Remodeling a β-peptide bundle†
Matthew A.
Molski
a,
Jessica L.
Goodman
b,
Fang-Chieh
Chou
c,
David
Baker
*d,
Rhiju
Das
*c and
Alanna
Schepartz
*ae
aDepartment of Chemistry, Yale University, New Haven, CT 06511, USA. E-mail: matthew.molski@yale.edu; Fax: +1 203-432-3486; Tel: +1 203-432-8276
bWhitehead Institute for Biomedical Research, Nine Cambridge Center, Cambridge, MA 02142, USA. E-mail: jgoodman@wi.mit.edu; Fax: +1 617-258-7226; Tel: +1 617-258-5184
cDepartment of Biochemistry, Stanford University, Stanford, CA 94305, USA. E-mail: fcchou@stanford.edu; rhiju@stanford.edu; Fax: +1 650-723-5976; Tel: +1 650-723-7310
dDepartment of Biochemistry, University of Washington, Seattle, WA 98195, USA. E-mail: dabaker@u.washington.edu; Fax: +1 206-685-1792; Tel: +1 206-543-1295
eDepartments of Chemistry and of Molecular, Cellular and Developmental Biology, Yale University, New Haven, CT 06511, USA. E-mail: alanna.schepartz@yale.edu; Fax: +1 203-432-3486; Tel: +1 203-432-5094
First published on 21st September 2012
Abstract
Natural biopolymers fold with fidelity, burying diverse side chains into well-packed cores and protecting their backbones from solvent. Certain β-peptide oligomers assemble into bundles of defined octameric stoichiometry that resemble natural proteins in many respects. These β-peptide bundles are thermostable, fold cooperatively, exchange interior amide N–H protons slowly, exclude hydrophobic dyes, and can be characterized at high resolution using X-ray crystallography – just like many proteins found in nature. But unlike natural proteins, all octameric β-peptide bundles contain a sequence-uniform hydrophobic core composed of 32 leucine side chains. Here we apply rational design principles, including the Rosetta computational design methodology, to introduce sequence diversity into the bundle core while retaining the characteristic β-peptide bundle fold. Using circular dichroism spectroscopy and analytical ultracentrifugation, we confirmed the prediction that an octameric bundle still assembles upon a major remodelling of its core: the mutation of sixteen core β-homo-leucine side chains into sixteen β-homo-phenylalanine side chains. Nevertheless, the bundle containing a partially β-homo-phenylalanine core poorly protects interior amide protons from exchange, suggesting molten-globule-like properties. We further improve stability by the incorporation of eight β-homo-pentafluorophenyalanine side chains, giving an assembly with amide protection factors comparable to prior well-structured bundles. By demonstrating that their cores tolerate significant sequence variation, the β-peptide bundles reported here represent a starting point for the “bottom-up” construction of β-peptide assemblies possessing both structure and sophisticated function.
Introduction
Natural biopolymers fold with fidelity, can exist as oligomers or discrete complexes, and possess kinetic and thermodynamic signatures that distinguish them from most non-biological polymers and smaller molecules. In 2007, we reported that certain oligomers of β3-amino acids (β-peptides) fold into bundles of defined stoichiometry that resemble natural proteins in many respects.1–9 The high-resolution structures of four β-peptide bundles3,4,8,9 reveal a shared octameric fold composed of parallel and anti-parallel 314-helices, a salt-bridge-rich exterior, and a close-packed hydrophobic core. These β-peptide bundles are thermostable, undergo cooperative folding transitions, exchange interior amide N–H protons slowly, and exclude hydrophobic dyes, but contain a sequence-uniform core of 32 leucine side chains. Eliminating this side chain uniformity is a critical step toward the “bottom-up” construction of heterogeneous β-peptide assemblies possessing defined sizes, reproducible structures, and sophisticated function.10–12 Computational methods have also recently been used to predict β-peptide sequences that assemble into stable quaternary assemblies.13 Although these oligomers are not yet characterized structurally at high resolution, their sequences imply that they too possess uniform hydrophobic cores.13–28 Structurally characterized bundles composed of both α- and β-amino acids have also been reported.17,18,25In this work, we applied the Rosetta software package29,30 to predict β-peptide sequences that could effectively recapitulate the structurally characterized β-peptide bundle core using a mixture of leucine and non-leucine side chains. One such sequence (Acid-1YFF), containing an equal number of core β-homo-phenylalanine and β-homo-leucine residues, assembles into a 314-helical, relatively thermostable, octameric bundle. Despite this stability, Acid-1YFF displayed two properties associated with a molten globule state: rapid amide NH exchange and conformational heterogeneity as judged by NMR, indicating the potential for improved side chain organization within the core. It is well known that mixtures of phenylalanine and penta-fluorophenylalanine can improve protein stability when introduced into an otherwise all-hydrocarbon protein core.31–40 To test whether the thermodynamic stability and structural uniqueness of Acid-1YFF could be improved by fluorocarbon substitution, we synthesized an analogue (Acid-1YFF★) of Acid-1YFF containing β-pentafluoro-homo-phenylalanine (F5βPhe) at position 8. Acid-1YFF★ displays improved folding properties, resulting in a more conformationally distinct and stable core as judged by NMR. Acid-1YFF and Acid-1YFF★ are the first β-peptide bundles containing mixed sequence hydrophobic cores, suggesting further use of Rosetta and rational design principles as tools to remodel novel β-peptide bundles.
Results and discussion
The Rosetta software package29,30 has been applied successfully to improve protein thermal stability,41 design novel protein folds42 and enzymes43 and predict protein structure. More recently, the Rosetta approach has been extended to model and design RNA, another natural biopolymer.44–46 We began this work by using Rosetta to evaluate the core residue preference of the β-peptide bundle Acid-1Y, which had been characterized previously using X-ray crystallography. Acid-1Y assembles into a D2 symmetric octamer with two β-peptides in the asymmetric unit, each with three symmetry mates (Fig. 1A).4 We stripped out the core side chains and performed a full side chain conformer search to evaluate potential variants containing any of the twenty canonical amino acid side chains at any of eight core positions (residues 2, 5, 8 and 11 on the structurally two non-equivalent beta-peptides; the other 24 of the 32 core positions were constrained by symmetry).47 Rosetta predicted a uniform, all-leucine core as the most stable bundle among these 20 (ref. 8) variants, recovering the known sequence of Acid-1Y. Moreover, the Rosetta-modeled side-chain conformers superimposed with those seen in the Acid-1Y crystal structure in atomic detail (Table S1†), supporting use of Rosetta for further modeling.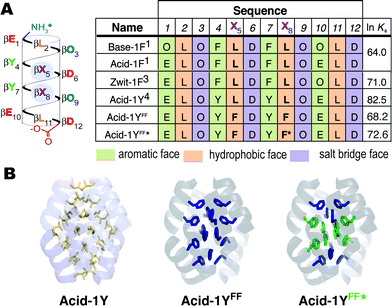 | ||
| Fig. 1 β-peptide bundles studied in this work. (A) Helical net diagram and sequences of Base-1F, Acid-1F, Zwit-1F, Acid-1Y, Acid-1YFF, and Acid-1YFF★. F represents β-homo-phenylalanine; F★ represents β-homo-pentafluorophenylalanine. Colors distinguish side chains on the aromatic (β-hY-containing), hydrophobic (β-hL containing) and salt bridge (β-hO- and β-hD-containing) faces. Also shown are ln Ka values characterizing each octameric assembly, as determined previously or in this work by SE-AU. (B) Ribbon representation of the crystal structure of Acid-1Y highlighting the packing of the leucine side chains, along with computationally predicted structure of Acid-1YFF and a color-coded guide to the locations of F5β-hPhe (green) in Acid-1YFF★. | ||
Next we searched for alternative side chains that could be accommodated in place of some or all of the β-homo-leucines within the bundle interior. While the fixed-backbone conformational search described above gave the all-leucine core as the optimal solution, a small void observed in the bundle center suggested that larger side chains might be tolerated at positions 5, 8 and/or 11 with minor backbone adjustments. We modeled the effects of introducing between four and eight alternative, proteinogenic side chains at these positions within Acid-1Y, and optimized the backbone torsion angles with a constraint potential tethering the angles to within ∼10° of their crystallographic values. Rosetta calculations predicted variant bundles would generally be poorer in energy than the starting bundle. However, a few were predicted to possess better hydrophobic packing of non-polar side-chains, as assessed by the sum of van der Waals and solvation energies. In particular, a variant containing β-homo-phenylalanine residues at positions 5 and 8 (Acid-1YFF, Fig. 1A) gave more favorable hydrophobic packing energy by ∼1 kT/monomer. Acid-1YFF contains 16 β-homo-leucine to β-homo-phenylalanine substitutions, placing an additional 48 carbon atoms into the bundle core. Nevertheless, the backbone atoms of the Acid-1YFF★ bundle were shifted by less than 0.8 Å RMSD from those in the starting bundle (Fig. 1B), suggesting that the bulky aromatic side chains in the interior could be accommodated without disrupting the octamer.
The β-peptide monomer Acid-1YFF was prepared using solid phase, microwave-assisted methods, purified to homogeneity by HPLC, and characterized initially using wavelength-dependent circular dichroism (CD) spectroscopy. As predicted, Acid-1YFF underwent a concentration-dependent increase in 314-helical structure (as judged by the molar residue ellipticity at 209 nm, MRE209)48 between 12 and 200 μM (Fig. 2a), consistent with an equilibrium between a partially structured monomer and a folded oligomer. A plot of MRE209vs. [Acid-1YFF] was first fit to a monomer–octamer equilibrium with ln Ka = 66.9 ± 0.5, suggesting that oligomerization of Acid-1YFF was less favorable than that of the Acid-1Y bundle (ln Ka = 82.5 ± 1.8).4 The fit, however, was imperfect [P = 2 × 10−10; see Table S3 and Fig. S1†], with a closer agreement at higher concentrations ([Acid-1YFF] > 50 μM) than at lower concentrations ([Acid-1YFF] < 50 μM). A plot of MRE209vs. [Acid-1YFF] was subsequently fit to alternative, three-state models containing either a dimeric (1–2–8) or tetrameric (1–4–8) intermediate (Fig. 2a). The resulting association constants for the 1–2–8 model were ln Ka1 = 18.4 ± 4.1 and ln Ka2 = 79.2 ± 9.8, and those for the 1–4–8 model were ln Ka1 = 36.8 ± 3.2 and ln Ka2 = 73.7 ± 11.2. Both three-state models fit the CD data substantially better than the original two-state monomer–octamer model (1–8), albeit with greater uncertainties in equilibrium constants due to the added fit parameters. Both fits gave predictions within the error of the data (Table S3 and Fig. S1†). Temperature-dependent CD studies also supported formation of a relatively stable Acid-1YFF bundle; the TM of a 200 μM solution of Acid-1YFF was 52 °C (Fig. 2b); this value is lower than that of Acid-1Y, whose TM was 82 °C at 150 μM.4 Taken together, these CD data established the relative stability of the Acid-1YFF bundle, but could not precisely define the stoichiometry of the putative intermediate or the difference in stability between Acid-1Y and Acid-1YFF.
![Self-association of designed β-peptide bundles. Circular dichroism spectra of Acid-1YFF (a and b) and Acid-1YFF★ (c and d) as a function of concentration (a and c) and temperature (b and d). Plots of MRE209 as a function of [β-peptide] were fit to a monomer–dimer–octamer (1–2–8) equilibrium (dotted line), a monomer–tetramer–octamer (1–4–8) equilibrium (dashed line) or monomer–octamer equilibrium (solid line). Inset: wavelength-dependent CD spectra of Acid-1YFF and Acid-1YFF★ (MRE in units of 103 deg cm2 dmol−1). The TM is defined as the maximum of a plot of δMRE209·δT−1versus temperature.](/image/article/2013/SC/c2sc21117c/c2sc21117c-f2.gif) | ||
| Fig. 2 Self-association of designed β-peptide bundles. Circular dichroism spectra of Acid-1YFF (a and b) and Acid-1YFF★ (c and d) as a function of concentration (a and c) and temperature (b and d). Plots of MRE209 as a function of [β-peptide] were fit to a monomer–dimer–octamer (1–2–8) equilibrium (dotted line), a monomer–tetramer–octamer (1–4–8) equilibrium (dashed line) or monomer–octamer equilibrium (solid line). Inset: wavelength-dependent CD spectra of Acid-1YFF and Acid-1YFF★ (MRE in units of 103 deg cm2 dmol−1). The TM is defined as the maximum of a plot of δMRE209·δT−1versus temperature. | ||
We therefore turned to sedimentation equilibrium analytical ultracentrifugation to more precisely characterize the stoichiometry and relative stability of the Acid-1YFF bundle. Sedimentation of Acid-1YFF at concentrations of 20, 80 and 200 μM was monitored at four speeds (36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 42000, 50000 and 60000 RPM). The AU data was fit to both two-state monomer-n-mer equilibrium models as well as three-state models proceeding through a dimer or tetramer intermediate (1–2–8 or 1–4–8, respectively). Poor fits with high RMSD values and larger and more systematic residuals were observed when n in the two-state model was set to any value other than 8 between 2 and 10 (see Fig. S2†). Both the 1–2–8 and the 1–4–8 model fit the AU data better than the two state 1–8 model (P < 10−6; F-test, see ESI†), in agreement with the CD analysis, and the 1–4–8 model produced the best fit. The ln Ka values calculated from the monomer–tetramer–octamer fit were ln Ka1 = 27.6 ± 0.2 and ln Ka2 = 68.2 ± 0.3, and the position of the monomer–octamer equilibrium (ln Ka2, 68.2 ± 0.3) agrees within error with the ln Ka2 value determined by CD (73.7 ± 11.2). Taken together, the CD and AU data suggest that Acid-1YFF assembly is a three state process involving a tetrameric intermediate towards a final octameric assembly, and that the oligomerization of Acid-1YFF is less favorable than that of Acid-1Y (ln Ka = 82.5 ± 1.8).4
000, 42000, 50000 and 60000 RPM). The AU data was fit to both two-state monomer-n-mer equilibrium models as well as three-state models proceeding through a dimer or tetramer intermediate (1–2–8 or 1–4–8, respectively). Poor fits with high RMSD values and larger and more systematic residuals were observed when n in the two-state model was set to any value other than 8 between 2 and 10 (see Fig. S2†). Both the 1–2–8 and the 1–4–8 model fit the AU data better than the two state 1–8 model (P < 10−6; F-test, see ESI†), in agreement with the CD analysis, and the 1–4–8 model produced the best fit. The ln Ka values calculated from the monomer–tetramer–octamer fit were ln Ka1 = 27.6 ± 0.2 and ln Ka2 = 68.2 ± 0.3, and the position of the monomer–octamer equilibrium (ln Ka2, 68.2 ± 0.3) agrees within error with the ln Ka2 value determined by CD (73.7 ± 11.2). Taken together, the CD and AU data suggest that Acid-1YFF assembly is a three state process involving a tetrameric intermediate towards a final octameric assembly, and that the oligomerization of Acid-1YFF is less favorable than that of Acid-1Y (ln Ka = 82.5 ± 1.8).4
Many de novo designed proteins exist as molten globules, and thus we hypothesized that the lower thermodynamic stability of the Acid-1YFF bundle compared to Acid-1Y might signal the presence of an undefined or heterogeneous hydrophobic core.49 Molten globules often bind and increase the fluorescence of dyes such as 1-anilino-8-naphthalenesulfonate (ANS) by factors ΔF > 100.49 By contrast, well-folded or unfolded proteins do not provide favorable ANS binding sites, and elicit little or no change in ANS fluorescence (ΔF < 10). All octameric β-peptide bundles reported thus far, including the Acid-1Y bundle, behave like well-folded proteins, causing minimal (<2-fold) changes in ANS fluorescence. Like previously characterized β-peptide bundles, even at concentrations as high as 200 μM (70% octamer), Acid-1YFF had little or no effect on the fluorescence of ANS (ΔF = 2) (Fig. S4†). This analysis suggests that the Acid-1YFF β-peptide bundle provides only limited access of solvent to its hydrophobic core.
Greater insight into the differences between the Acid-1YFF bundle relative to Acid-1Y was revealed by NMR experiments that monitored the rate of amide NH hydrogen/deuterium exchange. Unlike all previously characterized β-peptide bundles,4–6,9 the Acid-1YFF spectrum revealed no slowly exchanging amide NH protons; no amide NH resonances were visible as soon as 15 min after addition of D2O at 25 °C (Fig. 3a). In addition, relative to the NMR spectrum of Acid-1Y,4 the NMR spectrum of Acid-1YFF showed significant line broadening in the adjacent aromatic region. These data suggest that the aromatic sub-core of the Acid-1YFF bundle, although octameric and relatively thermostable, possesses significant conformational heterogeneity on the NMR time scale.
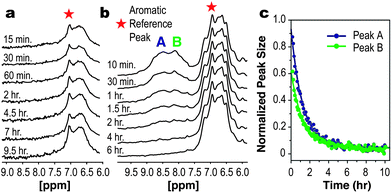 | ||
| Fig. 3 Selective substitution of β-homo-pentafluorophenylalanine improves β-peptide bundle stability as judged by NMR. (a) The amide NH regions of the Acid-1YFF and (b) Acid-1YFF★ β-peptide bundles after dissolution in D2O. In the case of Acid-1YFF, no amide NH resonances are visible within 15 min after addition of D2O. In the case of Acid-1YFF★, the amide signals persist for more than 1 h. (c) Integration of the resonances indicated in (B) normalized to the aromatic reference peak and fit to exponential decays to determine average exchange rate constants. | ||
Fluorocarbon side chains are more hydrophobic than their hydrocarbon counterparts50 and mixtures of phenylalanine and penta-fluorophenylalanine can improve protein stability when introduced into an otherwise all-hydrocarbon protein core.31–40 In certain cases, favorable face-to-face interactions between phenylalanine and pentafluorophenylalanine side chains account for improved stability,51–54 while in others steric and/or hydrophobic effects have been invoked.33,55 To evaluate whether the thermodynamic stability and structural uniqueness of Acid-1YFF could be improved by fluorocarbon substitution, we synthesized an analogue of Acid-1YFF containing β-pentafluoro-homo-phenylalanine (F5βPhe) at position 8 (Acid-1YFF★). Previous work has shown that β-peptide bundles containing β-homo-hexafluoroleucine at position 8 contain a discrete fluorous sub-domain and retain the characteristic β-peptide bundle fold.8
The Acid-1YFF★ sequence was synthesized and its assembly characterized by CD, SE-AU, ANS binding, and NMR to evaluate the effects of β-homo-pentafluorophenylalanine substitution on β-peptide bundle structure and stability. As expected, the CD spectrum of Acid-1YFF★ was characterized by concentration-dependent changes in MRE212, and the data was first fit to a monomer–octamer equilibrium with a ln Ka of 73.0 ± 0.5. Again the fits deviated from the experimental data in the lower concentration range ([Acid-1YFF★] < 25 μM (Fig. S1†) and could be improved by including a dimer or tetramer intermediate along the folding pathway. Fitting to a monomer–dimer–octamer equilibrium (1–2–8) resulted in ln Ka values of 18.4 ± 4.0 (ln Ka1) and 82.9 ± 2.3 (ln Ka2) (Fig. 2c), and fitting to a monomer–tetramer–octamer equilibrium (1–4–8) yielded ln Ka values of 41.4 ± 4.6 (ln Ka1) and 84.7 ± 3.0 (ln Ka2). Both fits agreed with the CD data within error (Fig. S1 and Table S3†). Although measured with large errors, the ln Ka2 values suggested that the Acid-1YFF★ octamer stability was greater than the initial Acid-1YFF design and now comparable to the starting Acid-1Y bundle with the all-leucine core (ln Ka of 82.5 ± 1.8). The temperature-dependence of the Acid-1YFF★ CD signal was consistent with improved stability; the TM of Acid-1YFF★ at 200 μM was 67 °C (Fig. 2d), a significantly higher value than the TM of 52 °C for Acid-1YFF at the same concentration.
We turned again to SE-AU to more precisely and accurately determine the relative association constants of the Acid-1YFF and Acid-1YFF★ bundles. The equilibrium sedimentation of Acid-1YFF★, performed at 20, 80 and 200 μM, was fit to two-state and three-state equilibrium models as described previously. Once again, the data fit best to a three-state model with a tetramer intermediate (1–4–8 model), although the 1–2–8 model was also an improvement over a two-state monomer–octamer equilibrium (P-value < 10−6 for both three state models according to the F-test; see ESI†). The ln Ka values calculated from the 1–2–8 fit were ln Ka1 = 11.6 ± 0.4 and ln Ka2 = 79.7 ± 1.3, and those from the 1–4–8 fit were ln Ka1 = 29.7 ± 0.2 and ln Ka2 = 72.6 ± 0.2 (Fig. S3†). Comparison to the AU analysis of the initial design indicates that, irrespective of model, the Acid-1YFF★ bundle is significantly more stable than the Acid-1YFF bundle.
The effects of perfluoro substitution on the aromatic β-peptide bundle core were studied further by monitoring the effect of Acid-1YFF★ on the intrinsic fluorescence of ANS (see ESI†). In previous work, designed α-amino acid proteins containing fluorous cores induced larger increases in ANS fluorescence than analogous proteins containing hydrocarbon cores; for example, the perfluorinated helical bundle α4-F2 increased ANS fluorescence by 5-fold.56 This value is greater than that observed in the presence of α4 (ΔF = 3), but less than that seen in the presence of classic molten globule states such as lactalbumin (ΔF ≥ 135).57,58 Whereas the Acid-1YFF bundle, like the Acid-1Y bundle, led to little or no increase in ANS fluorescence intensity (ΔF = 2-fold at [Acid-1YFF] = 200 μM), at an equivalent concentration the Acid-1YFF★ bundle caused ANS fluorescence to increase >19-fold (Fig. S4†). Interestingly, the ΔF of ANS observed in the presence of Acid-1YFF★ is comparable to that observed in the presence of the Zwit-8L*, in which eight leucine side chains were substituted by hexafluoroleucine (ΔF = 15), and whose structure is known at atomic detail.8 Thus, it may be that the increased fluorescence of ANS in the presence of the Acid-1YFF★ and Zwit-8L* results from an intrinsic affinity of ANS for fluorous domains, and not necessarily from a molten core.
Finally, H/D exchange NMR experiments gave the most incisive assessment of whether introduction of β-homo-pentafluorophenylalanine into the Acid-1YFF β-peptide bundle core would impart greater stability and structural uniqueness. Whereas the amide NH resonances in the Acid-1YFF bundle exchanged completely within 15 min, those of Acid-1YFF★ were better resolved and exchanged over the course of hours (Fig. 3b and c). The average rate of H/D exchange for two regions of the Acid-1YFF★ spectrum each fit a first-order decay function with a rate constant (kex) of 2.2 × 10−4 s−1. By comparison, the exchange rate constant for the random coil model poly-β-alanine (β3-homo-glycine) (krc) is 4.4 s−1. The ratio of these values (krc/kex), often defined as a protection factor P, is 2.0 × 104, a value comparable to that of previously reported β-peptide bundles (P between 9 × 103 and 6 × 104),5,6 including Acid-1Y (P = 6 × 104).4 This equivalence of the P values calculated for Acid-1YFF★ underscores the effect that the F5βPhe substitution has on the stability of the diversified hydrophobic core. The octamer bundle containing a mixed core of β-homo-leucine, β-homo-phenylalanine, and β-homo-pentafluorophenylalanine is characterized by monomer–octamer equilibrium constants and NMR amide hydrogen exchange rates that are comparable to previous bundles containing homogeneous cores.
Conclusions
The design of interfaces within a β-peptide bundle – a cooperatively folded structure that lacks even a single natural α-amino acid – is perhaps the most stringent test of our understanding of the principles that guide interactions between proteins. In this report, we apply the Rosetta computational algorithm and rational design to introduce sequence diversity into an octameric β-peptide bundle core that contains a uniform array of 32 leucine side chains. Using circular dichroism spectroscopy and analytical ultracentrifugation, we confirm that the Rosetta-remodeled bundle remains octameric upon changing sixteen core β-homo-leucine side chains into sixteen β-homo-phenylalanine side chains. We improve this design, bringing its stability to the level of the starting β-homo-leucine core, by introducing penta-fluorophenylalanine at selective positions within the bundle core. The repacking of de novo designed helical bundle proteins has been reported previously,59 although never in the context of β-peptides. This work represents the first example in which Rosetta is applied successfully to design a wholly non-biological polymer.60,61 By demonstrating that their hydrophobic cores will tolerate significant sequence variation, the β-peptide bundles reported here represent a starting point for the “bottom-up” construction of β-peptide assemblies possessing defined sizes, reproducible structures, and sophisticated function.Notes and references
- J. X. Qiu, E. J. Petersson, E. E. Matthews and A. Schepartz, J. Am. Chem. Soc., 2006, 128, 11338 CrossRef CAS.
- A. D. Bautista, C. J. Craig, E. A. Harker and A. Schepartz, Curr. Opin. Chem. Biol., 2007, 11, 685 CrossRef CAS.
- D. S. Daniels, E. J. Petersson, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 1532 CrossRef CAS.
- J. L. Goodman, E. J. Petersson, D. S. Daniels, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 14746 CrossRef CAS.
- E. J. Petersson, C. J. Craig, D. S. Daniels, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 5344 CrossRef CAS.
- J. L. Goodman, M. A. Molski, J. Qiu and A. Schepartz, ChemBioChem, 2008, 9, 1576 CrossRef CAS.
- E. J. Petersson and A. Schepartz, J. Am. Chem. Soc., 2008, 130, 821 CrossRef CAS.
- M. A. Molski, J. L. Goodman, C. J. Craig, H. Meng, K. Kumar and A. Schepartz, J. Am. Chem. Soc., 2010, 132, 3658 CrossRef CAS.
- C. J. Craig, J. L. Goodman and A. Schepartz, ChemBioChem, 2011, 12, 1035 CrossRef CAS.
- V. Nanda and R. L. Koder, Nat. Chem., 2009, 2, 15 CrossRef.
- J. W. Bryson, S. F. Betz, H. S. Lu, D. J. Suich, H. X. Zhou, K. T. O'Neil and W. F. DeGrado, Science, 1995, 270, 935 CAS.
- P. B. Harbury, J. J. Plecs, B. Tidor, T. Alber and P. S. Kim, Science, 1998, 282, 1462 CrossRef CAS.
- I. V. Korendovych, Y. H. Kim, A. H. Ryan, J. D. Lear, W. F. Degrado and S. J. Shandler, Org. Lett., 2010, 12, 5142 CrossRef CAS.
- S. Azeroual, J. Surprenant, T. D. Lazzara, M. Kocun, Y. Tao, L. A. Cuccia and J. M. Lehn, Chem. Commun., 2012, 48, 2292 RSC.
- L. Fischer and G. Guichard, Org. Biomol. Chem., 2010, 8, 3101 CAS.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. C. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240 CrossRef CAS.
- M. W. Giuliano, W. S. Horne and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9860 CrossRef CAS.
- W. S. Horne, J. L. Price, J. L. Keck and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 4178 CrossRef CAS.
- R. Kudirka, H. Tran, B. Sanii, K. T. Nam, P. H. Choi, N. Venkateswaran, R. Chen, S. Whitelam and R. N. Zuckermann, Biopolymers, 2011, 96, 586 CrossRef CAS.
- S. Kwon, A. Jeon, S. H. Yoo, I. S. Chung and H. S. Lee, Angew. Chem., Int. Ed., 2010, 49, 8232 CrossRef CAS.
- S. Kwon, K. Kang, A. Jeon, J. H. Park, I. S. Choi and H. S. Lee, Tetrahedron, 2012, 68, 4368 CrossRef CAS.
- S. Kwon, H. S. Shin, J. Gong, J. H. Eom, A. Jeon, S. H. Yoo, I. S. Chung, S. J. Cho and H. S. Lee, J. Am. Chem. Soc., 2011, 133, 17618 CrossRef CAS.
- I. M. Mandity, L. Fulop, E. Vass, G. K. Toth, T. A. Martinek and F. Fulop, Org. Lett., 2010, 12, 5584 CrossRef CAS.
- W. C. Pomerantz, V. M. Yuwono, R. Drake, J. D. Hartgerink, N. L. Abbott and S. H. Gellman, J. Am. Chem. Soc., 2011, 133, 13604 CrossRef CAS.
- J. L. Price, W. S. Horne and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 6376 CrossRef CAS.
- S. Segman-Magidovich, M. R. Lee, V. Vaiser, B. Struth, S. H. Gellman and H. Rapaport, Chem.–Eur. J., 2011, 17, 14857 CrossRef CAS.
- E. Torres, E. Gorrea, K. K. Burusco, E. Da Silva, P. Nolis, F. Rua, S. Boussert, I. Diez-Perez, S. Dannenberg, S. Izquierdo, E. Giralt, C. Jaime, V. Branchadell and R. M. Ortuno, Org. Biomol. Chem., 2010, 8, 564 CAS.
- E. Torres, J. Puigmarti-Luis, A. P. del Pino, R. M. Ortuno and D. B. Amabilino, Org. Biomol. Chem., 2010, 8, 1661 CAS.
- R. Das and D. Baker, Annu. Rev. Biochem., 2008, 77, 363 CrossRef CAS.
- A. Leaver-Fay, M. Tyka, S. M. Lewis, O. F. Lange, J. Thompson, R. Jacak, K. W. Kaufmann, P. D. Renfrew, C. A. Smith, W. Sheffler, I. W. Davis, S. Cooper, A. Treuille, D. J. Mandell, F. Richter, Y.-E. Ban, S. J. Fleishman, J. E. Corn, D. E. Kim, S. Lyskov, M. Berrondo, S. Mentzer, Z. Popovic, J. J. Havranek, J. Karanicolas, R. Das, J. Meiler, T. Kortemme, J. J. Gray, B. Kuhlman, D. Baker and P. Bradley, Methods Enzymol., 2011, 487, 545 CAS.
- B. Bilgicer and K. Kumar, Tetrahedron, 2002, 58, 4105 CrossRef CAS.
- B. Bilgicer, X. Xing and K. Kumar, J. Am. Chem. Soc., 2001, 123, 11815 CrossRef CAS.
- B. C. Buer, R. de la Salud-Bea, H. M. A. Hashimi and E. N. G. Marsh, Biochemistry, 2009, 48, 10810 CrossRef CAS.
- B. C. Buer, J. L. Meagher, J. A. Stuckey and E. N. G. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4810 CrossRef CAS.
- H. Y. Lee, K. H. Lee, H. M. Al-Hashimi and E. N. G. Marsh, J. Am. Chem. Soc., 2006, 128, 337 CrossRef CAS.
- K. H. Lee, H. Y. Lee, M. M. Slutsky, J. T. Anderson and E. N. G. Marsh, Biochemistry, 2004, 43, 16277 CrossRef CAS.
- A. J. Link, M. L. Mock and D. A. Tirrell, Curr. Opin. Biotechnol., 2003, 14, 603 CrossRef CAS.
- E. Neil and G. Marsh, Chem. Biol., 2000, 7, R153 CrossRef CAS.
- Y. Tang, G. Ghirlanda, W. A. Petka, T. Nakajima, W. F. DeGrado and D. A. Tirrell, Angew. Chem., Int. Ed., 2001, 40, 1494 CrossRef CAS.
- N. C. Yoder, D. Yuksel, L. Dafik and K. Kumar, Curr. Opin. Chem. Biol., 2006, 10, 576 CrossRef CAS.
- G. Dantas, B. Kuhlman, D. Callender, M. Wong and D. Baker, J. Mol. Biol., 2003, 332, 449 CrossRef CAS.
- B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, B. L. Stoddard and D. Baker, Science, 2003, 302, 1364 CrossRef CAS.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St.Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309 CrossRef CAS.
- P. Barth, J. Schonbrun and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15682 CrossRef CAS.
- R. Das and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14664 CrossRef CAS.
- R. Das, J. Karanicolas and D. Baker, Nat. Methods, 2010, 7, 291 CrossRef CAS.
- I. André, P. Bradley, C. Wang and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17656 CrossRef.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS.
- S. F. Betz, D. P. Raleigh and W. F. Degrado, Curr. Opin. Struct. Biol., 1993, 3, 601 CrossRef CAS.
- V. H. Dalvi and P. J. Rossky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13603 CrossRef CAS.
- G. Cornilescu, E. B. Hadley, M. G. Woll, J. L. Markley, S. H. Gellman and C. C. Cornilescu, Protein Sci., 2006, 16, 14 CrossRef.
- H. Zheng, K. Comeforo and J. M. Gao, J. Am. Chem. Soc., 2009, 131, 18 CrossRef CAS.
- H. Zheng and J. M. Gao, Angew. Chem., Int. Ed., 2010, 49, 8635 CrossRef CAS.
- B. C. Gorske and H. E. Blackwell, J. Am. Chem. Soc., 2006, 128, 14378 CrossRef CAS.
- M. G. Woll, E. B. Hadley, S. Mecozzi and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 15932 CrossRef CAS.
- R. Li, R. Gorelik, V. Nanda, P. B. Law, J. D. Lear, W. F. DeGrado and J. S. Bennett, J. Biol. Chem., 2004, 279, 26666 CrossRef CAS.
- K. J. Lumb and P. S. Kim, Biochemistry, 1995, 34, 8642 CrossRef CAS.
- K. H. Lee, H. Y. Lee, M. M. Slutsky, J. T. Anderson and E. N. Marsh, Biochemistry, 2004, 43, 16277 CrossRef CAS.
- J. W. Bryson, J. R. Desjarlais, T. M. Handel and W. F. DeGrado, Protein Sci., 1998, 7, 1404 CrossRef CAS.
- S. A. Sievers, J. Karanicolas, H. W. Chang, A. Zhao, L. Jiang, O. Zirafi, J. T. Stevens, J. Münch, D. Baker and D. Eisenberg, Nature, 2011, 475, 96 CrossRef CAS.
- P. D. Renfrew, E. J. Choi, R. Bonneau and B. Kuhlman, PLoS One, 2012, 7, e32637 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: β-Peptide design and synthesis and characterization using CD, SE-AU, and NMR. See DOI: 10.1039/c2sc21117c |
| This journal is © The Royal Society of Chemistry 2013 |