Inorganic topochemistry. Vapor-induced solid state transformations of luminescent, three-coordinate gold(I) complexes†
Sang Ho
Lim
,
Marilyn M.
Olmstead
and
Alan L.
Balch
*
Department of Chemistry, University of California, One Shields Avenue, Davis, CA, USA. E-mail: albalch@ucdavis.edu; Fax: +1 (530) 752 2820; Tel: +1 (530) 752 0941
First published on 10th September 2012
Abstract
The solid state interconversions of four different crystalline compounds containing the Au2(dppe)2I2 unit (dppe is bis-(diphenylphosphino)ethane) have been observed and characterized through X-ray diffraction and emission spectroscopy. Treatment of AuI in acetone with solid dppe yields the crystalline polymorphs: α-Au2(μ-dppe)2I2·2OCMe2 (1) with orange emission and β-Au2(μ-dppe)2I2·2OCMe2 (2) with green emission. Both polymorphs contain dimeric molecules with three-coordinate gold(I) centers that are further apart in the orange emitting (1) (Au⋯Au distance, 3.6720(2) Å) than in the green emitting (2) (Au⋯Au distance, 3.3955(2) Å). The acetone molecules do not bond to the gold centers in either polymorph. Crystals of α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) undergo reversible, single-crystal to single-crystal transformations. Exposure of (2) to acetone vapor converts it into (1), while (1) is converted into (2) by exposure to air for a short time. Upon loss of acetone, α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) are converted into a microcrystalline powder (3) with a green emission. Remarkably, exposure of (3) to acetone vapor converts it into acetone-free Au2(μ-dppe)2(μ-I)2 (4), which displays orange emission. The X-ray crystal structure of Au2(μ-dppe)2(μ-I)2 (4) shows that each gold is in a highly distorted tetrahedral environment with bonds to two phosphorus and two iodine atoms. The transformations between these four types of crystals are notable because of the unusual role of vapors in promoting structural changes within solids that do not necessarily change composition. Thus, the reversible interconversion of α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) occurs as vapor-stimulated single-crystal-to-single-crystal transformation between polymorphs. Likewise, the irreversible transformation of microcrystalline (3) into Au2(μ-dppe)2(μ-I)2 (4) appears to be another case of a transformation of one polymorph into another.
Introduction
Crystals are generally considered as fairly static, ordered arrays of atoms that provide us with some of the most detailed structural information on molecules, ionic solids and metals that can be obtained. However, it is also possible to observe structural changes and even chemical transformations within crystals. In regard to such topochemical changes, it has been postulated that reactions in crystals occur with a minimal amount of atomic movement.1 Generally, an appropriate stimulus is needed to instigate reactions in crystals. For example, ultraviolet irradiation can induce [2 + 2] cycloadditions between olefins in molecular crystals.2–7 Additionally, interconversions of polymorphs with attendant structural changes can be induced by thermal treatment of appropriate crystals.8 Application of mechanical pressure on certain crystalline metal complexes can produce marked changes in their luminescence, color and structure.9–11Exposure of a number of crystalline substances to vapors of water and/or volatile organic compounds can lead to alteration and sometimes destruction of the crystals. Usually, such processes are accompanied by the uptake of the vapor into the solid. For example, solid desiccants function by absorbing water. Solid anhydrous calcium sulfate (Drierite) forms the hemi-hydrate, CaSO4·0.5H2O, while crystalline P4O10 is converted into liquid phosphoric acid upon exposure to water vapor.12 Extensive efforts are being made to develop crystalline solids with persistent voids from organic or metal–organic frameworks that can be used for the uptake of various gases.13,14 Solid carboxylic acids will react with vapors of ammonia and other amines to form crystalline salts, again with uptake of the vapor.15,16 The magnetic properties of the spin-crossover iron complex, Fe(tris(2-pyridylmethyl)amine)(NCS)2, can be altered by the absorption of methanol vapor.17 Certain transition metal complexes, particularly those with metallophilic interactions between metal centers with d8 and d10 electronic configurations,18 change their luminescence or color in response to exposure to vapors of volatile organic compounds. For example, the orange form of cis-(p-EtC6H4CN)2Pt(CN)2 absorbs vapors of aromatic hydrocarbons like toluene to form yellow crystals of cis-(p-EtC6H4CN)2Pt(CN)2·0.5(toluene), a process that alters the interactions between the platinum ions in the solid.19 Similarly a platinum terpyridine complex undergoes a reversible red–orange luminescence change upon uptake and loss of methanol, which again alters the Pt⋯Pt separations.20 Metal chain complexes such as {Tl[Au(C6Cl5)2]}n are vapochromic due to coordination of the volatile compounds to the metal ions in the chain that alter the interactions between the metal ions.21–25 Such vapoluminescent and vapochromic materials have the potential to act as sensors for various volatile molecules.26,27
Here, we report the discovery of some luminescent gold complexes whose crystals undergo changes in luminescence and structure in response to exposure to organic vapors without the uptake of the vapor itself. This work utilizes the flexible, accordion-like structure of the Au2(μ-dppe)2X2 unit (dppe is bis-(diphenylphosphino)ethane).28 A number of other smaller bite diphosphines such as bis-(diphenylphosphino)methane (dppm) can position gold atoms in close proximity,29 but dppe shows a degree of flexibility in accommodating different Au⋯Au separations that is unusual. Previously, we documented the vapoluminescent behavior of crystals containing the dimer, Au2(μ-dppe)2Br2, and described processes that required the uptake and loss of dichloromethane or acetone. That work revealed that Au2(μ-dppe)2Br2 has the flexibility to accommodate Au⋯Au separations that range from 3.8479(3) to 3.0995(10) Å.20 Variations in the Au⋯Au separations signal changes in the aurophilic interactions between the metal centers30 and are well known to produce alterations in the luminescence of gold(I) complexes.31,32
Results and discussion
Structures and reversible interconversions of the polymorphs α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2)
Treatment of a suspension of AuI in acetone with solid dppe resulted in the formation of a colorless solution, which was filtered and evaporated to dryness. The resulting solid was redissolved in a minimum volume of acetone. The solution was transferred to a 5 mm diameter glass tube and layered with diethyl ether. Colorless crystals of α-Au2(μ-dppe)2I2·2OCMe2 (1), which have an orange emission (emission, λem, 607 nm, excitation, λex, 364 nm) grew along with colorless crystals of β-Au2(μ-dppe)2I2·2OCMe2 (2), which have a green emission (λem, 577 nm, excitation, λex, 368 nm). Thus, these crystals are concomitant polymorphs.33The structures of α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2), are shown in Fig. 1 and 2, respectively. Crystal data are given in Table 1. In both cases, the dimeric Au2(μ-dppe)2I2 molecule packs about a center of symmetry so that one half of the dimer resides in the asymmetric unit. The gold centers are three-coordinate with bonds to two phosphorus atoms of two different bridging ligands and to the terminal iodide ligand. The acetone molecules do not bond to the gold centers in either polymorph. The major differences between the two structures involve the Au⋯Au distances which are 3.6720(2) Å in the orange-glowing α-polymorph and significantly shorter, 3.3955(2) Å, in the green-glowing β-polymorph. At the shorter distance in (2), aurophilic interactions become significant and alter the luminescence of the complex. Other differences between the polymorphs include a lengthening of the Au–I distance in the β-polymorph, changes in the orientations of the phenyl rings and shifts in the positions of the acetone molecules. The structure and flexibility of Au2(μ-dppe)2I2 parallels that of Au2(μ-dppe)2Br2, which was reported earlier.20
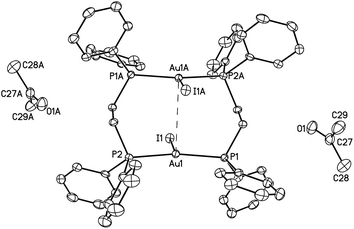 | ||
| Fig. 1 A drawing of the structure of orange-glowing, α-Au2(μ-dppe)2I2·2OCMe2 (1). Thermal ellipsoids are shown at the 50% probability level. The Au1⋯Au1A separation is 3.6720(2) Å. Other distances; Au1–I1, 2.9106(2); Au1–P1, 2.3157(6); Au1–P2, 2.3118(6) Å. | ||
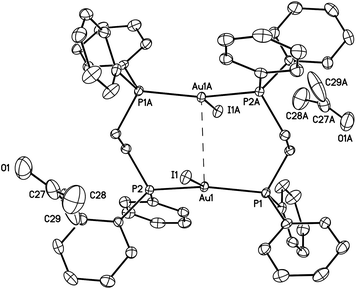 | ||
| Fig. 2 A drawing of the structure of green-glowing β-Au2(μ-dppe)2I2·2OCMe2 (2). Thermal ellipsoids are shown at the 50% probability level. The Au1⋯Au1A separation is 3.3955(2) Å, which is shorter than the corresponding separation (3.6720(2) Å) in the α-polymorph (I). Other distances; Au1–I1, 2.97590(18); Au1–P1, 2.3143(5); Au1–P2, 2.3177(5) Å. | ||
| α-Au2(μ-dppe)2I2·2OCMe2 (1) | β-Au2(μ-dppe)2I2·2OCMe2 (2) | Au2(μ-dppe)2(μ-I)2 (4) | |
|---|---|---|---|
| a For data with I > 2σ(I): R1 = ∑||Fo| − |Fc||/∑|Fo|. b For all data: wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | |||
| Formula | C58H60Au2I2O2P4 | C140H60N4NiSm2 | C52H40Au2I2P4 |
| Fw | 1560.67 | 1560.67 | 1436.45 |
| Color, habit | Colorless block | Colorless block | Colorless protopyramid |
| Crystal system | Monoclinic | Triclinic | Tetragonal |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
I41/a |
| a, Å | 10.6771(3) | 11.8300(3) | 16.0859(4) |
| b, Å | 22.8740(6) | 11.9725(3) | 16.0859(4) |
| c, Å | 12.0196(3) | 12.3591(4) | 36.7500(9) |
| α, deg | 90 | 65.098(2) | 90 |
| β, deg | 105.824(2) | 69.838(2) | 97.638(2) |
| γ, deg | 90 | 65.309(2) | 90 |
| V, Å3 | 2824.28(13) | 1411.99(7) Å | 9509.3(4) |
| Z | 2 | 1 | 8 |
| ρ calcd, Mg m−3 | 1.835 | 1.835 | 2.007 |
| T, K | 90(2) | 90(2) | 90(2) |
| Radiation (λ, Å) | Mo-Kα, 0.71073 Å | Mo-Kα, 0.71073 Å | Mo-Kα, 0.71073 Å |
| Unique data | 8659 [R(int) = 0.035] | 8630 [R(int) = 0.020] | 5464 [R(int) = 0.046] |
| Parameters | 309 | 309 | 137 |
| Obsd (I > 2σ(I)) data | 7303 | 7969 | 4428 |
| R 1 a (obsd data) | 0.034 | 0.019 | 0.039 |
| wR2b (all data) | 0.047 | 0.041 | 0.095 |
Remarkably, the α and β polymorphs of Au2(μ-dppe)2I2·2OCMe2 can be reversibly interconverted. Fig. 3 shows photographs of crystals of the α-polymorph of Au2(μ-dppe)2I2·2OCMe2 (1) under (a) ambient light and (b) under UV irradiation. Gentle drying of the crystals by simple exposure to air over a 30 minute period results in their transformation into the green-glowing β-polymorph as seen in (d). When these green-glowing crystals are exposed to acetone or dichloromethane vapor, they are converted back into the orange-glowing α-polymorph as seen in (c). As the photograph indicates these transformations are reproducible for an array of different crystals.
 | ||
| Fig. 3 Photographs of crystals of α-Au2(μ-dppe)2I2·2OCMe2 (1) under (a) ambient light and (b) under UV irradiation. After the crystals are briefly allowed to stand in air, the green-emissive sample shown in (d) was obtained. Exposure of that green-glowing sample to acetone vapor restores the orange emission as shown in (c). | ||
The interconversion of α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) has been monitored by X-ray powder diffraction as shown in Fig. 4. Traces (A) and (B) show powder pattern computed from the single-crystal data and the experimental pattern for a sample of α-Au2(μ-dppe)2I2·2OCMe2 (1). After the sample stood in air until a uniform green emission was observed, the sample was exposed to acetone vapor and the powder pattern shown in (C) was obtained. This result indicates that α-Au2(μ-dppe)2I2·2OCMe2 (1) was reformed after the sample had been converted into the green-glowing β-Au2(μ-dppe)2I2·2OCMe2 (2). Unfortunately, powder diffraction data could not be obtained for β-Au2(μ-dppe)2I2·2OCMe2 (2), since the finely divided material rapidly lost crystallinity. However, as the next paragraph shows, larger individual crystals of β-Au2(μ-dppe)2I2·2OCMe2 (2) could be examined.
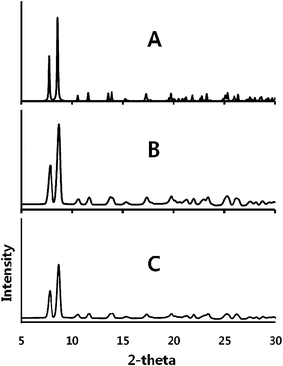 | ||
| Fig. 4 X-ray powder diffraction showing the interconversion of α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) and back to (1). (A) Pattern computed for α-Au2(μ-dppe)2I2·2OCMe2 (1) from single-crystal data. (B) Experimental data for α-Au2(μ-dppe)2I2·2OCMe2 (1). (C) Experimental data for the sample that produced the data in trace B after standing in air until the sample produced green emission to form (2) followed by exposure to acetone vapor, which resulted in orange emission. Experimental data were collected with Cu Kα radiation at 90(2) K. | ||
These transformations have also been monitored by single-crystal X-ray diffraction on an individual crystal. Initial diffraction data from a crystal of α-Au2(μ-dppe)2I2·2OCMe2 (1) confirmed that the crystal was monoclinic, space group P21/c. After removal from the diffractometer and subsequent air exposure, the diffraction data indicated that a single-crystal-to-single-crystal transformation had occurred and that the crystal now belonged the triclinic space group P, with cell parameters consistent with the transformation into β-Au2(μ-dppe)2I2·2OCMe2 (2). Subsequently, the crystal was again removed from the diffractometer and exposed to acetone vapor as described in the Experimental section. After remounting of the crystal on the diffractometer, conversion of the crystal back into the original monoclinic P21/c form was observed. An entire new data set was acquired at this point, and its solution agreed with that obtained previously for the α-polymorph.
Irreversible solvate loss from α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2)
A second type of transformation occurs when crystals of the α- or β-polymorphs are allowed to stand in air for several hours. Under these conditions, the crystals crumble into a colorless powder of compound (3), which displays a green luminescence (λem, 570 nm, excitation, λex, 395 nm). A comparison of the emission and excitation spectra of the three compounds, α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) and compound (3), is shown in Fig. 5. The process is accompanied by a loss of acetone from the crystals. The infrared spectrum of the α-polymorph shows a strong band at 1702 cm−1 due to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) from acetone. Those of the β-polymorph display a corresponding band at 1700 cm−1. Upon standing in air, these prominent features disappear and are absent in the infrared spectrum of the colorless, green-luminescent powder of compound (3).
O) from acetone. Those of the β-polymorph display a corresponding band at 1700 cm−1. Upon standing in air, these prominent features disappear and are absent in the infrared spectrum of the colorless, green-luminescent powder of compound (3).
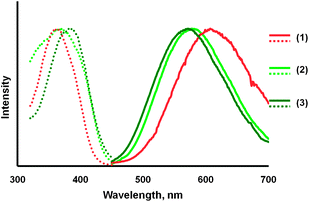 | ||
| Fig. 5 Emission (solid lines) and excitation (dotted lines) spectra for: orange-glowing, α-Au2(μ-dppe)2I2·2OCMe2 (1) in orange; green-glowing β-Au2(μ-dppe)2I2·2OCMe2 (2) in light green, and the green-glowing, colorless powder (3) obtained by drying α-Au2(μ-dppe)2I2·2OCMe2 for three hours in air in dark green. | ||
This second transformation has also been monitored by X-ray powder diffraction as shown in Fig. 6. Trace (A) shows the powder diffraction from a crushed sample of α-Au2(μ-dppe)2I2·2OCMe2 (1). This pattern agrees with the diffraction pattern computed from the single-crystal diffraction data as can be seen in Fig. 4. Upon prolonged exposure to air, the powder diffraction pattern seen in trace (B) of Fig. 6 is obtained. The sample, compound (3), shows evidence of crystallinity, but the observed diffraction data do not match those of either α- or β-Au2(μ-dppe)2I2·2OCMe2. Solvate-free compound (3) melts at 255–259 °C.
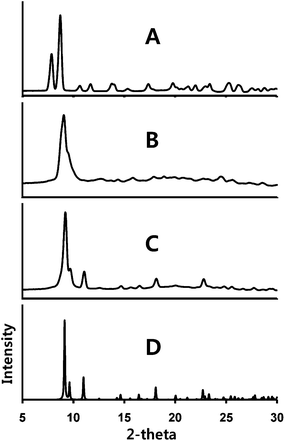 | ||
| Fig. 6 X-ray powder diffraction from: (A) a sample of α-Au2(μ-dppe)2I2·2OCMe2 (1); (B) the same sample after standing in air; (C) the sample from (B) after exposure to acetone vapor. (D) The computed powder pattern obtained from the single-crystal diffraction data for solvate-free Au2(μ-dppe)2(μ-I)2 (4). Experimental data were collected with Cu Kα radiation at 90(2) K. | ||
Exposure of the green-glowing powder (3) to acetone vapor results in a third transformation into an orange-luminescent powder. Acetone is not incorporated into this powder since the infrared spectrum does not show a band at ca. 1700 cm−1. The X-ray powder diffraction data for this form are shown in trace (C) of Fig. 4. The pattern is distinct from that of its immediate predecessor (trace (B)) and from those of either α- or β-Au2(μ-dppe)2I2·2OCMe2. However, the powder pattern does correspond to that of another crystalline complex, solvate-free Au2(μ-dppe)2(μ-I)2 (4).
In separate experiments, uniquely shaped, bipyramidal crystals of Au2(μ-dppe)2(μ-I)2 (4) shown in Fig. 7 were obtained by layering diethyl ether or n-pentane over a solution of Au(dppe)I in dichloromethane, acetone, chloroform, dimethylformamide or dimethyl sulfoxide. These crystals grew in the diethyl ether- or pentane-rich region near the upper part of the tube. Under UV irradiation, these colorless crystals produced an orange emission (λem, 602 nm, excitation, λex, 380 nm).
 | ||
| Fig. 7 Photographs of crystals of solvate-free Au2(μ-dppe)2(μ-I)2 (4) under (A) ambient light and (B) under UV light. | ||
The structure of Au2(μ-dppe)2(μ-I)2 (4) as determined by single-crystal X-ray diffraction is shown in Fig. 8. A crystallographic two-fold axis runs through I1 and I2. The gold ion is coordinated to two phosphorus atoms from different bridging ligands and by two iodide ions to form a highly distorted tetrahedral arrangement. The Au1⋯Au1A separation is 3.3926(5) Å. The Au–I distances (3.2116(5), 3.2030(5) Å) are nearly equivalent, but significantly longer than the corresponding distances (α, 2.9106(2); β, 2.97590(18) Å) in both polymorphs of three-coordinate Au2(μ-dppe)2I2·2OCMe2. In the related binuclear cation, [Au2(μ-dppm)2(μ-I)]+, the Au–I distances are 3.127(2) and 3.196(2) Å.34 Nevertheless, the iodide ligands in Au2(μ-dppe)2(μ-I)2 (4) must be considered bonded to the gold ions. The cation, [Au2(μ-dppe)2]2+, has been isolated as a salt with a non-coordinating anion and was found to have a much shorter Au⋯Au separation of 2.9220(3) Å and nearly linear P–Au–P angles.35
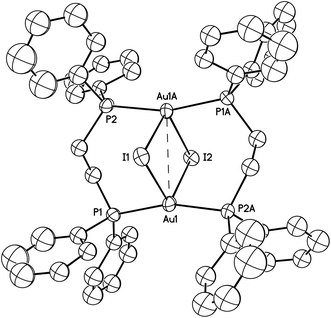 | ||
| Fig. 8 A drawing of the structure of orange-glowing Au2(μ-dppe)2(μ-I)2 (4). The Au1⋯Au1A separation is 3.3926(5) Å. Other distances and angles are: Au1–I1, 3.2116(5); Au1-I2, 3.2030(5); Au1–P1, 2.3060(16); Au1–P2, 2.3042(16) Å; Au1–I1–Au1A, 63.767(14); Au1–I2–Au1, 63.958(14); P1–Au1–P2A, 163.14(6). Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms are not shown for clarity. | ||
Crystals of Au2(μ-dppe)2(μ-I)2 (4) do not show any changes upon standing in air and are not altered by exposure to acetone or dichloromethane vapor. Solvate-free crystals of (4) melt at 277–280 °C. The higher melting point of (4) relative to (3) suggests that (4) is more stable than (3), a situation compatible with the observation that (3) is converted into (4) through exposure to acetone vapor.
Conclusions
Scheme 1 summarizes the transformations among crystals that we have observed. The interconversions of α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) polymorphs are reversible single-crystal-to-single-crystal transformations. Prolonged exposure of β-Au2(μ-dppe)2I2·2OCMe2 (2) to air results in the irreversible loss of acetone and conversion into microcrystalline (3). The detailed structure of (3) remains to be determined, but it is likely to contain the Au2(μ-dppe)2I2 unit with a short Au⋯Au contact, since its luminescence resembles that of β-Au2(μ-dppe)2I2 (2). Strangely, exposure of (3) to acetone or dichloromethane vapor results in its conversion into Au2(μ-dppe)2(μ-I)2 (4) without uptake of the vapor. If the structure of (3) contains Au2(μ-dppe)2I2 molecules with dimensions similar to those of β-Au2(μ-dppe)2I2·2OCMe2 (2), then the iodide ligands must reposition themselves between the two gold ions to form the structure shown in Fig. 5. To accomplish this transformation, the Au1–I1 distance in (4) (2.97590(18) Å) must lengthen while the I1–Au1A distance (3.7144(2) Å) must shorten to produce Au2(μ-dppe)2(μ-I)2 (4) with Au1–I1 and Au1–I2 distances of 3.2116(5) and 3.2030(5) Å, respectively. While the Au1⋯Au1A separations in β-Au2(μ-dppe)2I2·2OCMe2 (2) (3.3955(2) Å) and Au2(μ-dppe)2(μ-I)2 (4) (3.3926(5) Å) are similar, the bridging diphosphine ligands must undergo some twisting to reach the arrangement found in Au2(μ-dppe)2(μ-I)2 (4). Nevertheless, the key features in these solid state transformations reflect the ability of the bridging dppe ligand place the reacting centers close to one another and the flexibility of the Au2(μ-dppe)2 framework.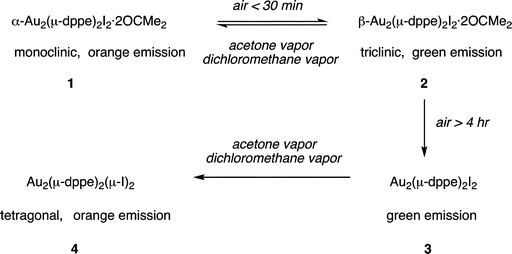 | ||
| Scheme 1 Solid state transformations of crystalline gold(I) complexes. | ||
The transformations between these four types of crystals are notable because of the unusual role of vapors in promoting structural changes within solids that do not necessarily change composition. Thus, the reversible interconversion of α-Au2(μ-dppe)2I2·2OCMe2 (1) and β-Au2(μ-dppe)2I2·2OCMe2 (2) occurs as vapor-stimulated single-crystal-to-single-crystal transformation between polymorphs. Likewise, the irreversible transformation of microcrystalline (3) into Au2(μ-dppe)2(μ-I)2 (4) appears to be another case of a transformation of one polymorph into another.
At this stage, we have uncovered and documented some remarkable solid state transformations of the accordion-like dimer Au2(μ-dppe)2I2. Additionally, we can offer some speculation in regard to how these transformations might occur. It appears likely that the vapor interacts with the surface of the crystals, perhaps by adsorption, to initiate the transformation. Finely ground samples of these crystals undergo these changes more rapidly than large crystals, an observation consistent with the idea that vapor may be transiently adsorbed on the surface of these crystals.
Three-coordinate gold(I) complexes, such as those encountered in this study, are frequently luminescent both in the solid state and in solution.36–38 For such three-coordinate gold(I) compounds, the excitation process is generally attributed to a transition from the filled, in-plane dx2−y2,dxy orbitals on gold to the empty, out-of-plane pz orbital. Emission occurs from a corresponding triplet state, which may undergo significant distortion for the ground state geometry.39 Several recent experimental40–42 and computational33,43 studies have examined the various types of structural changes that occur in the excited states of these three-coordinate complexes. Thus, the different emission seen in (1) and (2) results from the differences in the P–Au–P bond angles, the differences in Au–I distances and the differences in proximity of the two gold ions. It is entirely likely the nature of the emissive state changes significantly depending upon whether there is or is not an aurophilic interaction between the two gold ions.
Clearly, the luminescence from these otherwise colorless crystals aided in our ability to identify the changes that are reported here. One wonders whether this sort of behavior might be more widespread and other vapor-related polymorphic phase changes might be going unnoticed in non-luminescent crystals. Control of polymorph formation and polymorph stability is frequently a challenging issue.44–46 The phenomenon of disappearing polymorphs is one manifestation of these difficulties.47 Vapor exposure of certain crystalline polymorphs may be an important issue in determining their apparent stability.
Experimental section
Preparation of α-Au2(μ-dppe)2I2·2OCMe2, β-Au2(μ-dppe)2I2·2OCMe2, and solvate-free Au2(μ-dppe)2(μ-I)2
AuI (100 mg, 0.309 mmol) was suspended in 30 mL of acetone. While stirring, solid dppe 185 mg (0.463 mmol) was added. After additional stirring for 3 h, the solution was filtered, and then the solvent was removed in a vacuum. The white solid, Au(dppe)I, was collected and washed with diethyl ether. The yield was 180 mg (80.6%). Crystals suitable for X-ray structure determination were grown by slow diffusion of diethyl ether to a solution of the colorless solid in acetone in a 5 mm outside diameter glass tube. Colorless, orange and green emitting crystals of Au2(μ-dppe)2Br2·2OCMe2 appeared in the lower portion of the tube and were identified by their characteristic emission and external morphology. Distinctively shaped, colorless bipyramids of solvate-free Au2(μ-dppe)2(μ-I)2 grew in the upper, diethyl ether-rich region of the tube. The individual types of crystals were separated manually using the differences in emission color, external morphology and spatial separation during crystal growth to identify the various phases. Crystals of solvate-free Au2(μ-dppe)2(μ-I)2 could also be obtained from solutions of Au(dppe)I in dichloromethane, chloroform, dimethylformamide, and dimethyl sulfoxide that were layered with diethyl ether or pentane. In these cases the crystals of Au2(μ-dppe)2(μ-I)2 always grew in the upper, diethyl ether- or pentane-rich region of the tubes.Infrared spectra
Orange emissive α-Au2(μ-dppe)2Br2·2OCMe2: 3050w, 3002w, 2903w, 1700vs (ν(CO) from acetone), 1482m, 1435s, 1411m, 1224w, 1185m, 1120m, 1099m, 1070m, 1026m, 997w, 820w, 742s, 725m, 693s, 532m, 521m, 509s, 486m cm−1.
Green emissive β-Au2(μ-dppe)2Br2·2OCMe2: 3051w, 3003w, 2905w, 1702vs (ν(CO) from acetone), 1482m, 1435s, 1411m, 1307w, 1223m, 1184m, 1121m, 1099m, 1070m, 1026m, 998w, 871w, 820w, 742s, 725m, 693s, 532m, 521m, 510s, 486m cm−1.
Orange emissive Au2(μ-dppe)2(μ-I)2: 3069w, 3046w, 2927w, 2892w, 1478m, 1433s, 1098m, 1069w, 995w, 824m, 742m, 715w, 688s, 616m, 508s, 479m cm−1.
Physical measurements
Infrared spectra were recorded on a Bruker ALPHA FT-IR spectrometer. Fluorescence excitation and emission spectra were recorded on a Perkin-Elmer LS50B luminescence spectrophotometer.Interconversion of α-Au2(μ-dppe)2I2·2OCMe2 and β-Au2(μ-dppe)2I2·2OCMe2
Colorless, orange emitting crystals of α-Au2(μ-dppe)2I2·2OCMe2 were placed on a glass slide and allowed to stand in air for 30 min. During this period the crystals retained their clarity and shape, but were transformed into β-Au2(μ-dppe)2I2·2OCMe2 with green emission. The slide was then placed in a jar that contained a few drops of acetone that provide the vapor needed to convert α-Au2(μ-dppe)2I2·2OCMe2 into β-Au2(μ-dppe)2I2·2OCMe2.Single-crystal X-ray crystallography and data collection
The crystals were removed from the glass tubes in which they were grown together with a small amount of mother liquor and immediately coated with a hydrocarbon oil on a microscope slide. A suitable crystal of each compound was mounted on a glass fiber with silicone grease and placed in the cold stream of a Bruker SMART Apex II CCD with graphite monochromated Mo Kα radiation at 90(2) K. Crystal data can be found in Table 1.The structures were solved by direct methods and refined using all data (based on F2) using the software SHELX multi-scan method utilizing equivalents was employed to correct for absorptions.48 Hydrogen atoms were added geometrically and refined with a riding model.
Crystal disorder in solvate-free Au2(μ-dppe)2(μ-I)2
The structure has two kinds of disorder. There is a difference map peak that probably stems from rotation of the molecule by 90° and gives rise to an alternative Au⋯Au vector. The occupancy for the second orientation is only based on this one peak. Other atoms corresponding to this orientation are not likely to show up since the defect is only 3%. The positions of I1 and I2 are compatible with the alternative Au site. The refined occupancies for the two Au sites are: Au1/Au2 0.9688(7)/0.0312(7). The Au1⋯Au1A distance is 3.393 Å while that of Au2⋯Au2B is 3.504 Å. The second kind of disorder arises from rotational disorder of the phenyl rings bonded to P. Three of the four phenyl rings were modeled with a second orientation. Thus C1–C6 has disorder 0.707(11)/0.293(11), C15–C20 has disorder 0.571(12)/0.429(12) and C21–C26 has disorder 0.680(9)/0.320(9). In each case the six-membered ring was refined using AFIX 66 restraints. The carbon atoms were kept isotropic.X-ray powder diffraction data collection
Each sample was finely ground with a mortar and pestle as quickly as possible and immediately mixed with minimum amount of hydrocarbon oil. This mixture was mounted on a polymer micro-loop and placed in the 90(2) K cold stream of a Bruker Apex Duo. Powder diffraction data were collected with the use of Cu-Kα radiation and the program PILOT.49Acknowledgements
We thank the Petroleum Research Fund (Grant 37056-AC to ALB) and the U. S. National Science Foundation [CHE-1011760 to ALB and MMO]References
- M. D. Cohen and G. M. J. Schmidt, J. Chem. Soc., 1964, 1996–2000 RSC.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC.
- G. M. J. Schmidt, J. Chem. Soc., 1964, 2014–2021 RSC.
- L. R. MacGillivray, CrystEngComm, 2002, 4, 37–41 RSC.
- V. Enkelmann, G. Wegner, K. Novak and K. B. Wagener, J. Am. Chem. Soc., 1993, 115, 10390–10391 CrossRef CAS.
- X. Gao, T. Friščić and L. R. MacGillivray, Angew. Chem., Int. Ed., 2004, 43, 232–236 CrossRef CAS.
- I. Abdelmoty, V. Buchholz, L. Di, C. Guo, K. Kowitz, V. Enkelmann, G. Wegner and B. M. Foxman, Cryst. Growth Des., 2005, 5, 2210–2217 CAS.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2008 Search PubMed.
- D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002–4011 CrossRef CAS.
- A. L. Balch, Angew. Chem., Int. Ed., 2009, 38, 2641–2644 CrossRef.
- D. Braga and F. Grepioni, Chem. Commun., 2005, 3635–3645 RSC.
- W. L. F. Armarego and C. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Maryland Heights, MO, 6th edn, 2009, p. 28 Search PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1507 RSC.
- I. C. Paul and D. Y. Curtin, Science, 1975, 187, 19–26 CAS.
- D. Braga, F. Grepioni, M. Polito, M. R. Chierotti, S. Ellena and R. Gobetto, Organometallics, 2006, 25, 4627–4633 CrossRef CAS.
- B. Li, R.-J. Wei, J. Tao, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2010, 132, 1558–1566 CrossRef CAS.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef.
- C. E. Buss and K. R. Mann, J. Am. Chem. Soc., 2002, 124, 1031–1039 CrossRef CAS.
- T. J. Wadas, Q.-M. Wang, Y. Kim, C. Flashenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef CAS.
- E. J. Fernandez, J. M. Lopez-de-Luzuriaga, M. Monge, M. E. Olmos, J. Perez, A. Laguna, A. A. Mohamed and J. P. Fackler, Jr, J. Am. Chem. Soc., 2003, 125, 2022–2023 CrossRef CAS.
- E. J. Fernandez, J. M. Lopez-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos, J. Perez, A. Laguna, F. Mendizabal, A. A. Mohamed and J. P. Fackler, Jr, Inorg. Chem., 2004, 43, 3573–3581 CrossRef CAS.
- T. Lasanta, M. E. Olmos, A. Laguna, J. M. López-de-Luzuriaga and P. Naumov, J. Am. Chem. Soc., 2011, 133, 16358–16361 CrossRef CAS.
- A. Laguna, T. Lasanta, J. M. López-de-Luzuriaga, M. Monge, P. Naumov and M. E. Olmos, J. Am. Chem. Soc., 2010, 132, 456–457 CrossRef CAS.
- C. E. Strasser and V. J. Catalano, J. Am. Chem. Soc., 2010, 132, 10009–10011 CrossRef CAS.
- M. N. Fiddler, I. Begashaw, M. A. Mickens, M. S. Collingwood, Z. Assefa and S. Bililign, Sensors, 2009, 9, 10447–10512 CrossRef CAS.
- X. He and V. W. W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS.
- S. H. Lim, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2011, 133, 10229–10238 CrossRef CAS.
- (a) L.-S. Llou, C.-P. Liu and J.-C. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 538–540 CrossRef; (b) H. Schmidbaur, R. Herr, G. Müller and J. Riede, Organometallics, 1985, 4, 1208–1213 CrossRef CAS; (c) H. Schmidbaur, T. Pollok, G. Reber and G. Müller, Chem. Ber., 1988, 121, 1345–1348 CrossRef CAS; (d) J. B. Foley, S. E. Gay, M. J. Vela, B. M. Foxman, A. E. Bruce and M. R. M. Bruce, Eur. J. Inorg. Chem., 2007, 4946–4951 CrossRef CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- A. L. Balch, Struct. Bonding, 2007, 123, 1–40 CrossRef CAS.
- V. W. W. Yam and E. C. C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC.
- J. Bernstein, R. J. Davey and J.-O. Henck, Angew. Chem., Int. Ed., 1999, 38, 3440–3461 CrossRef.
- J. Shain and J. P. Fackler, Jr, Inorg. Chim. Acta, 1987, 131, 157–158 CrossRef CAS.
- C. E. Strasser, S. Cronje and H. G. Raubenheimer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m914 Search PubMed.
- R. Ziolo, S. Lipton and Z. Dori, Chem. Commun., 1970, 1124–1125 RSC.
- T. M. McCleskey and H. B. Gray, Inorg. Chem., 1992, 31, 1733–1734 CrossRef CAS.
- C. King, M. N. I. Khan, R. J. Staples and J. P. Fackler, Jr, Inorg. Chem., 1992, 31, 3236–3238 CrossRef CAS.
- K. A. Barakat, T. R. Cundari and M. A. Omary, J. Am. Chem. Soc., 2003, 125, 14228–14229 CrossRef CAS.
- M. Hoshino, H. Uekusa and Y. Ohashi, Bull. Chem. Soc. Jpn., 2006, 79, 1362–1366 CrossRef CAS.
- M. Hoshino, H. Uekusa, S. Sonoda, T. Otsuka and Y. Kaizu, Dalton Trans., 2009, 3085–3091 RSC.
- M. Hoshino, H. Uekusa, S. Ishii, T. Otsuka, Y. Kaizu, Y. Ozawa and K. Toriumi, Inorg. Chem., 2010, 49, 7257–7265 CrossRef CAS.
- P. Sinha, A. K. Wilson and M. A. Omary, J. Am. Chem. Soc., 2005, 127, 12488–12489 CrossRef CAS.
- R. W. Lancaster, L. D. Harris and D. Pearson, CrystEngComm, 2011, 13, 1775–1777 RSC.
- N. Blagden and R. J. Davey, Cryst. Growth Des., 2003, 3, 873–885 CAS.
- R. J. Davey, Chem. Commun., 2003, 1463–1467 RSC.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- SADABS 2.0, G. M. Sheldrick, based on a method of R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- PILOT, XRD2Eval, Bruker-Axs, Inc., 2012, Madison, WI Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files. CCDC 840562–840564. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc20820b |
| This journal is © The Royal Society of Chemistry 2013 |