Brondsted, Chem. Rev., 2004, 104, 5115 CrossRef.
- (a) K. Lahlil, A. Moradpour, C. Bowlas, F. Menou, P. Cassoux, J. Bonvoisin, J.-P. Launay, G. Dive and D. Dehareng, J. Am. Chem. Soc., 1995, 117, 9995 CrossRef CAS; (b) C. Wang, A. Ellern, V. Khodorkovsky, J. Y. Becker and J. Bernstein, J. Chem. Soc., Chem. Commun., 1994, 2115 RSC; (c) M. R. Bryce, Adv. Mater., 1999, 11, 11 CrossRef CAS; (d) C.-Y. Jia, S.-X. Liu, C. Tanner, C. Leiggener, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. Commun., 2006, 1878 RSC; (e) P.-T. Chiang, N.-C. Chen, C.-C. Lai and S.-H. Chiu, Chem.–Eur. J., 2008, 14, 6546 CrossRef CAS.
- (a) Y. N. Kreitsberga, E. E. Liepinsh, I. B. Mazheika and O. Y. Neilands, Z. Org. Khim. (Engl. Trans.), 1986, 22, 367 Search PubMed; (b) T. Devic, P. Batail and N. Avarvari, Chem. Commun., 2004, 1538 RSC; (c) O. Jeannin and M. Formigué, Chem.–Eur. J., 2006, 12, 1994 CrossRef.
- Y. Sonoda, Molecules, 2011, 16, 119 CrossRef CAS.
- C. Simão, M. Mas-Torrent, J. Casado-Montenegro, F. Otón, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2011, 133, 13256 CrossRef.
- (a) N. S. Hush, Trans. Faraday Soc., 1961, 57, 557 RSC; (b) N. S. Hush, Prog. Inorg. Chem., 1967, 8, 391 CrossRef CAS.
- S. V. Rosokha and J.-K. Kochi, Acc. Chem. Res., 2008, 41, 641 CrossRef CAS.
- (a) P. Day, Endeavour, 1970, 29, 45 CAS; (b) M. Robin and P. Day, Adv. Inorg. Chem., 1967, 10, 247 CrossRef CAS.
- (a) A. Y. Ziganshina, Y. H. Ko, W. S. Jeon and K. Kim, Chem. Commun., 2004, 806 RSC; (b) A. Coskun, J. M. Spruell, G. Barin, A. C. Fahrenbach, R. S. Forgan, M. T. Colvin, R. Carmieli, D. Benítez, E. Tkatchouk, D. C. Friedman, A. A. Sarjeant, M. R. Wasielewski, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 4538 CrossRef CAS.
- (a) J. C. Goetz and C. P. Kubiak, J. Phys. Chem. C, 2008, 112, 8114 CrossRef; (b) E. J. Pacsial, D. Alexander, R. J. Alvarado, M. Tomasulo and F. M. Raymo, J. Phys. Chem. B, 2004, 108, 19307 CrossRef CAS; (c) R. Yuge, A. Miyazaki, T. Enoki, K. Tamada, F. Nakamura and M. Hara, J. Phys. Chem. B, 2002, 106, 6894 CrossRef CAS.
- M. Mas-Torrent, C. Rovira, J. Veciana, Adv. Mater. DOI:10.1002/adma.201201510.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data, UV-Vis and EPR data of 2, CV of 4 and S-4, characterization data of SAM S-4. CCDC reference number 876433. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21134c |
| This journal is © The Royal Society of Chemistry 2013 |